

The link between glomerular leakage of protein and progressive renal damage is a recurrent but incompletely understood observation in many clinical and experimental studies. High urinary protein levels in patients with glomerular disease predicts an adverse outcome. These patients frequently demonstrate tubular and interstitial fibrosis on renal biopsy. From this has emerged the hypothesis, supported by animal studies, that proteinuria leads to a fibrotic response within the tubulointerstitial compartment. The exact mechanism by which proteinuria exerts this effect is unknown.
Proteinuria and progressive injury
Many glomerular diseases cause proteinuria. The outcome of these diseases is in part dependent on the degree of proteinuria and the extent of tubulointerstitial fibrosis. There is increasing evidence that the proteinuria may lead directly to interstitial fibrosis and inflammation. Therapeutic interventions that reduce the level of proteinuria also decrease the level of fibrosis in animal models, and in clinical studies slow down the loss of renal function in patients with progressive disease. Tubular epithelial cells are at the interface between high concentrations of protein in the urinary space and inflammatory and fibrotic reactants generated within the interstitium. It is probable, therefore, that the tubular epithelium has a critical role in determining events within the tubulointerstitial compartment. Epithelial responses may simply be modified by an excess of protein in the tubular lumen or by exposure to specific plasma proteins, for example transferrin and lipoproteins. The presence of complement proteins in the tubular lumen is one such influence that may affect fibrotic and inflammatory responses within the interstitium.
Complement proteins, including the complex C5b-9, can be detected in the urine of patients with proteinuria. The high molecular weight of C5b-9 means that it is unlikely to enter the urinary space by glomerular filtration. It could be derived from shedding from the glomerulus in immune complex diseases such as membranous nephropathy. However, C5b-9 can be found in the urine from proteinuric patients who do not have C5b-9 in their glomeruli [1]. This suggests strongly that complement components are reaching the tubular lumen and that C5b-9 is being assembled at this site. In human biopsy studies and animal models, deposition of complement components can be identified at sites of tubulointerstitial injury [2,3]. Such observations support the notion that complement activation in the tubular compartment may contribute to proteinuria-associated tubulointerstitial injury, but do not establish a causal relationship.
The effect of complement activation on the renal tubule
Activation of the complement cascade generates a number of proinflammatory products. The fragment C5a is chemotactic and activates leucocytes; and C5b-9, the membrane attack complex, inserts into cell membranes and induces injury. Nucleated cells are relatively resistant to lysis by C5b-9 but undergo functional and structural changes depending on cell type. In vitro, C5b-9-stimulated proximal tubular epithelial cells (PTEC) undergo cytoskeletal and structural changes and produce proinflammatory mediators, including arachadonic acid products, IL-1 and TNF-α[4]. Mesangial cells stimulated by C5b-9 produce several pro-fibrotic factors including TGF-β, b-FGF, PDGF and matrix components such as Collagen IV [5,6]. It is reasonable to assume that tubular epithelial cells and interstitial fibroblasts may react in a similar manner to stimulation by C5b-9 (Fig. 1).
Fig. 1.
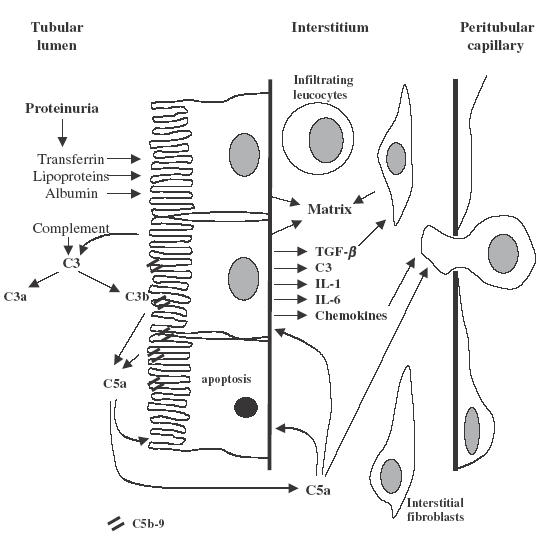
Schematic representation of the possible roles of complement in the generation of tubulointerstitial inflammation and fibrosis. Complement components derived from either the glomerular infiltrate or local synthesis by resident cells are activated on the luminal border of the PTECs with the generation of C5B-9 and C5a. These products activate PTECs leading to synthesis of new matrix, secretion of fibrogenic and proinflammatory cytokines and cell apoptosis. Additionally, C5a may have a direct chemoattractant effect.
What makes the proximal tubule vulnerable to attack by complement? The PTEC luminal border possesses convertase-like activity that leads to the enzymatic splitting of the pivotal component C3 [7]. Secondly, PTEC produce ammonia that can also activate C3 [8]. Finally, the luminal border of PTEC is relatively deficient in complement inhibitory proteins, proteins that inhibit convertase activity or block the assembly of C5b-9 [9]. All of these factors may contribute to the activation of complement by the renal epithelium.
