

Abstract
During an 8-year period of observation, defects of immune responses were characterized and monitored in 40 of 50 Polish children with Nijmegen breakage syndrome referred to the Children's Memorial Health Institute in Warsaw. The following parameters were determined at diagnosis: (1) concentrations of serum IgM, IgG, IgA; (2) concentrations of IgG subclasses; and (3) lymphocyte subpopulations. In addition, naturally acquired specific antibodies against Streptococcus pneumoniae were determined in 20 patients with a history of recurrent respiratory infections. During follow-up, total serum immunoglobulins and IgG subclasses were monitored systematically in 17 patients who did not receive immunomodulatory therapy. Moreover, anti-HBs antibody response was measured after vaccination of 20 children against HBV. We found that the immune deficiency in NBS is profound, highly variable, with a tendency to progress over time. Systematic monitoring of the humoral response, despite good clinical condition, is essential for early medical intervention.
Keywords: children, immune responses defects, Nijmegen breakage syndrome
INTRODUCTION
Nijmegen breakage syndrome (NBS) is a relatively rare chromosomal instability disorder clinically characterized by microcephaly, typical facial appearance, growth and mental retardation, immunodeficiency and a high predisposition to lymphoid malignancy [1]. NBS cells show spontaneous chromosomal instability, hypersensitivity to ionizing radiation and abnormal cell-cycle regulation after irradiation resulting in radioresistant DNA synthesis [1,2]. The disease is caused by mutations in the NBS1 gene, which is located on chromosome 8q21 and has recently been cloned [3,4]. The gene product, nibrin, is a part of the hMre11/hRad50 protein complex that plays a central role in the cellular DNA damage response. These proteins are involved in processing DNA double-strand breaks (DSBs), both those induced by such pathological agents as ionizing radiation (IR) or those that are part of normal processes such as meiotic recombination and mitotic V(D)J rearrangement in maturing lymphocytes [5]. Thus, the nibrin deficiency disrupts key regulatory processes in NBS cells and, in consequence, a specific combination of clinical and cellular symptoms may be found in affected patients.
One of the common features of NBS is dysregulation of both cellular and humoral arms of the immune system. The degree of its functional degradation is closely associated with enhanced susceptibility to recurrent bacterial and viral infections, mainly of the respiratory tract. In particular, low levels of serum IgG subclasses and/or defective synthesis of specific antibodies to common pathogens are considered to be important factors responsible for the pathogenesis of abnormally frequent or prolonged infections [6,7]. Thus, early recognition of immune response defects in NBS patients is of great importance for instituting suitable medical intervention to prevent serious complications of the disease.
Until now, over 100 patients with NBS have been identified worldwide; among them are 66 Polish children from 57 families, of whom 50 were diagnosed at the Children's Memorial Health Institute in Warsaw. All Polish patients tested to date are homozygous for the common 657del5 mutation in the NBS1 gene [8].
The individual course of the disease and variability of clinical symptoms, which are often the reason for delayed diagnosis of severe immune disturbances, prompted us to undertake longitudinal studies to characterize the immune status of our patients at diagnosis and to monitor the evolution of humoral defects with time. The high prevalence of new cases in our population offers a unique opportunity to gather clinical and laboratory data in a larger series of patients in a single medical centre. We wanted to assess whether monitoring of humoral parameters could be useful laboratory tool to detect changes preceding severe clinical complications.
MATERIALS AND METHODS
Patients
Between January 1992 and December 2000, serum samples were collected from 40 of 50 children (17 boys and 23 girls) from 34 families (six pairs of siblings and 28 unrelated) in whom detailed clinical, cytogenetic and biochemical investigations were performed before the diagnosis of NBS was established. All patients included in the study met the diagnostic criteria for NBS, and were confirmed by mutation analysis [4]. The median age of patients at the first examination was 6·33 years (range 2 months−18 years). A serum sample was obtained from each child at the time of diagnosis and then control sera were withdrawn systematically (at 3–6-month intervals) from patients who were in good clinical condition and did not receive any immunomodulating treatment or immunoglobulin administration. At each control visit infection data was collected on the basis of information received from parents and/or from hospital documentation given to patients at discharge. Most upper and lower respiratory tract infections were associated with common bacteria (mainly with Pseudomonas aeruginosa and Candida albicans) and viruses, while opportunistic organisms were not isolated. Moreover, chronic hepatitis B infection was found in four (10%) and chronic hepatitis C infection in three (7·5%) patients. In one child, both HBV and HCV infections co-existed.
