

Abstract
Although intestinal epithelial cells are known to up-regulate the expression of several chemokine genes in response to the stimulation with B. fragilis enterotoxin (BFT), there has been little understanding on the cellular mechanisms of BFT-induced mucosal inflammation. To test whether nuclear transcriptional factor-kappa B (NF-κB) is involved in the process, we stimulated intestinal epithelial cells with BFT, and evaluated the signalling NF-κB pathways. BFT increased signals of NF-κB in HT-29 and T84 epithelial cell lines as well as primary human colon epithelial cells. NF-κB molecules activated by BFT stimulation were composed of p65 and p50 heterodimers. In contrast, BFT decreased the signals of IκBα and IκBɛ, as assessed by immunoblot. Super-repressors of IκBα, IκB kinase (IKK)β, and NF-κB inducing kinase (NIK) inhibited an up-regulated transcription of downstream target gene (CXCL8) of NF-κB. Moreover, blocking the activation of NF-κB by MG-132 or antisense p50 oligonucleotide transfection resulted in down-regulated expression of chemokines such as CXCL1, CXCL8, and CCL2 in BFT-stimulated HT-29 cells. In addition, NF-κB inhibition suppressed the BFT-induced neutrophil transepithelial migration in T84 cells. These results indicate that NF-κB can be a central regulator of chemokine gene expression in BFT-stimulated intestinal epithelial cells and may be an important regulator of neutrophil migration.
Keywords: Bacteroides fragilis, chemokines, epithelial cells, IκB, NF-κB
INTRODUCTION
Enterotoxigenic Bacteroides fragilis (ETBF) has been associated with noninvasive diarrheal disease in animals, young children and adults [1,2]. The inflammatory response to ETBF is attributed to the expression of a ∼20 kD heat-labile metalloprotease toxin (B. fragilis enterotoxin, or BFT). BFT was reported to induce morphological changes of intestinal epithelial cells [3–6]. Histological examination revealed that the inoculation of ETBF or BFT to intestinal lumen caused mucosal inflammation characterized by the infiltration of neutrophils [6,7]. These studies suggest that mucosal inflammatory signals may be initiated from intestinal epithelial cells in response to BFT stimulation. Our recent report demonstrating that BFT could induce the expression and basolateral secretion of CXC chemokines such as CXCL8 (interleukin-8) and CXCL1 (growth-related oncogene-α) supports this hypothesis [8]. However, the exact mechanism of BFT-induced mucosal inflammation has not been clarified.
Relatively limited expression of CXC chemokine genes was found to be up-regulated in intestinal epithelial cells after stimulation with BFT [8,9]. This finding suggests that a common signal transduction pathway might underlie the up-regulated expression of the CXC chemokine genes that are activated in intestinal epithelial cells in response to BFT stimulation.
Many of the genes that are activated in intestinal epithelial cells after bacterial infection are target genes of the transcription nuclear factor-kappa B (NF-κB). NF-κB is a dimeric transcription factor composed of homodimers or heterodimers of Rel protein, of which there are five family members in mammalian cells (i.e. RelA (p65), c-Rel, Rel B, NF-κB1 (p50), and NF-κB2 (p52)) [10]. NF-κB dimers are held in the cytoplasm in an inactive state by inhibitory proteins, the IκBs [11]. IκBs are preferentially asso-ciated with various Rel family protein dimers (e.g. IκBα and IκBβ predominantly associated with p65/p50 and p50/c-Rel heterodimers, IκBɛ preferentially associated with p65 and c-Rel homodimers, and Bcl-3 associates with nuclear p50 and p52 homodimers). Heterodimers of p65 and p50 are the predominant NF-κB subunits that translocate to the nucleus after cytokine stimulation or enteroinvasive bacterial infection of intestinal epithelial cells [12,13]. However, the role of NF-κB in the BFT-induced signal transduction in intestinal epithelial cells has not been clarified.
The present study thus tested whether intestinal epithelial cells could use a NF-κB signal transduction pathway to activate the host's chemokine response to BFT stimulation. We report here that BFT stimulation can induce NF-κB signals in intestinal epithelial cells and the activated NF-κB signals may be a regulator of the activation of chemokine genes and neutrophil trans-epithelial migration.
MATERIALS AND METHODS
Cell culture and purification of B. fragilis enterotoxin
HT-29 human colon epithelial cells (ATCC HTB 38) were grown in DME (pH 7·4, Sigma Chemical Co., St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS, Gibco BRL, Grand Island, NY, USA), 2 mm glutamine, 25 mm HEPES, and antibiotics (100 unit/ml of penicillin and 100 μg/ml of streptomycin). T84 colon epithelial cells (ATCC CCL-248) were grown in 50% DMEM and 50% Ham's F-12 media supplemented with 5% FBS and 2·5 mm glutamine, as described before [14].
Human colon epithelial cells were obtained from normal-appearing mucosa of surgically resected colons from the individuals with colon cancer, as described previously [8]. Freshly isolated colon epithelial cells were cultured at 2 × 106/ml in RPMI-1640 media, supplemented with 10% FBS, 2 mm glutamine, and antibiotics (100 unit/ml of penicillin and 100 μg/ml of streptomycin). Epithelial cell preparations had less than 5% of contaminating B cells and monocytes/macrophages as assessed by flow cytometry using CD19/20 and CD14 as markers.
Human neutrophils were obtained from healthy volunteers by means of dextran sedimentation and Ficoll-Hypaque (Histopaque 1077, Sigma Chemical Co.) density gradient separation as previously described [15].
