

Abstract
It is generally accepted that the interaction between CD40 and its ligand (CD154) plays a decisive role in contact-dependent help for T and B cells. In CD154-deficient MRL/Mp-Faslpr (MRL/lpr) mice, however, high titres of IgG2a-type autoantibodies against small nuclear ribonucleoproteins (snRNPs) are observed. We successfully isolated two CD154-deficient MRL/lpr Th1 lines, which could provide B cell help for anti-snRNP antibody production. The proliferative responses of the Th1 cell lines were MHC class II (I-Ek)-restricted. Although syngeneic B cell proliferation was induced by Th1 lines in both a contact-dependent and -independent manner, the soluble form of TNF-α (sTNF-α) was not involved in contact-independent B cell proliferation. On the other hand, both anti-TNF-α and TNF-receptor 2 (TNF-R2, p75) monoclonal antibody (MoAb) blocked contact-dependent B cell proliferation, suggesting that the transmembrane form of TNF-α (mTNF-α)–TNF-R2 co-stimulation participates in B cell activation. Similarly, anti-TNF-α and TNF-R2 MoAb inhibited anti-snRNP antibody production in vitro, but anti-CD154 or TNF-R1 MoAb did not. These results indicate that the interaction of mTNF-α on activated Th1 cells with TNF-R2 on B cells may be involved in the autoimmunity seen in MRL mice, and that the blockade of CD40-CD154 co-stimulation may not always be able to suppress some Th1-related manifestations of lupus.
Keywords: autoantibodies, CD154, co-stimulation, lupus, TNF-α
INTRODUCTION
Human systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies against many nuclear antigens such as chromatin, DNA and certain RNA-associated proteins, including anti-small nuclear ribonucleoproteins (anti-snRNP) antibodies. Several mouse lupus models display autoimmune manifestations reminiscent of SLE, including the MRL/Mp (MRL-+/+) strain that develops a syndrome nearly indistinguishable from the human disease, including marked humoral autoimmunity such as anti-Sm antibody [1]. MRL/Mp-Faslpr (MRL/lpr) mice also develop spontaneously a severe disease resembling SLE, especially immune-complex glomerulonephritis, with the generation of IgG autoantibodies to self-antigens such as chromatin, dsDNA, snRNPs and IgG (rheumatoid factors; RF) [2].
A large number of investigations have established that the pathogenesis of MRL lupus mainly requires CD4+αβ T cells, which help to stimulate autoreactive B cells [3–5]. These studies have demonstrated that a lack of such cells cause substantial reductions in autoantibody production and glomerulonephritis. In addition, CD4+αβ T cells that help autoantibody production have been isolated from other mouse strains [6,7]. Autoantibodies produced by MRL mice display evidence of Ag selection [8], suggesting that autoreactive CD4+αβ T cells drive autoantigen-specific autoantibody production. This notion is supported by the finding that genetic elimination of autoantigen-specific αβ T cells in MRL mice eliminate autoantibody production and end-organ disease [9].
The binding of CD40 ligand (CD154), a type II membrane protein transiently expressed on activated CD4+ T cells [10], to CD40 on B cells plays a critical role in T–B collaboration, including the initiation of immunoglobulin (Ig) synthesis and class switching in response to thymus-dependent (TD) antigens [11–14], as well as playing a role in the formation of germinal centres and the development of memory B cells [14,15]. In CD154-deficient mice, antigen-specific T cell priming is impaired as a result of the failure to initiate a specific T cell immune response to TD antigens [16]. Along with the apparent requirement for CD4+αβ T cells, CD154 appears to be instrumental in IgG autoantibody production and end-organ disease in murine and probably human lupus. Support for this notion comes from the observations that administration of anti-CD154 antibody to autoimmune SNF1 and (NZB × NZW) F1 lupus-prone mice inhibits anti-dsDNA synthesis and glomerulonephritis [17,18], and that human subjects with SLE show increased CD154 levels on both T and B cells [19,20].
We previously characterized CD154-deficient lupus-prone MRL/lpr mice and found that these animals lacked anti-dsDNA and RF as well as lacking fully developed glomerulonephritis. Surprisingly, however, the CD154-deficient MRL/lpr mice underwent Ig class switching to IgG2a with Ig levels much higher than those of control, non-autoimmune mice, and showed partial maintenance of IgG2a anti-snRNP antibody responses [21]. These results indicate that CD40–CD154 co-stimulation is required to provide contact-dependent help for anti-dsDNA antibody production in MRL mice, although IgG class switching and the generation of IgG anti-snRNP antibody do not necessarily rely upon T cell help mediated via CD154 in MRL/lpr mice.
The study presented here demonstrates that the autoreactive CD4+αβ Th1 cells generated by us have the capacity to provide B cell help for the production of IgG anti-snRNP antibody in vitro, and that the transmembrane form of TNF-α (mTNF-α)–TNF receptor (TNF-R) system can be substituted for the CD40–CD154 co-stimulation required for the production of IgG autoantibodies. These observations suggest that mTNF-α expression on activated T cells is involved in lupus autoimmunity and that blockade of CD40–CD154 co-stimulation might be insufficient to suppress some manifestations related to the Th1 response of lupus.
MATERIALS AND METHODS
Mice
MRL/Mp-Faslpr mice (MRL/lpr mice, purchased from the Jackson Laboratory, Bar Harbor, ME, USA) were bred with female CD154-deficient 129/SvJ × C57BL/6 (129 × B6 CD154-deficient) mice [13] to produce F1 offspring heterozygous for lpr (mutant Fas gene) and for CD154. The animals were then back-crossed to the MRL background to the N8 generation, followed by intercrossing and analysis for wild-type and mutant CD154 and Fas by PCR [21]. These mice were bred and housed in specific pathogen-free facilities at the Yale School of Medicine.