In this issue Welch and coworkers suggest an additional mechanism by which complement activation contributes to proteinuria-associated tubulointerstitial injury [10]. If C5b-9 is being formed C5a will also be released. Proteinuric mice that are deficient in the C5a receptor have a reduced interstitial inflammatory response, including fewer infiltrating cells. This suggests that C5a is an important interstitial chemoattractant mediating cellular infiltration and tissue damage. The situation is, however, perhaps more complex than this. C5a receptor is expressed on the tubular epithelium [11] and C5a may therefore act directly the PTEC to induce release of other proinflammatory molecules and induce apoptosis (Fig. 1).
What is the source of complement within the tubule?
Most of the proteins required for complement activation have a molecular weight of 80–200 kDa, falling well within the range of proteins filtered in non-selective proteinuria. Therefore, one probable source of tubular complement proteins in proteinuric patients is the glomerular filtrate.
What has emerged more recently is the capability of the kidney itself to synthesize the complement proteins required to activate both the classical and alternative pathways [12]. A number of renal cell types can synthesize complement components, with a potential further contribution from infiltrating leucocytes. Tissue injury increases the level of C3 and C4 transcripts within the kidney [13,14]. Evidence of increased glomerular gene expression is seen in several of the immune-mediated glomerulonephritides, including IgA nephropathy and membranous nephropathy. However, it is clear from many studies of glomerular disease that the principal site of complement gene expression is in fact the renal tubule [15,16]. Synthesis may be stimulated by cytokines [17], secreted locally or from activated glomerular cells. Alternatively, non-physiological exposure of filtered proteins could stimulate C3 production by PTEC [18].
An unknown factor is the amount of complement the kidney produces in proteinuric states. Studies of transplant recipients receiving kidneys from donors with a different C3 allotype have indicated that up to 12% of circulating C3 may be of renal origin [19]. In this instance the most prominent source of C3 seems to be the tubular epithelium. The tubular cells therefore have significant capability to synthesize complement that may contribute to local injury.
Evidence form animal models
It follows that if complement plays a role in mediating proteinuria-associated tubulointerstitial injury, complement inhibition should reduce the extent of this injury. Complement inhibition using either cobra venom factor or soluble complement receptor-1 reduces tubulointerstitial injury and functional impairment in two models of proteinuria in the rat [20,21]. An alternative approach has been to use rats that have an inherited deficiency in C6 and therefore are unable to generate C5b-9. C6-deficient mice are protected from proteinuria-associated tubulointerstitial injury [22,23]. This confirms specifically the importance of the membrane attack complex, C5b-9, in the generation of injury as these rats will generate C5a normally. A final approach has been to reduce expression of complement inhibitors to determine whether this increases the degree of injury. Blocking the expression of the rat complement inhibitor crry with antisense oligonucleatides did indeed result in increased proteinuria-associated tubulointerstial injury [24].
In summary, a growing number of studies indicate that complement proteins, derived either from the glomerular filtrate or from local production, reach the tubular compartment. Complement activation, both in the urinary space and interstitium, generates the complement effector proteins C5b-9 and C5a. Consequently, complement activation at this site enhances the production of proinflammatory and pro-fibrotic cytokines, leucocyte infiltration and resident cell apoptosis, contributing to progressive tubulointerstitial injury and decline in renal function. The increasing availability of molecules that can block complement activation raises the possibility that this may be a target for future therapeutic intervention.
REFERENCES
- 1.Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol. 2000;11:700–7. doi: 10.1681/ASN.V114700. [DOI] [PubMed] [Google Scholar]
- 2.Mosolitis S, Magyarlaki T, Nagy J. Membrane attack complex and membrane cofactor protein are related to tubulointerstitial inflammation in various human glomerulopathies. Nephron. 1997;75:179–85. doi: 10.1159/000189529. [DOI] [PubMed] [Google Scholar]