The study was approved by the Ethics Committee of the Children's Memorial Health Institute and informed consent was obtained from all parents.
Serum immunoglobulins and IgG subclasses
Total serum IgM, IgG and IgA were determined by nephelometry (Array 360 System; Beckman Instruments, Brea, CA, USA). The specificity and sensitivity of the method is regularly checked by the Bio-Rad External Quality Assurance Services-Immunoassay Program (Bio-Rad, Irvine, CA, USA).
The determination of IgG subclasses was performed in 38 of 40 patients by an enzyme-linked immunosorbent assay (ELISA) according to the method described elsewhere [9] and individual results were compared with normal ranges established in age-matched groups of healthy children. In two children the determinations were not performed because of extremely low levels of IgG (<0·4 g/l).
Specific antibodies to Streptococcus pneumoniae (serotypes 3, 19, 23)
Naturally acquired immunity to most invasive pneumococcal polysaccharides (serotypes 3, 19, 23) of IgG isotype was evaluated in 20 patients aged 4–16 years with a history of recurrent respiratory tract infections. Specific antibodies were determined by an ELISA according to Zielen et al. [10] with a minor modification, i.e. results were expressed as the reciprocal of the highest dilution that gave an optical density (O.D.) value of more than two standard deviations above the mean calculated for 40 negative samples.
Specific antibodies to hepatitis B antigen
The determination of antibody to hepatitis B antigen (anti-HBs) was performed in 20 patients vaccinated against HBV with Engerix B (SmithKline Beecham Biologicals, Rixensart, Belgium) according to the manufacturer's instructions (0, 1, 6 months). A serum sample was withdrawn from each child 1–3 months after vaccination and the quantitative determination of anti-HBs antibodies was performed by microparticle enzyme immunoassay with IMx AUSAB kits (Abbott Laboratories, Chicago, USA). The geometric mean of antibody titres (GMT) was calculated.
IgG anti-HBs subclasses and IgM anti-HBs were determined by an ELISA as described previously [11]. Levels of IgM anti-HBs were measured in those serum samples in which IgG anti-HBs were not detected.
Lymphocyte subpopulations
Total B, T and T cell subsets (CD19, CD3, CD4, CD8) and natural killer cells (NK-CD56) were assessed by double-colour flow cytometric analysis (Coulter Flow Cytometer, Miami, FL, USA) using MoAbs conjugated with phycoerythrin (PE) or fluorescein isothiocyanate (FITC) (Becton Dickinson, San Jose, CA, USA) according to a method described elsewhere [12]. Results were expressed in absolute numbers, i.e. cells/µl.
Statistical analysis
The correlation between parameters of humoral response and particular cell subsets was analysed by non-parametric Spearman's rank order correlations, and individual results were compared by non-parametric sign tests for dependent samples. The GMT value of anti-HBs antibodies was accompanied by 95% confidence intervals (CI). A P-value <0·05 was considered significant.
RESULTS
Humoral immunity and lymphocyte subpopulations in NBS patients at the time of diagnosis
Serum immunoglobulins and IgG subclasses
Normal levels of total serum IgM, IgG and IgA were found in eight out of 40 patients (20%), while in the remainding 32 (80%) deficiency of one or more Ig isotypes was found. The most common was a combined deficit of IgG and IgA found in 10 of 32 deficient children (31·3%), followed by an isolated IgG deficiency (9/32, i.e. 28·1%), and then by combined deficiency of all three isotypes (7/32, i.e. 21·9%). On the other hand, in five of 40 children (12·5%), markedly elevated concentrations of IgM (ca. 1·4–2·9 times above the upper normal limit for age) were found. The frequency of particular immunoglobulin deficiencies found in the group as a whole (i.e. 40 patients) is shown in Fig. 1a.