Generation of autoreactive T cell lines from CD154-deficient MRL/lpr mice
Autoreactive T cells were isolated and cloned according to Naiki's method [7] with some modifications. A single cell suspension of lymph node cells was prepared from three different 4-month-old, anti-snRNP antibody-positive CD154-deficient MRL/lpr mice [21]. The cells were treated with red blood cell lysis buffer (Sigma Chemical Co., St Louis, MO, USA). Initially, 4 × 106/ml of cells were cultured in 24-well tissue culture plates with Click's medium (Irvine Scientific, Santa Ana, CA, USA) supplemented with 10% FCS, antibiotics, l-glutamine and 2-ME. The irradiated (3000 rad) syngeneic APC (2 × 106/ml) were added weekly along with 10 µ/ml mouse rIL-2 (R&D systems, Minneapolis, MN, USA). After 1 month, cells were transferred to 96-well plates at a concentration of five cells/well for limiting dilution. Growing cells were harvested and expanded in 24-well plates for further study. In some experiments, autoreactive Th1 and Th2 cell lines (5-1 and 4-1), which were derived from CD154-intact MRL/lpr mice using the same method and not reactive for specific antigens, were used as controls.
Flow cytofluorometric analysis
T cell lines were analysed by flow cytometry using anti-TCR-Cβ (H57-597-FITC), anti-TCRγδ (GL3-PE), anti-CD4 (H129·19-FITC) and anti-CD8 (53–6·7-PE). For detection of CD154 or mTNF-α, cells after stimulation with plate-bound anti-CD3ɛ monoclonal antibody (MoAb) (5 µg/ml) were stained with PE-conjugated anti-CD154 (MR1, PharMingen, San Diego, CA, USA) or anti-TNF-α (G281-2626, PharMingen). Stained cells were analysed with a FACScanTM flow cytometer and using CellQuestTM software (Becton Dickinson, Mountain View, CA, USA).
Proliferation assays
For T cell proliferation assays, T cell lines (1 × 105/well) were co-cultured with 5 × 105 irradiated (3000 rad) splenocytes from CD154-deficient MRL/lpr mice as APC in triplicate for 3 days in round-bottomed, 96-well microtitre plates. Before the initiation of culture, APC were incubated with either crude ENA (extractable nuclear antigen, 1 mg/ml) prepared from murine Ehrlich ascites cells as described previously [22] or with medium (control) at 37°C for 2 h. [3H]-labelled thymidine (1 µCi, Amersham, Arlington Heights, IL, USA) was added to each well during the last 16 h of culture, and cells were harvested with a semi-automatic cell harvester (Skatron Instruments, Sterling, VA, USA). The incorporated radioactivity was measured with a β-plate scintillation counter (Beckman Instruments, Fullerton, CA, USA).
For B cell proliferation assays, MRL/lpr B cells were purified from splenocytes of CD154-deficient MRL/lpr mice using CellectTM Mouse B (Biotex Laboratories Inc., Edmonton, Canada) followed by further T cell depletion using anti-Thy1·2 antibody (HO-13–4) and Low-Tox-M rabbit complement (Accurate Chemical & Scientific Corporation, Westbury, NY, USA). The B cell purity was>95% as determined by flow cytometry. Purified B cells (5 × 104 for 96-well plates and 25 × 104 for 24-well plates) were co-cultured with irradiated (1500 rad) T cell lines (2 × 104 for 96-well plates and 10 × 104 for 24-well plates) for 3 days. One µCi of [3H]-labelled thymidine was added for the last 16 h of culture, cells were harvested and the incorporated radioactivity measured. A membrane insert (0·4 µm pore size, Becton Dickinson Labware, Franklin Lakes, NJ, USA) was used in some experiments to prevent contact between T cell lines and B cells. The separated T and B cells were co-cultured in 24-well culture plates for 48 h, followed by the addition of 4 µCi of [3H]-labelled thymidine. After 16 h, cells above and below the membrane were mixed, and harvested immediately after transfer from 24-well plates to 96-well plates.
For blockade of T or B cell proliferative responses, monoclonal antibodies to I-Ek (14-4-4S, PharMingen, dialysed before use), I-Ak (10–3·6, PharMingen, dialysed before use), CD154 (MR1, NA/LE, PharMingen), IFN-γ (R4–6A2, NA/LE, PharMingen), IL-2 (Genzyme, Cambridge, MA, USA), TNF-α (G281-2626, NA/LE, PharMingen), TNF-R1 and R2 (p55[55R-170·1] and p75 [TR75-54·7, a gift from R.D. Schreiber, Washington University School of Medicine]) [23] were used.
In vitro helper assay
T cell lines (5 × 105 cells/well) were co-cultured with 2 × 106 purified MRL/lpr B cells in 24-well plates for 1 week. Culture supernatants were then harvested, and the concentration of anti-snRNP antibodies determined by ELISA (described below). To determine if antibody production was enhanced via soluble factors (e.g. cytokines), a membrane insert was used to prevent contact between the T cell lines and B cells. Non-autoreactive controls consisted of a T cell line derived from antipigeon cytochrome c (PCC) TCR transgenic TCR-α-/–TCRβ-/- MRL/lpr mice [9] and purified B cells of PCC-immunized MRL/lpr mice.
Determination of anti-snRNP antibodies by ELISA
For detection of anti-snRNP antibody, mouse snRNPs (1 µg/ml) in carbonate buffer, pH 9·6, were coated on Serocluster ‘U’ Vinyl Plates (Costar, Cambridge, MA, USA) overnight at 4°C. Mouse sera were diluted 1 : 100 with PBS containing 3% BSA and incubated at room temperature for 2 h, followed by detection of bound IgG with alkaline phosphatase-conjugated antimouse IgG (Southern Biotechnology Associates Inc., Birmingham, AL, USA) at O.D.405nm in a microtitre ELISA reader. Anti-dsDNA antibodies were measured with Rubin's methods [24].