BFT was purified from a highly toxigenic strain of ETBF as previously described [8]. Briefly, B. fragilis was grown anaerobically at 37°C in prereduced brain heart infusion broth for 18 h. BFT was purified from culture supernatants by sequential ammonium sulphate precipitation, ion-exchange chromatography on Q-Sepharose (Pharmacia Biotech, Brussels, Belgium), and hydrophobic interaction chromatography on phenyl-agarose (Sigma Chemical Co.). Purification of the toxin was monitored by its cytotoxic effect on HT-29 cells characterized by cell rounding [3,5]. Buffers were prepared using LPS-free water (Baxter Healthcare Corp., Deerfield, IL, USA). The activity of lipopolysaccharide (LPS) in BFT solutions (1 mg/ml) was less than one endotoxin unit/ml (quantitative chromogenic limulus amebocyte lysate; BioWhittaker, Walkersville, MD, USA). BFT was frozen at − 20°C in aliquots immediately after purification.
Electrophoretic mobility shift assays
Cells were harvested, and nuclear extracts were prepared as described [8,16,17] The concentrations of proteins in the extracts were determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Electrophoretic mobility shift assays (EMSA) were performed according to the protocol of the manufacturer (Promega, Madison, WI, USA). In brief, 5 μg of nuclear extracts were incubated for 30 min at room temperature with γ32P-labelled oligonucleotide probe corresponding to a consensus NF-κB binding site. After incubation, bound and free DNAs were resolved on 5% native polyacrylamide gels as described previously [8,16].
Supershift EMSA
Supershift assays were used to identify the specific members of the NF-κB family which could be activated by stimulation of BFT. EMSA was performed as described above except that rabbit antibodies (1 μg/reaction) against that NF-κB proteins p50, p52, p65, c-Rel, and Rel B (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were added during the binding reaction period.
Immunoblots for IκBs
Confluent monolayers in 6-well plates were washed with ice-cold PBS, and lysed in a 0·5-ml/well lysis buffer (150 mm NaCl, 20 mm Tris, pH 7·5, 0·1% Triton X-100, 1 mm PMSF, 10 μg/ml aprotonin) as described previously [16,17]. Protein concentrations in the lysates were determined by the Bradford method (Bio-Rad). Fifteen μg protein/lane was size-fractionated on a denaturing, nonreducing 6% polyacrylamide minigel (Mini-PROTEIN II; Bio-Rad), and electrophoretically transferred to a nitrocellulose membrane (0·1-μm pore size). Specific proteins were detected using mouse antihuman IκBα and IκBɛ (Santa Cruz Biotechnology) as a primary antibody, and peroxidase-conjugated antimouse IgG (Transduction Laboratories, Lexington, Ky, USA) as a secondary antibody. Specifically bound peroxidase was detected by enhanced chemiluminescence (ECL system; Amersham Life Science, Buckinghamshire, England) and exposure to X-ray film (XAR5; Eastman Kodak Company, Rochester, NY, USA) for 10–30 s.
Plasmids and transfections
A mammalian expression vector encoding a haemagglutinin (HA) epitope-tagged mutant IκBα (IκBα-AA) having substitutions of serine residues at positions 32 and 36 with alanine residues (a gift of Joseph A. DiDonato, Cleveland Clinic Foundation, Cleveland, OH) [17] and an expression vector encoding FLAG-tagged IκB kinase (IKK)α in which lysine at position 44 was replaced by alanine (IKKα-AA), a gift of Dr Mercurio (Signal Pharmaceuticals, San Diego, CA, USA) [18], were used to block NF-κB activation as described before [8]. An expression vector for a NF-κB-inducing kinase (NIK) catalytic mutant that has a double replacement of alanine residues to lysine residues at positions 429 and 430 and lacks kinase activity (NIK-AA) was a gift of Dr Karin, of the University of California, San Diego [12]. pCXCL8-luciferase, p2x NF-κB-luciferase, pβ-actin-luciferase and pRSV-β-galactosidase transcriptional reporters were kindly provided by Dr Kagnoff of the University of California, San Diego [12] Cells in six-well dishes were transfected with 1·5 μg of plasmid DNA, using Lipofectamine Plus (Gibco BRL), according to the manufacturer's instructions [16]. The transfected cells were incubated for 48 h at 37°C in a 5% CO2 incubator. Cells were incubated with BFT or recombinant human TNF-α (R & D Systems, Minneapolis, MN, USA) as a control for 6 h, after which cells were harvested and whole cell lysates were prepared as described before [8]. Briefly, cells were lysed at 4°C for 25 min in a whole cell lysis buffer (0·1 m KPO4, 0·1 m DTT, 0·5% Triton X-100, pH 7·8). Luciferase activity was determined and normalized relative to β-galactosidase expression in accordance with the manufacture's instruction (Tropix Inc., Bedford, MA, USA). Light release was quantified for 10 s using a luminometer (MicroLumat Plus, Berthold GmbH & Co. KG, Bad Wildbad, Germany). β-galactosidase activity was determined using the chemilumines-cent substrate AMPGD (3-(4-methoxyspiro1,2-dioxetane-3, 2′-tricy1o3.3.1.1]decan]-4-yl) phenyl-β-d-galactopyranoside; Tropix Inc.) as described before [19]. Light release was induced by the addition of 50 μl 0·2 N NaOH containing 10% Emerald enhancer (Tropix Inc.), and quantified for 10 s in a luminometer. Increased transfection from pCXCL8, p2x NF-κB and pβ-actin promoters in response to BFT stimulation was calculated by comparing ratios of luciferase to β-galactosidase activities in the cells cotransfected with pCXCL8-luciferase and pRSV-β-galactosidase, p2x NF-κB- luciferase and pRSV-β-galactosidase, or pβ-actin-luciferase and pRSV-β-galactosidase, respectively. Non-transfected cells were used as a background control [12,20].