- 3.Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. J Clin Invest. 1985;76:667–75. doi: 10.1172/JCI112020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David S, Biancone L, Caserta C, Bussolati B, Cambi V, Camussi G. Alternative pathway complement activation induces proinflammatory activity in human proximal tubular epithelial cells. Nephrol Dial Transplant. 1997;12:51–6. doi: 10.1093/ndt/12.1.51. [DOI] [PubMed] [Google Scholar]
- 5.Lovett DH, Haensch GM, Goppelt M, Resch K, Gemsa D. Activation of glomerular mesangial cells by the terminal membrane attack complex of complement. J Immunol. 1987;138:2473–80. [PubMed] [Google Scholar]
- 6.Torbohm I, Schonermark M, Wingen AM, Berger B, Rother K, Hansch GM. C5b-8 and C5b-9 modulate the collagen release of human glomerular epithelial cells. Kidney Int. 1990;37:1098–104. doi: 10.1038/ki.1990.91. [DOI] [PubMed] [Google Scholar]
- 7.Biancone L, David S, Della Pietra V, Montrucchio G, Cambi V, Camussi G. Alternative pathway activation of complement by cultured human proximal tubular epithelial cells. Kidney Int. 1994;45:451–60. doi: 10.1038/ki.1994.59. [DOI] [PubMed] [Google Scholar]
- 8.Clark EC, Nath KA, Hostetter MK, Hostetter TH. Role of ammonia in progressive interstitial nephritis. Am J Kidney Dis. 1991;17:15–9. [PubMed] [Google Scholar]
- 9.Ichida S, Yuzawa Y, Okada H, Yoshioka K, Matsuo S. Localization of the complement regulatory proteins in the normal human kidney. Kidney Int. 1994;46:89–96. doi: 10.1038/ki.1994.247. [DOI] [PubMed] [Google Scholar]
- 10.Welch TR, ch TR, Frenzke M, Witte D, Davis AE., III C5a is important in the tubulointerstitial component of experimental immune complex glomerulonephritis. Clin Exp Immunol. 2002;130:43–48. doi: 10.1046/j.1365-2249.2002.01957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zahedi R, Braun M, Wetsel RA, et al. The C5a receptor is expressed by human renal proximal tubulart epithelial cells. Clin Exp Immunol. 2000;121:226–33. doi: 10.1046/j.1365-2249.2000.01249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sacks SH, Zhou W, Sheerin NS. Complement synthesis in the injured kidney: does it have a role in immune complex glomerulonephritis? J Am Soc Nephrol. 1996;7:2314–9. doi: 10.1681/ASN.V7112314. [DOI] [PubMed] [Google Scholar]
- 13.Sacks SH, Zhou W, Andrews PA, Hartley B. Endogenous complement C3 synthesis in immune complex nephritis. Lancet. 1993;342:1273–4. doi: 10.1016/0140-6736(93)92362-w. [DOI] [PubMed] [Google Scholar]
- 14.Welch TR, Beischel LS, Witte DP. Differential expression of complement C3 and C4 in the human kidney. J Clin Invest. 1993;92:1451–8. doi: 10.1172/JCI116722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montinaro V, Gesualdo L, Ranieri E, Monno R, Grandaliano G, Schena FP. Renal cortical complement C3 gene expression in IgA nephropathy. J Am Soc Nephrol. 1997;8:415–25. doi: 10.1681/ASN.V83415. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki O, Zhou W, Miyazaki M, et al. Intraglomerular C3 synthesis in rats with passive Heymann nephritis. Am J Pathol. 1997;151:1249–56. [PMC free article] [PubMed] [Google Scholar]
- 17.Brooimans RA, Stegmann AP, van Dorp WT, et al. Interleukin 2 mediates stimulation of complement C3 biosynthesis in human proximal tubular epithelial cells. J Clin Invest. 1991;88:379–84. doi: 10.1172/JCI115314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang S, Sheerin NS, Zhou W, Brown Z, Sacks SH. Apical proteins stimulate complement synthesis by cultured human proximal tubular epithelial cells. J Am Soc Nephrol. 1999;10:69–76. doi: 10.1681/ASN.V10169. [DOI] [PubMed] [Google Scholar]
- 19.Tang S, Zhou W, Sheerin NS, Vaughan RW, Sacks SH. Contribution of renal secreted complement C3 to the circulating pool in humans. J Immunol. 1999;162:4336–41. [PubMed] [Google Scholar]
- 20.Morita Y, Nomura A, Yuzawa Y, et al. The role of complement in the pathogenesis of tubulointerstitial lesions in rat mesangial proliferative glomerulonephritis. J Am Soc Nephrol. 1997;8:1363–72. doi: 10.1681/ASN.V891363. [DOI] [PubMed] [Google Scholar]
- 21.Nomura A, Morita M, Maruyama S, et al. Role of complement in acute tubulointerstial injury of rats with aminoglycoside nephrosis. Am J Pathol. 1997;151:539–47. [PMC free article] [PubMed] [Google Scholar]
- 22.Nangaku M, Pippin J, Couser WG. Complement membrane attack complex (C5b-9) mediates interstitial disease in experimental nephrotic syndrome. J Am Soc Nephrol. 1999;10:2323–31. doi: 10.1681/ASN.V10112323. [DOI] [PubMed] [Google Scholar]
- 23.Nangaku M, Pippin J, Couser WG. C6 mediates chronic progression of tubulointerstitial damage in rats with remnant kidneys. J Am Soc Nephrol. 2002;13:928–36. doi: 10.1681/ASN.V134928. [DOI] [PubMed] [Google Scholar]
- 24.Hori Y, Yamada K, Hanafusa N, et al. Crry, a complement regulatory protein, modulates renal interstitial disease induced by proteinuria. Kidney Int. 1999;56:2096–106. doi: 10.1046/j.1523-1755.1999.00765.x. [DOI] [PubMed] [Google Scholar]