RESULTS
Antinuclear antibody production and CD4+ab T cells in CD154-deficient MRL/lpr mice
The levels of anti-dsDNA and -snRNP antibody, as determined by ELISA using purified autoantigens as substrates, were significantly higher in CD154-intact MRL/lpr mice than in their CD154-deficient counterparts; however, a number of the latter mice produced comparable amounts of anti-snRNP antibody to those of the CD154-intact animals [21]. The subclass of the higher titre anti-snRNP antibody in CD154-deficient mice was IgG2a, whereas IgG1 and IgG3 anti-snRNP antibody were also detected in CD154-intact mice [21].
The CD44highCD45RBhigh MRL T cells, which were assured to be effectors, developed in CD154-deficient MRL/lpr mice, while CD4+ naive (CD44lowCD45RBintermed) αβ T cells remained in the spleen (unpublished data). The development of memory T cells in these mice, however, was severely impaired because the number was much lower than that in CD154-intact 129 × B6 or MRL/lpr of the same age (data not shown).
T cell lines derived from CD154-deficient MRL/lpr mice
Twenty-six T cell lines were isolated by limiting dilution from three anti-snRNP antibody-positive MRL/lpr mice deficient in CD154. Five of these T cell lines were selected for further studies (Table 1) as they expressed both αβ TCR and CD4 and proliferated in response to crude ENA as a source of snRNPs. These lines all lacked CD154 expression after stimulation with PMA (10 ng/ml) and ionomycin (500 ng/ml)/anti-CD3∈ MoAb (5 µg/ml). The background stimulation indexes (SI) for proliferation of these Th lines were calculated by averaging their responses to histone, dsDNA or medium. The mean values of background SI + 2 s.d. for these five T cell lines that responded to ENA were 1·49 for C2, 1·42 for G1, 1·33 for G2, 1·52 for P5 and 1·72 for E7. The five lines selected showed increased proliferative responses when co-cultured with syngeneic APC with ENA (Table 1) compared with background levels, strongly suggesting that these lines recognize some self-murine ENA. T cell proliferative responses were MHC class II-restricted, as anti-I-Ek MoAb selectively inhibited the proliferation of four lines (C2, G1, G2, P5). Proliferation of the E7 line also appeared to be I-Ek-restricted, although inhibition was incomplete.
Table 1.
Autoreactive T cell lines derived from CD154-deficient MRL/lpr mice
| T cell proliferation with APC/ENAb | ||||||
|---|---|---|---|---|---|---|
| Isolated T cell lines | T cell proliferation with APC/ENA (SI)a | + IgG2a | anti-Ek | anti-Ak | %inhibition (anti-Ek/IgG2a) | TCR Vβusage |
| C2 | 1·52 | 14 531 | 3 004 | 12 964 | 77 | Vβ6 |
| G1 | 1·74 | 39 582 | 3 343 | 36 788 | 92 | Vβ8·1/8·2 |
| G2 | 1·36 | 66 534 | 3 952 | 56 638 | 95 | Vβ10 |
| P5 | 3·45 | 26 136 | 4 837 | 18 765 | 81 | Vβ8·3 |
| E7 | 3·88 | 31 249 | 14 468 | 33 245 | 54 | Vβ14 |
[3H]-thymidine incorporation (cpm) in the presence of extractantibodyle nuclear antigen (ENA)/[3H]-thymidine incorporation (cpm) in the absence of ENA.
[3H]-thymidine incorporation (cpm) in the presence of ENA with the antibodies shown. Mean cpm of triplicate culture is given; s.d. within each experiment were <10%.
In the presence of irradiated splenocytes from CD154-deficient MRL/lpr mice as a source of APC, the lines released either IFN-γ or IL-4 as respective indicators of Th1 or Th2 profiles (data not shown). Four of the five lines (C2, G1, G2, and P5) released IFN-γ after 24 and 72 h incubation, suggesting they had a Th1 phenotype. The last line, E7, released a small amount of IL-4 after 72 h incubation (indicating a Th0 or Th2 phenotype). Additionally, after stimulation with anti-CD3ɛ MoAb, the soluble form of TNF-α (sTNF-α) was detected in the culture supernatant of CD154-deficient Th1 lines (G1 and P5) and the CD154-intact autoreactive Th1 line (5-1), but not the control Th2 line (4-1).
mTNF-α and CD154 expression was examined on autoreactive Th1 or Th2 lines (Fig. 1). CD154 expression was observed on both Th1 and Th2 cells at 12 h, and later continued expression was seen in the Th2 line (4-1). Maximum expression of mTNF-α was observed after 24 h stimulation. mTNF-α but not CD154 expression was observed after 48 h stimulation in the 5-1 and P5 lines (data not shown) indicating that the expression of mTNF-α was predominantly induced on Th1 cells and followed CD154 expression.
Fig. 1.