Antisense p50 oligonucleotide transfection
The experiments using antisense p50 oligonucleotide transfection were performed as described before [21]. Phosphothioate- modified single-stranded p50 oligonucleotides were produced commercially (Gibco BRL). p50 antisense oligonucleotide and sense oligonucleotide targeted the ATG start codon of the p50 mRNA. The sequence of the p50 antisense oligonucleotide was 5′-GGA TCA TCT TCT GCC ATT CTG-3′. The sequence of the p50 sense oligonucleotide was 5-CAG AAT GGC AGA AGA TGA TCC-3′. HT-29 monolayers were treated with oligonucleotide (final concentration 0·5 μm) using a cationic liposome, a commercially available transfection reagent, DOTAP (N-1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethyl ammonium methylsulphate; 15 μl/ml; Boehringer-Mannheim, Mannheim, Germany) for 60 h, to improve stability and intracellular delivery of oligonucleotide, as described before [21]. Cells were trypsinized and plated again at a density 5 × 105 cell/ml in 24-well plates. A final concentration of 0·5 μm of oligonucleotide was added to the cells and incubated for an additional 18 h. The sense sequence has been shown previously to be unable to inhibit NF-κB activity [21]. The transfected monolayers were stimulated with BFT for 12 h and the culture medium was collected. Each chemokine was measured using enzyme-linked immunosorbent assay (ELISA).
Quantitative RT-PCR analysis and chemokine ELISA
Confluent monolayers of HT-29 cells in 24-well plates were stimulated with BFT for the indicated times, after which total cellular RNA was extracted from the cells using an acid guanidinium thiocyanate-phenol-chloroform method [22,23]. Oligonucleotide primers used for PCR amplification and the sizes of the PCR products obtained from target cellular RNA and synthetic standard RNA are described in the previous report [24]. Quantitative RT-PCR using internal standard was used to quantify chemokine mRNA levels, as assessed previously [8,22,23]. Synthetic standard RNA was kindly provided by Dr Kagnoff of the University of California, San Diego. PCR amplification consisted of 35 cycles of 1-min denaturation at 95°C, 2·5-min annealing and extension at either 60°C (CXCL1 and CXCL8), 65°C (CCL2) or 72°C (β-actin). Chemokine mRNA levels of 5 × 103 molecules/μg of total RNA were considered positive. Although lower levels could be detected and quantified, they were considered unlikely to be biologically meaningful as they would on average reflect <1 mRNA transcript/20 cells [25].
Chemokines in culture supernatants were assayed by ELISA. Before chemokine proteins were measured, the supernatants were filtered through a 0·22-μm filter to remove any contaminants. The levels of human GRO-α, IL-8 and MCP-1 were determined by Quantikine immunoassay kits (R & D Systems). Chemokine proteins were tested in triplicate. The detection limit is the same for the three chemokines, 15 pg/ml.
Chemotaxis assay for neutrophil transepithelial migration
The physiologically directed (basolateral-to-apical) neutrophil transepithelial migration assay has been detailed previously [26,27]. Before stimulation with BFT, T84 monolayers in basal surface of transwell filter (5 μm-pore size, 0·33 cm2 of surface area, Costar, Cambridge, Mass., USA) were washed and kept for 3 h in serum-free medium. In this step, the baseline electrical resistance across T84 monolayers was 1434 ± 208 Ω× cm2 (mean ± SD; n = 5). The monolayers stimulated with BFT for 6 h were then transferred, apical side down, into a 24-well tissue culture tray with 1·0 ml of Hanks’ balanced salt solution (HBSS) containing 0·1% glucose. Isolated human neutrophils (106 in 50 μl) plus 150 μl of HBSS were added to the basolateral side (upper reservoir) of each monolayer and incubated 2 h at 37°C. Positive controls for transmigration assay were represented by the transmigration response to 1 μm formyl-Met-Leu-Phe (fMLP).
Transmigration of neutrophils was assessed by quantification of myeloperoxidase as described previously [26,27]. Briefly, at the end of the experiment, T84 monolayers were cooled to 4°C, washed with HBSS and solubilized in HBSS containing 0·5% Triton X-100 to determine the number of monolayers-associated neutrophils. Myeloperoxidase activity was determined by an ELISA kit in accordance with the manufacture's instruction (R and D Systems). To quantify the number of neutrophils that had completely transmigrated the epithelial monolayers and were present in the lower reservoir, Triton X-100 was added directly to the reservoir and myeloperoxidase activity was measured as described above. Standard curves were generated for each experiment. T84 monolayers were found to have no significant myeloperoxidase activity in the absence of neutrophils.
In our preliminary studies, neutrophil transmigration by BFT stimulation (100 ng/ml) was ∼22% as effective as that which occurred in response to the potent chemoattractant 100 nm fMLP (8·5 ± 1·1) × 104 and (37·8 ± 7·2) × 104 neutrophils equivalents/monolayers for BFT and fMLP, respectively; mean ± SEM, n = 5]. This number represents neutrophils that have completely transmigrated the monolayers and are present in the opposite reservoir from which they were added as well as neutrophils that have infiltrated the epithelial monolayers but not yet impaled the tight junctions, thus remaining associated with the monolayer. The number of neutrophils that remained membrane-associated at the end of the experiment was quite low (1·6 ± 0·7) × 104 neutrophils equivalents/monolayers; mean ± SEM, n = 10].
Statistical analysis
Data are presented as the mean ± standard deviation (SD) for quantitative RT-PCR, and the mean ± standard error of the means (SEM) for ELISA and neutrophil transepithelial migration assay. Wilcoxon's rank sum test was used for statistical analysis. A P-value less than 0·05 was considered statistically significant.