Fig. 1.
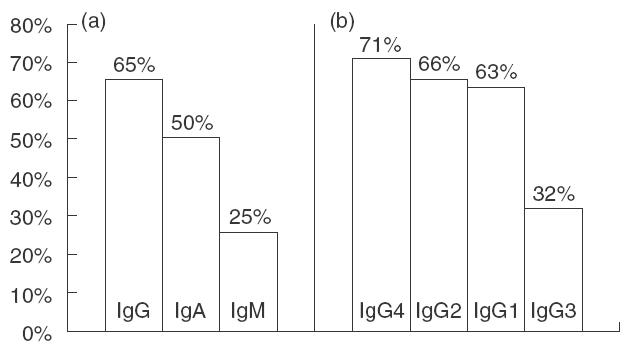
Frequency of particular serum immunoglobulin (a) and IgG subclass (b) deficiencies at the time of NBS diagnosis. (a) Total IgG; (b) IgG subclasses.
At diagnosis, severe hypogammaglobulinaemia with very low concentrations of IgG (<2·0 g/l) was found in eight of 40 patients (20%). In five of them, extremely low concentrations of IgG (<1·0 g/l) with low or absent IgA and decreased IgM were observed. These patients and the other 15 (37·5%) with moderate hypogammaglobulinaemia (i.e. over 2·0 g/l, but below the lower limit for their age) who suffered from frequent infections had undergone protective therapy and were excluded from the follow-up study.
The most characteristic feature of the humoral abnormalities was deficiency of at least one or more IgG subclasses found in all children studied (38/38). In 14 of them (36·8%) IgG subclass deficiencies were ‘masked’ by normal levels of total IgG. The most common was a combined deficiency of all four IgG subclasses found in eight of 38 patients (21·1%), followed by an association of IgG1, IgG2 and IgG4 deficiency (7/38, i.e. 18·4%), and by combined IgG2 and IgG4 deficiency (7/38, i.e. 18·4%). Figure 1b shows the frequency of particular IgG subclass deficiencies.
Specific antibody levels
Naturally acquired class IgG specific antibodies to all three pneumococcal polysaccharides (serotypes 3, 19, 23) were present in five of 20 patients studied (25%). All of them had normal levels of total serum IgG. The range of titres of specific antibodies against serotype 3 was: 200–400; serotype 19: 400–6400; and serotype 23: 200–3200. In the rest of the patients, polysaccharide IgG antibodies directed against single serotypes were found in low titres (100–400) or were not detected at all. This finding correlated with markedly decreased levels of IgG2 (P < 0·05).
Total anti-HBs titres and specific IgG subclass profiles
In the group of 20 children vaccinated with Engerix B, the GMT value of total anti-HBs antibodies measured 1–3 months post-immunization in responding patients was 75 IU/l (95% CI, 50–113 IU/l). Five patients (25%) developed anti-HBs of the IgG class, engaging exclusively two IgG subclasses: IgG1 was present in all serum samples, while IgG3 was found in one.
Thirteen children (65%) responded only with IgM anti-HBs, and in the other two (10%) specific antibodies were not detected, in spite of normal or only moderately decreased levels of total IgG in eight of them.
Lymphocyte subpopulations
The mean values of absolute numbers of total B, T and T cell subsets, and NK cells (CD19, CD3, CD4, CD8, CD56) accompanied by 95% CI are summarized in Table 1. The most evident defects were reduced absolute numbers of CD3+ T cells found in 37 of 40 NBS patients (92·5%), reduced number of CD4+ T cells found in 38/40 (95%) and CD8+ T cells in 32/40 patients (80%). A decreased ratio of CD4+/CD8+ was present in 70% of cases (28/40). On the other hand, in 25/40 NBS patients (62·5%) considerable elevation of NK cells was observed.
Table 1.