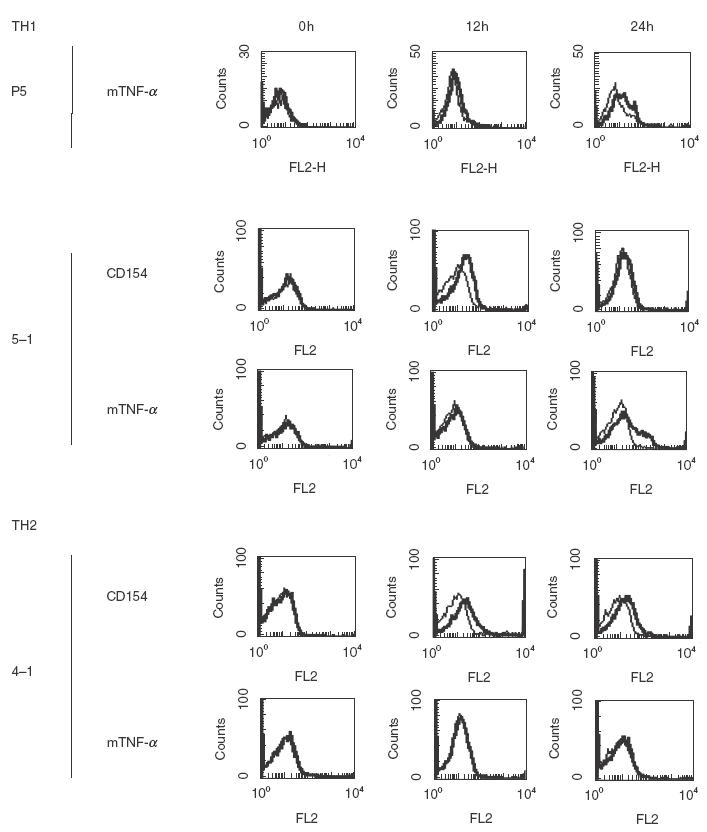
CD154 and the transmembrane form of TNF-α (mTNF-α) on activated MRL/lpr T cell lines. CD154-intact autoreactive Th1 (5-1), and Th2 (4-1) lines and CD154-deficient Th1 (P5) were stimulated with plate-bound anti-CD3ɛ MoAb (5 µg/ml) for the indicated time. CD154 was not expressed on P5 during 0–48 h stimulation with anti-CD3ɛ MoAb. Bold lines show staining with anti-CD154 or TNF-α MoAb, and regular lines show isotype control.
CD154-deficient T cell lines promote contact-dependent and -independent B cell proliferation
B7-2 (CD86) expression on MRL B cells was enhanced after co-culture with the CD154-deficient Th1 line (G1) for 24 h and 48 h (data not shown). All the lines induced B cell proliferative responses, which anti-CD154 MoAb (MR1) could not block (see Fig. 2a for a representative result). Anti-CD154 MoAb inhibited the B cell proliferation induced by the CD154-intact Th1 line 5-1, but not that induced by either of the CD154-deficient Th1 lines G1 or P5. To determine if B cell proliferation is contact-dependent, a membrane insert was used to prevent contact by the T cell lines with purified B cells (Fig. 2b). The CD154-deficient line (P5) induced substantial proliferation of B cells either with or without a membrane insert between irradiated P5 and purified B cells, indicating that P5 stimulates a B cell proliferative response in both a contact-dependent and -independent manner. In addition, under the same conditions, anti-IFN-γ or IL-2 MoAb partially inhibited B cell proliferation but anti-TNF-α did not. These results indicate that contact-independent T cell-mediated B cell proliferation is partially dependent on IFN-γ and IL-2, but not on sTNF-α.
Fig. 2.
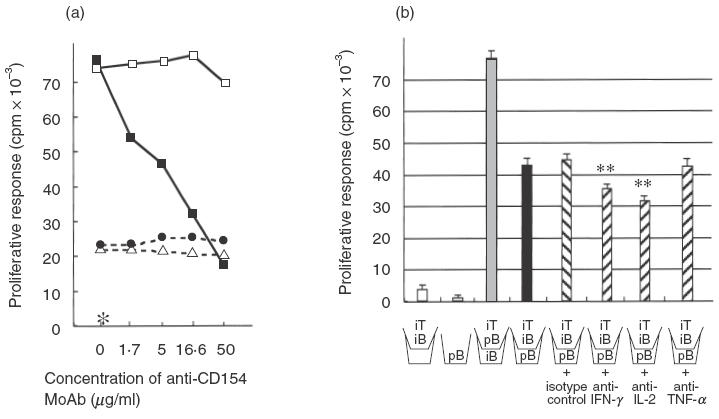
B cell proliferative responses to autoreactive Th1 lines. (a) Anti-CD154 MoAb (MR1) dramatically inhibited B cell proliferation induced by the CD154-intact line (5-1), but not that by the CD154-deficient line (G1 or P5). (b) A membrane insert (\_/) was used to prevent contact by T cell line (P5) with B cells. Irradiated (1500 rad) T cells, which do not proliferate themselves, enhanced purified B cell proliferation. Irradiated (3000 rad) B cells are thought to promote cytokine production from the T cell line as an APC. CD154-deficient lines induce B cell proliferation both in a contact-dependent and -independent manner; however, contact-independent B cell proliferation (black bar) was not inhibited at all by anti-TNF-α MoAb, but was inhibited by anti-IFN-γ or IL-2 MoAb (hatched bar). Concentration of MoAb or isotype control: 25 µg/ml. This figure shows the representative results of three experiments. iT; irradiated T cells (P5), pB; purified B cells, iB; irradiated B cells. **P <0·01 (versus isotype control). (a) □, 5-1 (with hamster IgG); •, G1; ▵, P5; ▪, 5-1; *B cells alone.
Involvement of mTNF-a and TNF-R in CD154-independent B cell activation
Next, to clarify whether the mTNF-α-TNF-R pathway participates in contact-dependent B cell proliferation, anti-TNF-α or TNF-R MoAb was added to the co-cultures of irradiated lines and purified B cells (Fig. 3). Anti-TNF-α MoAb induced a reduction of about 50% in the B cell proliferative responses induced by a CD154-deficient line (G1 or P5). As sTNF-α was not involved in contact-independent B cell proliferation, this result indicates that the anti-TNF-α MoAb completely abrogates contact-dependent B cell proliferation, and suggests that mTNF-α is responsible for triggering B cell proliferation. Anti-TNF-R2 MoAb also inhibited the proliferative responses induced by both CD154-deficient and intact Th1 lines. Anti-TNF-R1 MoAb inhibited B cell proliferation, but only at a high concentration (= 25 µg/ml).
Fig. 3.