RESULTS
B. fragilis enterotoxin induces NF-κB activation and IκB degradation in human colon epithelial cells
The transcription factor NF-κB plays a major role in the transcriptional activation of chemokine mRNA expression which is known to be increased by infection of human colon epithelial cells with enteric pathogens [12,16,28]. To determine whether BFT activates NF-κB in human colon epithelial cells, DNA binding studies were performed using cell extracts after stimulation of HT-29 or primary intestinal epithelial cells with BFT. Stimulation of these cells with BFT increased NF-κB DNA binding, as shown by EMSA (Fig. 1a,c).
Fig. 1.

NF-κB activation and IκB degradation in human intestinal epithelial cells stimulated with B. fragilis enterotoxin. Human colon epithelial HT-29 cell line (a) and primary colon epithelial cells (c) were stimulated with B. fragilis enterotoxin (100 ng/ml). NF-κB DNA binding activity was assessed by EMSA at the indicated times. Immunoblots for concurrent IκBα and IκBɛ levels under the same condition for HT-29 cells are provided beneath each EMSA time point (b). The results are representative of five repeated experiments. (+) represents positive control in which HT-29 cells were treated with TNF-α (20 ng/ml) for 1 h (−) represents negative control.
The magnitude of the NF-κB response was dependent on the concentration of stimulating BFT per epithelial cell. The stimulation of HT-29 cells with increasing concentrations of BFT was paralleled by increased luciferase activity in HT-29 cells transfected with 2 × NF-κB promoter plasmid. At the concentrations of 1, 10, 100 and 1000 ng/ml, reporter gene activity of 2 × NF-κB increased 1·2, 3·6, 7·2 and 5·8-fold at 6 h after stimulation, respectively, relative to those of nonstimulated controls (mean value, n = 3).
One of the major pathways for NF-κB activation involves the phosphorylation of IκBs, which is followed by IκBs degradation and the subsequent migration of NF-κB dimers from the cytoplasm to the nucleus. Therefore, we assayed the kinetics of IκBα and IκBɛ degradation by immunoblot analysis in HT-29 cells after BFT stimulation (100 ng/ml). As shown in Fig. 1b, degradation of IκBα (0 h, 100%; 1 h, 74·8%; 3 h, 65%; 6 h, 62·7%; 12 h, 51·5%) and IκBɛ (0 h, 100%; 1 h, 64·8%; 3 h, 66·5%; 6 h, 79·9%; 12 h, 73·1%) was observed in BFT-stimulated HT-29 cells (% means the band intensities measured by image analyser (Vilber Lourmat Bio1D V6·32, Bio-rad)).
NF-κB exists as dimeric complexes, either homo- or hetero-dimers. To identify the specific NF-κB subunits that comprise the NF-κB signal detected by EMSAs in BFT-stimulated HT-29 cells, supershift assay was performed. Specific antibodies to p50, p52, p65, c-Rel, and Rel B were used for these experiments. Supershift studies demonstrated that antibodies to p65 shifted the entire signal and that antibodies to p50 also caused a significant shift. However, antip52, antic-Rel, or anti-Rel B antibodies did not shift the NF-κB signals (Fig. 2). Similar findings were found with T84 cells (data not shown). These results suggest that NF-κB activation by BFT stimulation may be mediated predominantly by heterodimers of p65/p50.
Fig. 2.
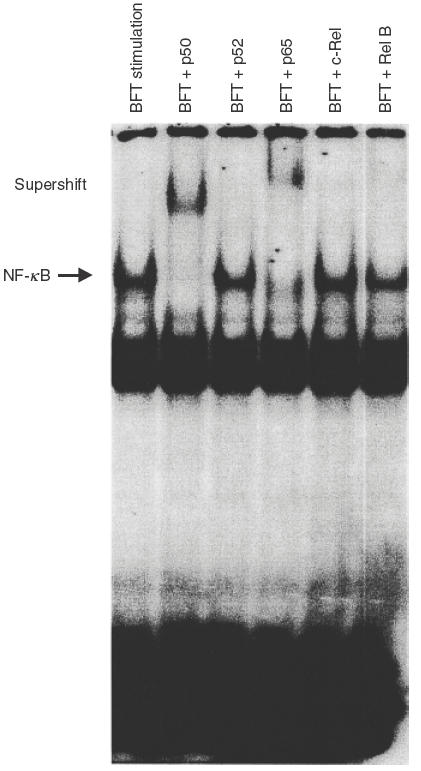
Activation of specific NF-κB subunits in HT-29 cells stimulated with B. fragilis enterotoxin. Supershift assays were performed using antibodies to p50, p52, p65, c-Rel, and Rel B. Antibodies to p50 and p65 shift the entire NF-κB signals. Anti-p50, c-Rel and Rel B did not show the shifts. The results are representative of three repeated experiments.
B. fragilis enterotoxin-induced activation of NF-κB and CXCL8 reporter genes was inhibited by IκBα, IKKβ and NIK super-repressors
We previously demonstrated that stimulation with BFT significantly increased luciferase activity in HT-29 cells transfected with CXCL8 and 2 × NF-κB promoter plasmids [8]. In addition, we reported that transfection of dominant-negative IκBα or IKKβ inhibit the activity of NF-κB in HT-29 cells stimulated with BFT. Based on these results, we asked whether activation of NIK was also a component of signalling pathway. Activation of the CXCL8 and NF-κB transcriptional reporters was inhibited in the cells cotransfected with the IκBα, IKKβ or NIK super-repressor plasmids (Fig. 3), but not in the cells cotransfected with control plasmid (data not shown). However, the inhibition of CXCL8 and NF-κB transcriptional reporters by NIK super-repressor was less than that by IκBα or IKKβ super-repressors. These results suggest that NF-κB activation and CXCL8 expression of intestinal epithelial cells by BFT stimulation may be involved in other pathways although NIK can, in partially, be involved.