Absolute number of peipheral blood lymphocyte subpopulations in NBS at diagnosis
| NBS patients cells/µl | Normal values cells µl | ||||
|---|---|---|---|---|---|
| Lymphocyte subpopulations | Mean | Range | Mean | Range | P-level |
| T cells | |||||
| CD3+ | ↓704 | 568–872 | 2416 | 2236–2610 | P < 0·001 |
| CD4+ | ↓364 | 296–450 | 1513 | 1376–1659 | P < 0·001 |
| CD8+ | ↓334 | 259–431 | 852 | 787–920 | P < 0·001 |
| CD4/+CD8+ | ↓1·4 | 0·16–4·68 | 2·1 | 1·6–2·6 | – |
| NK cells | |||||
| CD56+ | ↑409 | 311–536 | 267 | 239–300 | P < 0·05 |
| B cells | |||||
| CD19+ | ↓82 | 54–124 | 388 | 345–435 | P < 0·001 |
The number of B cells (CD19) was normal in five children (12·5%), reduced in 30 (75%) and elevated, ca. 1·4–2 times over the upper limit for age, in five patients (12·5%). Generally, in the group as a total, tendency toward low B cell count was observed, but no significant correlation was found between the pattern or degree of humoral response defects and the number of B cells (P = n.s.).
Evaluation of abnormalities in humoral immune response during follow-up
In 17 NBS patients who did not receive immunomodulatory therapy, concentrations of serum IgM, IgA, IgG and IgG subclasses were checked systematically over 2–8 years of observation (as a rule, at 3–6-month intervals). At the end of the study, two groups of children could be selected on the basis of the humoral defects pattern. In eight of 17 patients (47%) with moderate defects of the humoral response at diagnosis, no evident changes of particular parameters were observed over the course of time and infections were not more frequent than in healthy children.
In contrast, in nine of 17 patients (53%) humoral immunodeficiency showed evident progression with time. In six of them, in addition to the previous defects of IgG2 and IgG4, concentrations of total IgG and IgG1 systematically decreased; in the other two patients more profound decreases of IgG2 and IgG4 concentrations were found, while in the last child total IgG and IgG3 were elevated ca. 1·5 times above the upper normal limit. Frequent and prolonged infections of the respiratory tract observed in six patients were the reason for introducing protective therapy.
The most interesting observation in the group as a whole was the initially sudden and subsequently gradual increase of IgM concentrations (ca. 1·5–9·5-fold above the normal upper limit for age) observed in 16 of 40 (40%) patients. Until now, in 10 children available for study, immunofixation electrophoresis revealed µ bands with restricted electrophoretic mobility and corresponding κ or λ bands. These abnormalities preceded by 2 years the development of lymphomas, mainly of B cell origin, in all patients. Liver function tests performed at the beginning of IgM elevation were normal (manuscript in preparation). An example of changes in humoral parameters, fatal for the patient who was lost from systematic follow-up after 15 months of observation, is shown in Fig. 2.
Fig. 2.
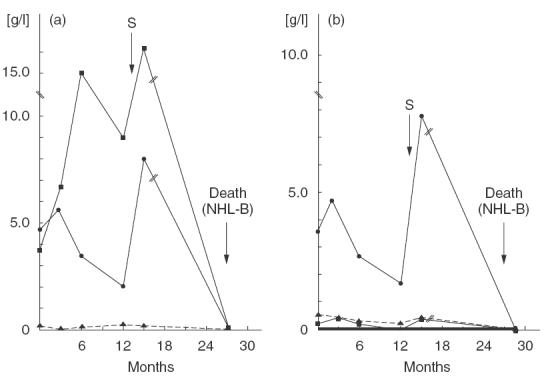
Example of progression of humoral immunity defects in a single NBS patient over the course of time. (a) Total immunoglobulins. ▪, IgM; •, IgG; p, IgA. (b) IgG subclasses. •, IgG1; p, IgG2; ▪, IgG3; —, IgG4. // Between months 15 and 28 after diagnosis the patient did not present for control investigations. S: substitution with intravenous immunoglobulins.