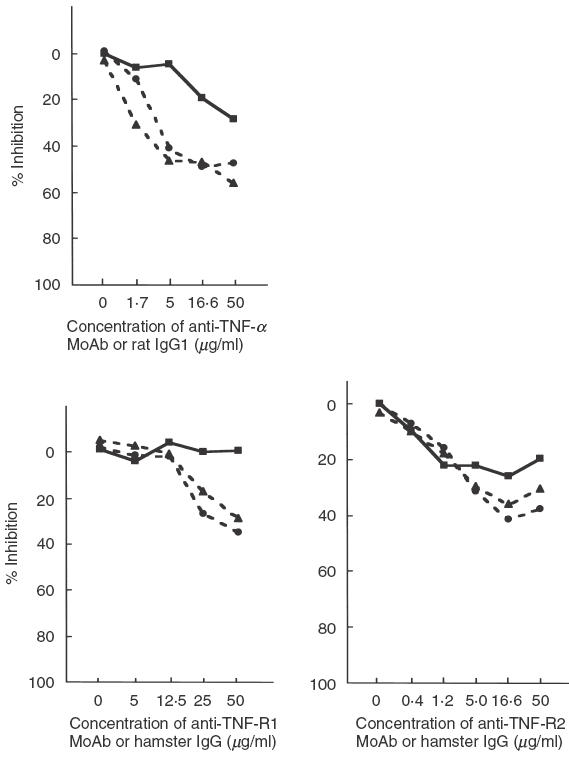
Inhibition by anti-TNF-α/TNF-R MoAb of B cell proliferative responses to CD154-intact (5-1) or -deficient (G1 and P5) Th1 cell lines. Anti-TNF-α MoAb and anti-TNF-R2 MoAb partially inhibit B cell proliferative responses, and are therefore thought to be mediated via contact-dependent T cell help. Anti-TNF-R1 MoAb also seems to block proliferation by G1 and P5, although a high concentration (>25 µg/ml) was required. Rat IgG1 and hamster IgG constitute isotype controls of anti-TNF-α and anti-TNF-R1 or R2 MoAb, respectively. % Inhibition = (1-{[3H]-thymidine uptake with blocking MoAb (cpm)/[3H]-thymidine uptake with isotype control(cpm)}) × 100. Mean cpm of triplicate cultures was used for calculations; s.d. for each experiment was less than 10%. ▪, 5-1; •, G1; ▴, P5.
We next examined whether our CD154-deficient Th1 cell lines had the capacity to provide B cell help for autoantibody production in vitro (Fig. 4a). While anti-dsDNA antibody was not released from CD154-deficient B cells after co-cultures with the lines (data not shown) two of the five lines, G1 and P5, induced high levels of SI, especially in the absence of the membrane insert, suggesting that both provide contact-dependent help for anti-snRNP antibody production. With the membrane insert, the SI of CD154-deficient lines was the same as that of PCC-Tg T cells, which is thought to constitute contact-independent help [9]. Moreover, in in vitro helper assays, both anti-TNF-α and TNF-R2 MoAb inhibited anti-snRNP antibody production but anti-CD154 and TNF-R1 MoAb did not (Fig. 4b). Anti-IFN-γ MoAb also inhibited anti-snRNP antibody production, indicating that IFN-γ is also involved in autoantibody production in a contact-independent manner.
Fig. 4.
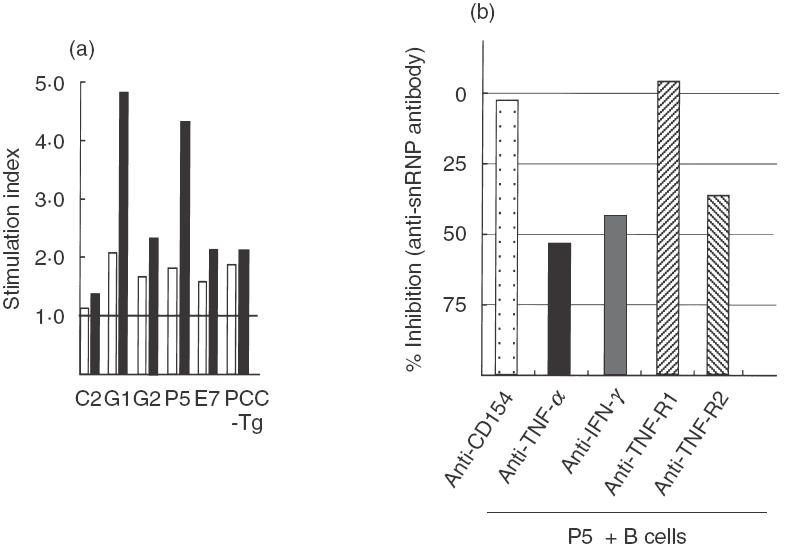
In vitro B cell helper assay. (a) A membrane insert was used to prevent contact by the T cell line with B cells (white bar). In the absence of the membrane, G1 and P5 could induce more than the standard level of SI. Stimulation index = (concentration in culture supernatant of experimental wells)/(concentration in culture supernatant of B cells alone). The figure shows representative results of three experiments. (b) Comparison of levels of anti-snRNP antibody in the supernatant of T cell line (P5)–B cell co-culture with the blocking antibody and isotype control determined by ELISA. Anti-TNF-α, IFN-γ, and TNF-R2 MoAb inhibited anti-snRNP antibody production, while anti-TNF-R1 MoAb did not. % Inhibition = (1-{titre of anti-snRNP antibody with blocking MoAb (O.D.405nm)/anti-snRNP antibody with isotype control (O.D.405nm)}) × 100. Concentrations of blocking antibody and isotype control: 25 µg/ml. This figure shows the representative results of three experiments. □, With membrane insert; ▪, without membrane insert.
Discussion
We successfully isolated two autoreactive CD4+αβ Th1 cell lines from CD154-deficient MRL/lpr mice. These lines (G1 and P5) have the capacity to provide B cell help for IgG anti-snRNP antibody production in a contact-dependent manner. Co-stimulation of mTNF-α, which is expressed predominantly on Th1 cells, and TNF-R2 was mainly involved in the production of IgG autoantibodies. In a previous study, we demonstrated that CD154-deficient MRL/lpr mice exhibited IgG2a class switching similar to that seen in wild-type MRL/lpr mice, and produced anti-snRNP, but not anti-dsDNA antibody [21]. These results indicate that CD154 is required for a high titre of anti-dsDNA antibody production, but that IgG class switching and the genesis of IgG2a anti-snRNP antibody are not necessarily dependent upon the pathway mediated via CD154 in lupus-prone MRL/lpr mice.