Fig. 3.
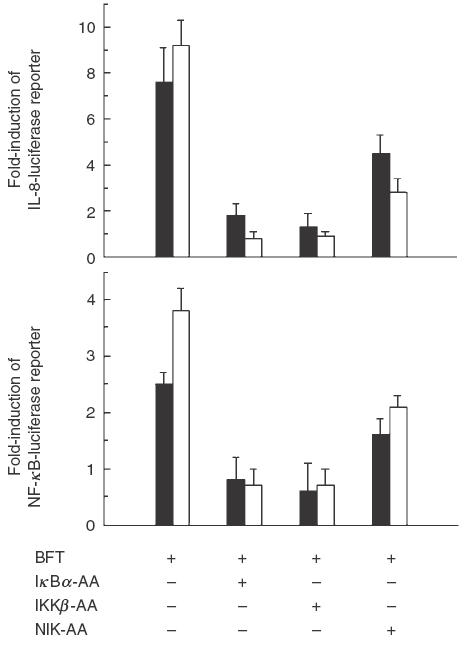
The effects of IκBα, IKKβ and NIK super-repressors on reporter gene activation in B. fragilis enterotoxin-stimulated HT-29 cells. HT-29 cells were transfected with pCXCL8- or p2x NF-κB-luciferase transcriptional reporters together with IκBα-AA, IKKβ-AA, or NIK-AA expression vectors, as indicated. 48 h later, the transfected cells were stimulated with BFT (100 ng/ml, ▪) or TNF-α (20 ng/ml, □) for 6 h. Data are expressed as the mean fold induction ± SEM in luciferase activity relative to nonstimulated controls (n = 7)
The inhibition of NF-κB suppresses chemokine expression in intestinal epithelial cells stimulated with B. fragilis enterotoxin
Given that chemokine genes are known to be major components of the epithelial cell response to BFT stimulation, we further evaluated whether blocking the activation of NF-κB could influence the expression of chemokine genes. HT-29 cells were pretreated with chemical NF-κB inhibitor MG-132 (50 μm in DMSO, Calbiochem-Novabiochem Corp., San Diego, CA, USA) for 1 h and were stimulated with BFT (100 ng/ml). MG-132-treated cells almost completely inhibited the up-regulated expression of CXCL8, CXCL1 and CCL2 mRNA by BFT stimulation (Table 1). In these experiments, the β-actin mRNA levels in the BFT-stimulated cells remained relatively constant in the presence or absence of MG-132 (~5 × 106 transcript/μg RNA).
Table 1.
Quantification of chemokine mRNA in HT-29 intestinal epithelial cells stimulated with B. fragilis enterotoxin in the presence of pretreating MG-132*
| Control | BFT-stimulated | Pretreated MG-132 + BFT | |
|---|---|---|---|
| CXCL8 | 9·4 ± 6·8 | 103·4 ± 23·9 | 26·2 ± 14·1† |
| CXCL1 | 1·0 ± 0·7 | 9·7 ± 5·1 | 1·4 ± 1·1† |
| CCL2 | <0·5 | 2·5 ± 1·8 | <0·5† |
| β-actin | 500 ± 80 | 530 ± 100 | 560 ± 100 |
Confluent HT-29 monolayers in 24-well plates were pretreated with MG-132 (50 μm) for 1 h and then the cells were stimulated with B. fragilis enterotoxin (BFT, 100 ng/ml) for 6 h. Each mRNA expression was assayed by quantitative RT-PCR using internal RNA standard. The values represent the number of mRNA transcripts (x 104)/μg total RNA, and are expressed as the mean ± SD of 5 repeated experiments.
The values are significantly different in comparison with the BFT-stimulated alone (P < 0·05).
To confirm that BFT-activated NF-κB was directly related to BFT-induced chemokine production, p50 antisense oligo-nucleotide experiments were also performed. Oligonucleotides consisting of either the antisense or sense p50 mRNA binding sequence were taken up by cells. The presence of antisense oligonucleotide in the cell cytoplasm prevents p50 mRNA transcription and NF-κB activation, resulting in the inhibition of NF-κB-dependent events [21]. As shown in Fig. 4, p50 antisense oligonucleotide significantly decreased BFT-induced production of chemokine CXCL8, CXCL1 and CCL2. Conversely, sense oligonucleotide sequence had no significant effect. These results demonstrate a direct connection between BFT-induced NF-κB activation and chemokine production.
Fig. 4.
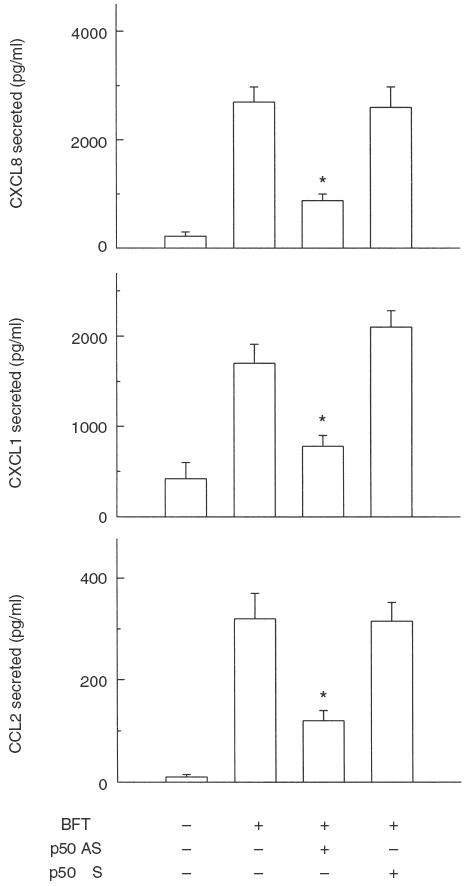
Effect of p50 antisense oligonucleotide on B. fragilis enterotoxin-induced chemokine production. HT-29 cells were transfected with either p50 antisense (AS) or sense (S) oligonucleotides using DOTAP. The transfected cells were then stimulated with B. fragilis enterotoxin (BFT, 100 ng/ml) for 18 h. Protein levels of each chemokine in culture supernatants were determined by ELISA. Data are mean ± SEM (n = 5). Asterisks indicate values with BFT + antisense oligonucleotide sequence that are significantly different from those of BFT alone (P < 0·05).