DISCUSSION
This is the first longitudinal study performed on 40 NBS patients in a single medical centre. It extends and supplements earlier information on immune defects, evaluated on the basis of single determinations [13,14]. Our study shows the considerable variability of immunodeficiency seen among different patients, as well as in the same individual over the course of time. In fact, at diagnosis, extremely low concentrations of IgG were found in 20% of patients, accompanied mainly by low (or not detectable) IgA and decreased IgM, while the other 20% had normal concentrations of all three major immunoglobulin classes. It is worth noting, however, that in 36·8% of patients, normal levels of total serum IgG masked deficiency of IgG subclasses (mainly IgG2 and/or IgG4). This finding points to the importance of determining IgG subclasses in NBS patients, especially in those suffering from frequent infections. The most common observation in our patients was decreased or undetectable levels of IgG4 (73·7% of cases) although this defect, especially when selective, seems to have no clinical significance. The next were deficiencies of IgG2 (65·8%), IgG1 (63·2%) and IgA (50%). This is in contrast to the pattern of humoral abnormalities reported in ataxia–telangiectasia (a genetic disease with overlapping phenotype), where IgA and IgG2 deficiency dominates in most patients (on average, 60% and 80%, respectively) [15] and confirms that the humoral defects in NBS are more profound than those found in other known chromosome instability syndromes [13,16,17].
Interestingly, IgM and especially IgG3, were rarely affected, despite cases with severe hypogammaglobulinaemia. These observations may suggest that the process of Ig class switching to CH genes downstream of γ3 (γ1, α1, γ2, γ4, ɛ and α2) is frequently blocked in NBS. One of the possible explanations is that maturation of T and B cells may be impaired when the NBS1 gene is lacking because the gene product, nibrin, as an integral part of the Mre11/Rad50/NBS1 nuclease complex, is involved in the repair of DNA double-strand breaks (DSBs) [4,5,18]. The V(D)J recombination, occurring during early B and T cell development, and class switch recombination, occurring exclusively in mature B cells, are two physiological forms of intentional double-strand DNA breakage and rejoining that appear during lymphoid differentiation [19]. Thus, one might predict that mutations in the NBS1 gene, which functions in recombination and/or repair processes in the immune system, can result in chromosomal translocations or loss of genetic information at the site of breakage. In other words, inherited dysfunction of nibrin may result in severe combined immune defects and may predispose to cancer, both phenomena observed in NBS. Recently, however, it was shown that nibrin is not involved in V(D)J recombination, but rather may play a role in keeping chromosomal breaks tethered together for assuring fidelity of the end joining process [20]. Thus, as the V(D)J recombination in NBS cells is normal the immune deficiencies associated with this disease may arise from defects in cell-cycle regulation which, in turn, may result in the reduction of certain lymphocyte subpopulations. In fact, highly reduced numbers of the CD4+ T cell subset found in 95% of our patients and a decreased CD4+/CD8+ ratio present in 70%, may be responsible for the lack of a second signal provided by activated CD4+ T cells and T cell-derived factors, both required for class switching [21]. In addition, decreased number of B lymphocytes found in 75% of children may also contribute to humoral defects present in NBS patients. On the other hand, the normal or even elevated number of B cells found in 25% of patients, despite the presence of antibody deficiencies, may indicate an intrinsic defect of B cell antibody class switching. In any case, these defects of immune responses, when present, may be responsible for the block in the switch from IgM to another class (IgG, IgA or IgE).
As the IgG subclasses are major serum immunoglobulins important in secondary antibody responses, they are responsible for immunological memory and long-term protection against viral and bacterial infections. Each of the four IgG subclasses is encoded by a separate C region gene and endowed with unique biological functions that are important for an efficient humoral response to a given pathogen. In adults, antibody responses to protein antigens are restricted mainly to IgG1, IgG3 or both; IgG2 is generally stimulated by carbohydrate antigens, whereas IgG4 has been associated with chronic antigenic stimulation [22]. Therefore, the severity of clinical manifestations of antibody deficiency syndromes, such as recurrent infections, is related to the kind of isotype deficiency. In our study, considerable variability of IgG subclass deficiency was found in all NBS patients, despite the normal level of total IgG in some of them. Moreover, absence or low titres of specific antibodies, especially after vaccination against HBV when 75% of patients did not develop anti-HBs antibodies of a protective IgG isotype, in spite of the normal or slightly decreased concentrations of total IgG, indicate the existence of a humoral defect at the specific antibody level.