B cells or MHC class II has a critical role in T cell activation during the generation of systemic autoimmunity [25,26]. The MHC class II molecule (I-Ek) was required for the proliferation and IFN-γ secretion of CD154-deficient MRL Th cells. Superantigens including endogenous Mtvs (from mouse mammary tumour virus), however, do not seem to be involved in the proliferation of our Th1 lines because the proliferation of the G1 and P5 lines was increased in the presence of extractable nuclear antigens. Papiernik et al. showed that clonal deletion of Vβ6+ CD4+ T cells by Mtv-6-SAG was avoided in MRL/lpr mice [27]. The influence of known Mtvs on MRL/lpr T cell receptor should be determined to exclude involvement of the maternally inherited superantigen-induced T cell response.
On the other hand, the dramatic inhibition by anti-CD154 MoAb of B cell proliferation with the CD154-intact line indicates that CD40-CD154 signalling is profoundly involved in B cell up-regulation even after T cells have been primed [28]. CD154-deficient lines, nevertheless, still induced B7-2 expression, syngeneic B cell proliferation and anti-snRNP secretion, indicating that MRL Th1 cells have the capacity to provide B cell help for autoantibody production in the absence of CD154. The insertion of a membrane to separate Th1 and B cells reduces the B cell proliferation and autoantibody production, suggesting that the Th1 cell lines stimulate B cells both in both a contact-dependent and -independent manner.
mTNF-α is processed to generate sTNF-α by a disintegrin metalloproteinase, known as the TNF-α-converting enzyme (TACE) [29,30]. Aversa et al. reported that mTNF-α was rapidly expressed on human CD4+ Th2 cells activated with Con A and that the mTNF-α-TNF–R1 interaction can provide a contact-dependent signal for the production of IgG4 and IgE [31]. In our study, we demonstrated that mTNF-a, which was expressed on the Th1 line 12 h after stimulation, was involved in B cell activation mediated via TNF-R2. This observation is consistent with a previous report that mTNF-α is the prime activating ligand for TNF-R2 [32]. mTNF-α is known to be involved in autoimmune disorders [33–35], but this is the first report to show the extent of mTNF-α-mediated autoimmune T-B contact help and autoantibody production. Macchia et al. demonstrated that HIV-infected human T cell clones did not express CD154 but did express mTNF-α, which induced polyclonal B cell activation in a contact-dependent manner [36]. This report also suggested that under certain pathological conditions mTNF-α may be up-regulated independently of CD154 expression and activate B cells. As reported previously, defective TNF function may cause autoimmunity [37]. In our preliminary experiments, the G1 line failed to induce renal diseases suggesting that mTNF-α-expressing cells may stimulate anti-snRNP antibody, but not severe organ involvement, in lupus. mTNF-α expressing recombinant virus-infected cells, however, had no effect on B cell proliferation [38], suggesting that other molecules such as CD44H or ICAM-1 [39,40] may be required for mTNF-α-mediated B cell activation.
Recently, some co-stimulatory molecules such as CD70/CD27 or CD134/CD134L have been reported to be associated with T cell-dependent B cell responses [41]. CD70 (CD27L) has also been reported to have the capacity to activate B cells through engagement with CD27 [42]. CD27–CD70 co-stimulation then contributes to enhancement of the antigen-presenting capacity of B cells or plasma cell differentiation after CD40-dependent activation [43,44]. There may also be other candidates participating in Th1-type T cell responses [45] in the absence of the CD40–CD154 system. Although our B cell activation assay with the Fab fragment derived from anti-CD27 MoAb (clone: LG.3A10) [45] was performed under the same conditions, this MoAb did not affect B cell proliferation/IgG2a secretion (data not shown). mTNF-α and CD70 therefore appear to be associated with different stages in T-cell dependent B cell activation.
How and to what extent CD154-inactive T cells are involved in autoantibody production in human SLE remains unclear. Serum levels of anti-dsDNA antibody in human lupus fluctuate with the intensity of lupus manifestations, and appear to be sensitive to the effects of immunomodulatory agents. On the other hand, it is unusual for anti-snRNP (or Sm) antibody to disappear completely [46] or show marked responsiveness to therapy. The mechanism of anti-snRNP antibody production thus appears to be different from that of anti-dsDNA antibody, and not be independent of CD154. The investigation of co-stimulatory molecules including CD154 and mTNF-α in peripheral lymphocytes from active and inactive lupus patients who have both anti-DNA and RNP antibody is a future project of ours. A recent study using (NZBxNZW)F1 mice indicated that CD4+ T lymphocytes with preformed CD154 could not provide a proliferation signal or reduce the activation threshold in B cells [47], suggesting that the direct target cells for CD154-expressing T cells may not be anti-dsDNA antibody-producing B cells. It is thus possible that the mechanisms of anti-dsDNA antibody and anti-snRNP antibody production are different in the target cells of autoantigen-reactive T cells. Our preliminary results show that autoantibodies to the whole chromatin molecule, which are thought to represent the primary immune response, could sometimes be detected in CD154-deficient MRL/lpr mice. Anti-dsDNA antibody production may be the secondary immune response, for which CD40–CD154 engagement is essential, while antichromatin and anti-snRNP antibody are the primary response, for which CD154 may not be essential.