Inhibition of NF-κB signals decreases neutrophil transepithelial migration in T84 cells stimulated with B. fragilis enterotoxin
Intestinal epithelial cells are structurally and functionally polarized in vivo. Since a significant infiltration of neutrophils occurs in vivo in response to enterotoxigenic B. fragilis infection [6,7], we investigated whether this phenomenon could be mediated by NF-κB activation using a polarized intestinal epithelial T84 cell line. By using an inverted monolayer system [26,27], the apical surface of the cultured monolayers was stimulated with BFT and isolated human neutrophils were added to the physiologically appropriate (basolateral) surface. As shown in Fig. 5, stimulation of BFT to the apical surface of T84 cells induced the transmigration of neutrophils from basolateral to apical direction. However, the pretreatment with NF-κB inhibitor MG-132 decreased neutrophil transmigration in response to BFT stimulation by ~62% (8·5 × 104 vs. 3·2 × 104 transmigrating neutrophils in the absence and presence of MG-132, respectively) (Fig. 5), indicating that NF-κB induction may be responsible for the neutrophil chemotaxis.
Fig. 5.
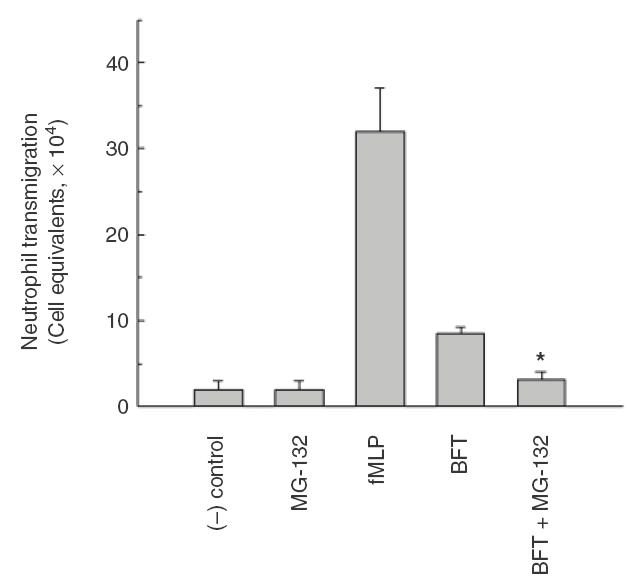
Transepithelial migration of neutrophils across T84 monolayers in response to apical stimulation by B. fragilis enterotoxin. T84 monolayers in transwell chamber were apically pretreated with NF-κB inhibitor MG-132 (50 μm) for 1 h, and then stimulated with B. fragilis enterotoxin (BFT, 100 ng/ml) for 6 h. The stimulated monolayers were transferred, apical side down, into a 24-well tissue culture tray. Isolated human neutrophils (106) were added to the basolateral side (upper reservoir) of each monolayer and incubated 2 h at 37°C. The number of neutrophils that had transmigrated was quantified by the myeloperoxidase assay. The potent chemotactic peptide fMLP (100 nm) was used as a positive control for these experiments. The negative control consisted of basolaterally positioned neutrophils in the absence of chemoattractive gradients. Data represent the mean ± SEM (n = 7). An asterisk indicates value significantly different from BFT-stimulated T84 monolayers without pretreated MG-132 (P < 0·05).
DISCUSSION
Human intestinal epithelial cells stimulated with BFT were shown to express and secrete CXCL1 and CXCL8 that can chemoattract and activate neutrophils [8], In this study, we found that CCL2 which can function mainly as chemoattractants of monocytes also up-regulates in intestinal epithelial cells stimulated with BFT. Moreover, the studies herein demonstrated that NF-κB may play a role as a central regulator for the activation of those chemokine genes of the intestinal epithelial cell innate immune response following stimulation with BFT.
NF-κB was activated in the human colon epithelial cells stimulated with BFT. Activation of NF-κB in the cytoplasm involves the inducible phosphorylation of IκBs, which then undergoes ubiquitin-mediated proteolysis, thereby releasing NF-κB dimers to translocate to the nucleus [29]. Our supershift studies showed that BFT stimulation induced primarily p65 and p50 heterodimers. In addition, we noted IκBα and IκBɛ degradation in BFT-stimulated HT-29 cells. This finding parallels that reported for NF-κB activation and IκBα degradation after enteroinvasive bacterial infection of the same cell lines [12,13], in which case incomplete degradation of IκBα was also sufficient for significant activation of NF-κB.
The various IκBs can differentially associate and regulate the activation of NF-κB and the transcriptional activation of various NF-κB target genes by virtue of binding to different populations of NF-κB dimers in the cytoplasm. IκBɛ, widely expressed in different human tissues, is known to interact preferentially with p65 and c-Rel members and appears to affect a subset of NF-κB genes apparently regulated by p65 homodimers [30,31]. Our finding of partial IκBɛ degradation after BFT stimulation of intestinal epithelial cells suggests a role for IκBɛ in the activation of several NF-κB target genes that are up-regulated in response to BFT stimulation of intestinal epithelial cells.