Systematic, longitudinal observations of 17 available patients showed a progressive deterioration of the immune system in 53% of them. Successive Ig isotypes, previously in the normal range, were affected gradually, reaching a risky low level in some cases and there was no correlation between the kind of immunodeficiency profile (or its severity) and duration of the disease, age or sex of NBS patients studied (data not shown).
One of the most striking observations in the group as a whole was the sudden and systematic elevation of IgM concentrations found during follow-up in 40% of patients. This phenomenon prompted us to investigate the characteristics of these abnormalities in relation to cancer susceptibility. We found that ∼63% of these patients developed lymphomas, mainly of B cell origin, during a few months to even years after elevation of the IgM level and appearance of monoclonal gammopathy. Thus, the rise in IgM may suggest some block in isotype switching with accumulation of IgM-producing clones that later undergo malignant transformation. The hypothesis that Epstein–Barr virus (EBV) infection may be one of the candidates responsible for uncontrolled proliferation of B cells in NBS needs further investigation. Recently, Akha et al. [23] reported a high incidence of hypergammaglobulinaemia (38·8%) in patients with ataxia–telangiectasia. Of these, 23·3% was an isolated increase of IgM, but also elevated IgG and/or IgA were found. In contrast to our current observations, they found oligo-/monoclonal gammopathy that involved all major immunoglobulin isotypes in 8% of cases, but there was no correlation with the emergence of neoplastic disease.
In summary, this study provides the first complex data on immune defects in 40 NBS patients studied in a single medical centre. We have shown that disturbances of the immune system in NBS were variable and profound, with a tendency to progress over time. We postulate that systematic monitoring of the humoral response in NBS patients is of practical importance because it offers a simple opportunity for early detection of immunodeficiency progression or appearance of lymphomas. This, in turn, may allow clinicians to implement early and suitable medical intervention to prevent serious complications of the disease. Moreover, the low frequency of protective IgG antibodies to HBsAg after vaccination with Engerix B indicates the presence of a very weak specific response, and suggests the necessity of applying another, more intensive vaccination scheme in NBS patients [24].
Acknowledgments
This work was supported by grant no. 4P05E07314 from the Committee for Scientific Research, Warsaw, Poland. We thank B. Wol´owska, H. Zajec, E. Balas, E. Kostecka for their excellent technical assistance and E. Gorecka for secretarial support.
REFERENCES
- 1.Wegner RD, Chrzanowska K, Sperling K, Stumm M. Ataxia–telangiectasia variants (Nijmegen breakage syndrome) In: Ochs HD, Smith CIE, Puck JM, editors. Immunodeficiency disorders: a molecular and genetic approach. Oxford: Oxford University Press; 1999. pp. 324–34. [Google Scholar]
- 2.Digweed M, Reis A, Sperling K. Nijmegen breakage syndrome. consequences of defective DNA double strand break repair. Bioassays. 1999;21:649–56. doi: 10.1002/(SICI)1521-1878(199908)21:8<649::AID-BIES4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 3.Matsuura S, Weemaes C, Smeets D, et al. Genetic mapping using microcell-mediated chromosome transfer suggests a locus for Nijmegen breakage syndrome at chromosome 8q21–24. Am J Hum Gen. 1997;60:1487–94. doi: 10.1086/515461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–76. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 5.Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–86. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 6.De Baets F, Kint J, Pauwels R, Leroy J. IgG subclass deficiency in children with recurrent bronchitis. Eur J Pediatr. 1992;151:274–8. doi: 10.1007/BF02072228. [DOI] [PubMed] [Google Scholar]