CD154-deficient MRL/lpr mice sometimes present with a much milder renal disease but severe skin disease [21]. These results are consistent with the previous observation that the pathogenesis of skin disease induced by αβ CD4+ T cells may be different from that induced by γδ T cells that need the CD40–CD154 signal [48]. A recent study found that the blockade of the CD40–CD154 interaction is not sufficient for complete suppression of lupus, and that additional blockade of the CD28/B7 molecule may be more beneficial [49]. Our findings also suggest that anti-CD154 MoAb therapy could suppress CD154-mediated severe lupus manifestations such as anti-dsDNA antibody synthesis and severe glomerulonephritis. Some manifestations, especially those related to the Th1-mediated immune response, however, may be resistant to anti-CD154 MoAb treatment. We therefore suggest that it is advantageous to treat murine lupus with a combined blockade of CD154 and TNF-R2.
Acknowledgments
We thank Saeed Fatenejad for the kind gift of TNF-R2-deficient MRL/lpr mice and helpful comments, Jean Zhang and Ping Zhu for their technical assistance with mice management, and Tom Taylor for technical assistance with flow cytometry. This work was supported by a grant from the Donaghue Foundation (DF-96–087).
REFERENCES
- 1.Eisenberg RA, Tan EM, Dixon FJ. Presence of anti-Sm reactivity in autoimmune mouse strains. J Exp Med. 1978;147:582–7. doi: 10.1084/jem.147.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonsson R, Tarkowski A, Backman K, Holmdahl R, Klareskog L. Sialadenitis in the MRL-l mouse: morphological and immunohistochemical characterization of resident and infiltrating cells. Immunology. 1987;60:611–6. [PMC free article] [PubMed] [Google Scholar]
- 3.Koh D-R, Ho A, Rahemutulla A, Fung-Leung W-P, Griesser H, Mak T-W. Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur J Immunol. 1995;25:2558–62. doi: 10.1002/eji.1830250923. [DOI] [PubMed] [Google Scholar]
- 4.Peng SL, Madaio MP, Hughes DPM, et al. Murine lupus in the antibodysence of αβ T cells. J Immunol. 1996;156:4041–9. [PubMed] [Google Scholar]
- 5.Chesnutt MS, Finck BK, Killeen N, Connolly MK, Goodman H, Wolfsy D. Enhanced lymphoproliferation and diminished autoimmunity in CD4-deficient MRL/lpr mice. Clin Immunol Immunopathol. 1998;87:23–32. doi: 10.1006/clin.1997.4492. [DOI] [PubMed] [Google Scholar]
- 6.Datta SK, Patel H, Berry D. Induction of a cationic shift in IgG anti-dsDNA autoantibodies: role of T helper cells with classical and novel phenotypes in three murine models of lupus nephritis. J Exp Med. 1987;165:1252–68. doi: 10.1084/jem.165.5.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naiki M, Chiang BL, Cawley D, et al. Generation and characterization of cloned T helper cell lines for anti-DNA responses in NZBH-2bm12 mice. J Immunol. 1992;149:4109–15. [PubMed] [Google Scholar]
- 8.Fatenejad S, Brooks W, Schwartz A, Craft J. Pattern of anti-small nuclear ribonucleoprotein antibodies in MRL/Mp-lpr/lpr mice suggests that the intact U1 snRNP particle is that autoimmunogenic target. J Immunol. 1994;152:5523–31. [PubMed] [Google Scholar]
- 9.Peng SL, Fatenejad S, Craft J. Induction of nonpathologic, humoral autoimmunity in lupus-prone mice by a class II-restricted, transgenic αβ T cell. Separation of autoantigen-specific and - nonspecific help. J Immunol. 1996;157:5225–30. [PubMed] [Google Scholar]
- 10.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA. 1992;89:6550–4. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van den Alfons JM, Noelle RJ, Roy M, et al. In vitro CD40–gp39 interactions are essential for thymus-dependent humoral immunity. I. In vivo expression of CD40 ligand, cytokines, and antibody production delineates sites of cognate T–B cell interactions. J Exp Med. 1993;178:1555–65. doi: 10.1084/jem.178.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foy TM, Scepherd DM, Durie FH, Aruffo A, Ledbetter JA, Noelle RJ. In vivo CD40–gp39 interactions are essential for thymus-dependent humoral immunity. II. Prolonged suppression of the humoral immune response by an antibody to the ligand for CD40, gp39. J Exp Med. 1993;178:1567–75. doi: 10.1084/jem.178.5.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Foy TM, Lamen JD, et al. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–34. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 14.Renshow BR, Fanslow WC, III, Armitage RJ, et al. Humoral immune responses in CD40 ligand-deficient mice. J Exp Med. 1994;180:1889–900. doi: 10.1084/jem.180.5.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, Noelle RJ. gp39–CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med. 1994;180:157–63. doi: 10.1084/jem.180.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grewal IS, Xu J, Fravell R. Impairment of antigen-specific T cell priming in mice lacking CD40 ligand. Nature. 1995;378:617. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 17.Mohan C, Shi Y, Lamen JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol. 1995;154:1470–80. [PubMed] [Google Scholar]
- 18.Early GS, Zhao W, Burns CM. Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand Black × New Zealand White mice. J Immunol. 1996;157:3159–64. [PubMed] [Google Scholar]
- 19.Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97:2063–73. doi: 10.1172/JCI118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. 1996;98:826–37. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Xu J, Madaio MP, et al. Autoimmune lpr/lpr mice deficient in CD40 ligand. Spontaneously Ig class switching with dichotomy of antoantibody responses. J Immunol. 1996;157:417–26. [PubMed] [Google Scholar]