Infection of intestinal epithelial cells with enteroinvasive bacteria or stimulation of cells with proinflammatory cytokines such as IL-1 and TNF-α, activates a signalling cascade that culminates in the phosphorylation of IκBs [12,16,32]. Two IκB kinases, described as IκB kinase (IKK)α and IKKβ, directly phosphorylate IκBs on serine residues and are components of a high molecular weight cytoplasmic IKK complex [18,32]. Activation of IKK appears to be mediated through the mitogen-activated protein-3 (MAP-3) kinase, NIK, and MEK kinase 1 (MEKK-1). Phosphorylation of IκBs on conserved serine residues targets the IκBs for subsequent ubiquitination and degradation [33,34]. These free dimers of NF-κB (e.g. p65/p50) translocate to the nucleus where they trans-activate NF-κB target genes. Our observations that an IKKβ or NIK super-repressor alone was sufficient to inhibit the activation of IL-8 transcriptional reporters indicate that both IKK and NIK could be involved in the epithelial cell signalling transduction pathway, leading to NF-κB activation following BFT stimulation. These findings are consistent with the previous reports that NIK and IKKβ play a physiological role such as IκB phosphorylation and NF-κB activation [18,35,36]. However, the finding that the inhibition of IL-8 and NF-κB transcriptional reporters by NIK super-repressor was less than that by IκBα or IKKβ super-repressors suggest that NF-κB activation and IL-8 expression of intestinal epithelial cells by BFT stimulation may be involved in other pathways.
In this study, the inhibition of BFT-induced NF-κB signals suppressed the expression of CXC chemokines such as IL-8 and GRO-α as well as CC chemokine MCP-1. In addition, transfection of p50 antisense oligonucleotide showed that BFT-induced NF-κB activation and CXC and CC chemokine production were directly linked. These results indicate that NF-κB is an essential transcription factor for integrating the host epithelial cell chemokine response to stimulation with BFT.
Since the infiltration of neutrophils occurs in vivo in response to enterotoxigenic B. fragilis [6,7], we investigated whether this phenomenon could be mediated by NF-κB activation in BFT-stimulated intestinal epithelial cells. Our results revealed that BFT stimulation of the apical membrane of intestinal epithelial cells induced neutrophil transmigration. In this model, inhibition of NF-κB by the pretreatment with MG-132 decreased the neutrophil transmigration. These results suggest that BFT-induced NF-κB signalling may be primarily involved in the neutrophil migration in the infected tissues.
In summary, intestinal epithelial cells stimulated with BFT produce several chemokines that can activate the host's mucosal inflammatory responses. The data herein demonstrate that NF-κB is an essential transcription factor for integrating the host epithelial cell chemokine response to infection with enterotoxigenic B. fragilis and may be an important regulator of neutrophil migration.
Acknowledgments
We thank Dr Hee-Bok Oh and Gyung-Tae Chung for purification of B. fragilis enterotoxin We also thank Dr Martin F. Kagnoff and Dr Joseph A. DiDonato for gifts of standard RNAs and several plasmids.
REFERENCES
- 1.Niyogi SK, Dutta P, Mitra U, Pal DK. Association of enterotoxigenic Bacteroides fragilis with childhood diarrhoea. Indian J Med Res. 1997;105:167–9. [PubMed] [Google Scholar]
- 2.Zhang G, Svenungsson B, Karnell A, Weintraub A. Prevalence of enterotoxigenic Bacteroides fragilis in adult patients with diarrhea and healthy controls. Clin Infect Dis. 1999;29:590–4. doi: 10.1086/598639. [DOI] [PubMed] [Google Scholar]
- 3.Mundy LM, Sears CL. Detection of toxin production by Bacteroides fragilis: assay development and screening of extraintestinal clinical isolates. Clin Infect Dis. 1996;23:269–76. doi: 10.1093/clinids/23.2.269. [DOI] [PubMed] [Google Scholar]
- 4.Pantosti A, Cerquetti M, Colangeli R, D’Ambrosio F. Detection of intestinal and extra-intestinal strains of enterotoxigenic Bacteroides fragilis by the HT-29 cytotoxicity assay. J Med Microbiol. 1994;41:191–6. doi: 10.1099/00222615-41-3-191. [DOI] [PubMed] [Google Scholar]
- 5.Chambers FG, Koshy SS, Saidi RF, Clark DP, Moore RD, Sears CL. Bacteroides fragilis toxin exhibits polar activity on monolayers of human intestinal epithelial cells (T84 cells) in vitro. Infect Immun. 1997;65:3561–70. doi: 10.1128/iai.65.9.3561-3570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obiso RJ, Jr, Lyerly DM, Van Tassell RL, Wilkins TD. Proteolytic activity of the Bacteroides fragilis enterotoxin causes fluid secretion and intestinal damage in vivo. Infect Immun. 1995;63:3820–6. doi: 10.1128/iai.63.10.3820-3826.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myers LL, Shoop DS, Collins JE, Bradbury WC. Diarrheal disease caused by enterotoxigenic Bacteroides fragilis in infant rabbits. J Clin Microbiol. 1989;27:2025–30. doi: 10.1128/jcm.27.9.2025-2030.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JM, Oh YK, Kim YJ, Oh HB, Cho YJ. Polarized secretion of CXC chemokines by human intestinal epithelial cells in response to Bacteroides fragilis enterotoxin: NF-κB plays a major role in the regulation of IL-8 expression. Clin Exp Immunol. 2001;123:421–7. doi: 10.1046/j.1365-2249.2001.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanfilippo L, Li CK, Seth R, Balwin TJ, Menozzi MG, Mahida YR. Bacteroides fragilis enterotoxin induces the expression of IL-8 and transforming growth factor-beta (TGF-β) by human colonic epithelial cells. Clin Exp Immunol. 2000;119:456–63. doi: 10.1046/j.1365-2249.2000.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 11.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–32. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 12.Elewaut D, DiDonato JA, Kim JM, Truong F, Eckmann L, Kagnoff MF. NF-kappa B is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J Immunol. 1999;163:1457–66. [PubMed] [Google Scholar]