- 7.Gregorek H, Pietrucha B, Janowicz W, Najberg E, Imielska D, Madalin´ski K. IgG subclass deficiencies in children with recurrent or severe respiratory tract infections. Pol J Immunol (Warsaw) 1993;18:319–28. [Google Scholar]
- 8.Varon R, Seemanowa E, Chrzanowska K, et al. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur J Hum Genet. 2000;8:900–2. doi: 10.1038/sj.ejhg.5200554. [DOI] [PubMed] [Google Scholar]
- 9.Gregorek H, Imielska D, Górnicki J, Mikol´ajewicz J, Przeradzka B, Madalin´ski K. Development of IgG subclasses in healthy Polish children. Arch Immunol Ther Exp. 1994;42:377–82. [PubMed] [Google Scholar]
- 10.Zielen S, Broker M, Strnad N, et al. Simple determination of polysaccharide specific antibodies by means of chemically modified ELISA plates. J Immunol Meth. 1966;193:1–7. doi: 10.1016/0022-1759(96)00033-6. [DOI] [PubMed] [Google Scholar]
- 11.Gregorek H, Madalin´ski K, Woynarowski M, Mikol´ajewicz J, Syczewska M, Socha J. The IgG subclass profile of anti-HBs response in vaccinated children and children seroconverted after natural infection. Vaccine. 2000;18:1210–7. doi: 10.1016/s0264-410x(99)00394-1. [DOI] [PubMed] [Google Scholar]
- 12.Michal´kiewicz J, Stachowski J, Chrzanowska K, Gregorek H, Krajewska-Walasek M, Madalin´ski K. Characterization of lymphocyte surface markers in the Nijmegen breakage syndrome. Pol J Immunol (Warsaw) 1994;19:213–9. [Google Scholar]
- 13.Chrzanowska KH, Kleijer WJ, Krajewska-Walasek M, et al. Eleven Polish patients with microcephaly, immunodeficiency, and chromosomal instability: the Nijmegen breakage syndrome. Am J Med Genet. 1995;57:462–71. doi: 10.1002/ajmg.1320570321. [DOI] [PubMed] [Google Scholar]
- 14.International Nijmegen Breakage Syndrome Study Group. Nijmegen breakage syndrome. Arch Dis Child. 2000;82:400–6. doi: 10.1136/adc.82.5.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Requeiro JR, Porras O, Lavin M, Gatti A. Ataxia–telangiectasia: a primary immunodeficiency revisited. Immunol Allergy Clin N Am. 2000;20:177–206. [Google Scholar]
- 16.Weemaes CMR, The TH, Van Munster JJ, Bakkeren JAJM. Antibody responses in vivo in chromosome instability syndromes with immunodeficiency. Clin Exp Immunol. 1984;57:529–34. [PMC free article] [PubMed] [Google Scholar]
- 17.Weemaes CMR, Smeets DFCM, van der Burgt CJAM. Nijmegen breakage syndrome: a progress report. Int J Radiat Biol. 1994;6:S185–8. [PubMed] [Google Scholar]
- 18.Haber JE. The many interfaces of Mre11. Cell. 1998;95:583–6. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- 19.Lieber MR. Pathological and physiological double-strand breaks: roles in cancer, aging, and the immune system. Am J Pathol. 1998;153:1323–32. doi: 10.1016/s0002-9440(10)65716-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harfst E, Cooper S, Neubauer S, Distel L, Grawunder U. Normal V (D) J recombination in cells from patients with Nijmegen breakage syndrome. Mol Immunol. 2000;37:915–29. doi: 10.1016/s0161-5890(01)00008-6. [DOI] [PubMed] [Google Scholar]
- 21.Fleisher TA, Bleesing JJH. Immune function. Pediatr Clin N Am. 2000;47:1197–209. doi: 10.1016/s0031-3955(05)70267-4. [DOI] [PubMed] [Google Scholar]
- 22.Jefferis R, Pound JD. Immunoglobulins. In: Gallin JI, Goldstein IM, Snyderman R, editors. Inflammation: basic principles and clinical correlates. 2. New York: Raven Press; 1992. pp. 11–32. [Google Scholar]
- 23.Akha AAS, Humphrey RL, Winkelstein JA, Loeb DM, Lederman HM. Oligo-/monoclonal gammopathy and hypergammaglobulinemia in ataxia-telangiectasia. Medicine. 1999;78:370–81. doi: 10.1097/00005792-199911000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Madalin´ski K, Gregorek H, Zembrzuska-Sadkowska E, et al. Immunogenicity of Engerix B vaccine in 786 children with risk of hepatitis B infection. Hepatologia Polska (Warsaw) 1998;5(Suppl 1):93–8. [Google Scholar]