- 13.Jobin C, Haskill S, Mayer L, Panja A, Sartor RB. Evidence for altered regulation of IκBα degradation in human colonic epithelial cells. J Immunol. 1997;158:226–34. [PubMed] [Google Scholar]
- 14.Kim JM, Eckmann L, Savidge TC, Lowe DC, Witthoft T, Kagnoff MF. Apoptosis of human intestinal epithelial cells after bacterial invasion. J Clin Invest. 1998;102:1815–23. doi: 10.1172/JCI2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JM, Kim JS, Jung HC, Song IS, Kim CY. Apoptosis of human gastric epithelial cells via caspase-3 activation in response to Helicobacter pylori infection: Possible involvement of neutrophils through tumor necrosis factor-alpha and soluble Fas ligands. Scand J Gastroenterol. 2000;35:40–8. doi: 10.1080/003655200750024515. [DOI] [PubMed] [Google Scholar]
- 16.Kim JM, Oh YK, Kim YJ, Cho SJ, Ahn MY, Cho YJ. Nuclear factor-kappa B plays a major role in the regulation of chemokine expression of HeLa cells in response to Toxoplasma gondii infection. Parasitol Res. 2001;87:758–63. doi: 10.1007/s004360100447. [DOI] [PubMed] [Google Scholar]
- 17.DiDonato JA, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine- activated IκB kinases essential for NF-kappa B activation. Science. 1997;278:860–6. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 19.Jain VK, Magrath IT. A chemiluminescent assay for quantitation of β-galactosidase in the femtogram range: application to quantitation of β-galactosidase in lacZ-transfected cells. Anal Biochem. 1991;199:119–24. doi: 10.1016/0003-2697(91)90278-2. [DOI] [PubMed] [Google Scholar]
- 20.Eckmann L, Reed SL, Smith JR, Kagnoff MF. Entamoeba histolytica trophozoites induce an inflammatory cytokine response by cultured human cells through the paracrine action of cytolytically released interleukin-1α. J Clin Invest. 1995;96:1269–79. doi: 10.1172/JCI118161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim JW, Kim H, Kim KH. Nuclear factor-κB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Laboratory Invest. 2001;81:349–60. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- 22.Jung HC, Kim JM, Song IS, Kim CY. Helicobacter pylori induces an array of pro-inflammatory cytokines in human gastric epithelial cells: quantification of mRNA for interleukin-8-1α/β, granulocyte-macrophage colony-stimulating factor, monocyte chemoattractant protein-1 and tumour necrosis factor-α. J Gastroenterol Hepatol. 1997;12:473–80. doi: 10.1111/j.1440-1746.1997.tb00469.x. [DOI] [PubMed] [Google Scholar]
- 23.Kim JM, Jung HC, Jin DZ, Im K, Song IS, Kim CY. Cytokine genes are down-regulated when attachment of Entamoeba histolytica to HT-29 colon epithelial cells is prevented. Scand J Immunol. 1997;45:613–7. doi: 10.1046/j.1365-3083.1997.d01-442.x. [DOI] [PubMed] [Google Scholar]
- 24.Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka- Wroblewska E, Kagnoff MF. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JM, Jung HC, Im K, Song IS, Kim CY. Synergy between Entamoeba histolytica and Escherichia coli in the induction of cytokine gene expression in human colon epithelial cells. Parasitol Res. 1998;84:509–12. doi: 10.1007/BF03356595. [DOI] [PubMed] [Google Scholar]
- 26.Parkos CA, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured intestinal epithelium. Dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. J Clin Invest. 1991;88:1605–12. doi: 10.1172/JCI115473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savkovic SD, Koutsouris A, Hecht G. Attachment of a noninvasive enteric pathogen, enteropathogenic Escherichia coli, to cultured human intestinal epithelial monolayers induces transmigration of neutrophils. Infect Immun. 1996;64:4480–7. doi: 10.1128/iai.64.11.4480-4487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savkovic SD, Koutsouris A, Hecht G. Activation of NF-κB in intes-tinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- 29.May MJ, Ghosh S. Signal transduction through NF-κB. Immunol Today. 1998;19:80–8. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Nabel GJ. A new member of the IκB protein family, IκBɛ, inhibits RelA (p65)-mediated NF-κB transcription. Mol Cell Biol. 1997;17:6184–90. doi: 10.1128/mcb.17.10.6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simeonidis S, Liang S, Chen G, Thanos D. Cloning and functional characterization of mouse IκBɛ. Proc Natl Acad Sci USA. 1997;94:14372–7. doi: 10.1073/pnas.94.26.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine- responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 33.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 34.Verma IM, Stevenson JK, Schwarz EM, van Antwerp D, Miyamoto S. Rel/NF-κB/ IκB family. intimate tales of association and dissociation. Genes Dev. 1995;9:2723–35. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 35.Mercurio F, Murray BW, Shevchenko A, et al. IκB kinase (IKK)- associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–38. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zandi E, Chen Y, Karin M. Direct phosphorylation of IκB by IKKα and IKKβ discrimination between free and NF-κB-bound substrate. Science. 1998;281:1360–3. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]