- 22.Fatenejad S, Mamula M, Craft J. Role of intrastructural B and T cell determinants in the diversification of autoantibodies to ribonucleoprotein particles. Proc Natl Acad Sci USA. 1993;90:12010–4. doi: 10.1073/pnas.90.24.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheehan KCF, Pinckard JK, Arthur CD, Dehner LP, Goeddel DV, Schreiber RD. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: identification of a novel in vivo role for p75. J Exp Med. 1995;181:607–17. doi: 10.1084/jem.181.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubin RL. Enzyme-linked immunosorbent assay for anti-DNA and antihistone antibodies including anti-(H2A-H2B) In: Rose NR, Demacario EC, Fathey JL, Friedman H, Penn GM, editors. Manual of clinical laboratory immunology. 4. Washington DC: American Society for Microbiology; 1992. pp. 735–40. [Google Scholar]
- 25.Jevnikar AM, Grusby MJ, Glimcher LH. Prevention of nephritis in major histocompatibility complex class II-deficient MRL-lpr mice. J Exp Med. 1994;179:1137–43. doi: 10.1084/jem.179.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. 1998;160:51–9. [PubMed] [Google Scholar]
- 27.Papiernik M, Pontoux C, Golstein P. Non-exclusive Fas control and age dependence of viral superantigen-induced clonal deletion in lupus-prone mice. Eur J Immunol. 1995;25:1517–23. doi: 10.1002/eji.1830250607. [DOI] [PubMed] [Google Scholar]
- 28.Jacquot S, Kobata T, Iwata S, Morimoto C, Schlossman SF. CD154/CD40 and CD70/CD27 interactions have different and sequential functions in T cell-dependent B cell responses. Enhancement of plasma cell differentiation by CD27 signaling. J Immunol. 1997;159:2652–7. [PubMed] [Google Scholar]
- 29.Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 30.Moss ML, Jin S-LC, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature. 1997;385:733–6. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 31.Aversa G, Punnonen J, de Vries JE. The 26-kDa transmembrane form tumor necrosis factor α on activated CD4+ T cell clones provides a co-stimulatory signal for human B cell activation. J Exp Med. 1993;177:1575–85. doi: 10.1084/jem.177.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grell M, Douni E, Wajant H, et al. The transmembrane form of tumour necrosis factor is prime activating ligand of the 80 kDa tumour necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 33.Kusters S, Tiegs G, Alexopoulou L, et al. In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol. 1997;27:2870–5. doi: 10.1002/eji.1830271119. [DOI] [PubMed] [Google Scholar]
- 34.Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgene mice. J Immunol. 1997;158:438–45. [PubMed] [Google Scholar]
- 35.Georgopoulos S, Plows D, Kollias G. Transmembrane TNF is sufficient to induce localized tissue toxicity and chronic inflammatory arthritis in transgene mice. J Inflamm. 1996;46:86–97. [PubMed] [Google Scholar]
- 36.Macchia D, Almerigogna F, Parronchi P, Ravina A, Maggi E, Romagnani S. Membrane tumor necrosis factor-α is involved in the B cell activation induced by HIV-infected human T cells. Nature. 1993;363:464–6. doi: 10.1038/363464a0. [DOI] [PubMed] [Google Scholar]
- 37.Kontoyiannis D, Kollias G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur J Immunol. 2000;30:2038–47. doi: 10.1002/1521-4141(200007)30:7<2038::AID-IMMU2038>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 38.Jumper MD, Nishioka Y, Davis LS, Lipsky PE, Meek K. Regulation of human B cell function by recombinant CD40 ligand and other TNF-related ligands. J Immunol. 1995;155:2369–78. [PubMed] [Google Scholar]
- 39.Guo Y, Wu Y, Shinde S, Sy M-S, Aruffo A, Liu Y. Identification of a co-stimulatory molecule rapidly induced by CD40L as CD44H. J Exp Med. 1996;184:955–61. doi: 10.1084/jem.184.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shinde S, Wu Y, Guo Y, et al. CD40L is important for induction of, but not response to, co-stimulatory activity. ICAM-1 as the second co-stimulatory molecule rapidly up-regulated by CD40L. J Immunol. 1996;157:2764–8. [PubMed] [Google Scholar]
- 41.Morimoto S, Kanno Y, Tanaka Y, et al. CD134L engagement enhances human B cell Ig production. CD154/CD40, CD70/CD27, and CD134/CD134L interactions coordinately regulate T cell-dependent B cell responses. J Immunol. 2000;164:4097–104. doi: 10.4049/jimmunol.164.8.4097. [DOI] [PubMed] [Google Scholar]
- 42.Kobata T, Jacquot S, Kozlowski S, Agematsu K, Schlossman SF, Morimoto C. CD27–CD70 interactions regulate B-cell activation by T cells. Proc Natl Acad Sci USA. 1995;92:11249–53. doi: 10.1073/pnas.92.24.11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ranheim EA, Cantwell MJ, Kipps TJ. Expression of CD27 and its ligand, CD70, on chronic lymphocytic leukemia B cells. Blood. 1995;85:3556–65. [PubMed] [Google Scholar]
- 44.Hartwig UF, Karlsson L, Peterson PA, Webb SR. CD40 and IL-4 regulate murine CD27L expression. J Immunol. 1997;159:6000–8. [PubMed] [Google Scholar]
- 45.Gravestein LA, Nieland JD, Kruisbeek AM, Borst J. Novel mAbs reveal potent co-stimulatory activity of murine CD27. Int Immunol. 1995;4:551–7. doi: 10.1093/intimm/7.4.551. [DOI] [PubMed] [Google Scholar]
- 46.Fisher DE, Reeves WH, Wisniewolski R, Lahita RG, Chiorazzi N. Temporal shifts from Sm to ribonucleoprotein reactivity in systemic lupus erythematosus. Arthritis Rheum. 1985;28:1348–55. doi: 10.1002/art.1780281206. [DOI] [PubMed] [Google Scholar]
- 47.Lettesjö H, Burd GP, Mageed RA. CD4+ T lymphocytes with constitutive CD40 ligand in preautoimmune (NZB × NZW) F1 lupus-prone mice. Phenotype and possible role in autoreactivity. J Immunol. 2000;165:4095–104. doi: 10.4049/jimmunol.165.7.4095. [DOI] [PubMed] [Google Scholar]
- 48.Peng SL, McNiff JM, Madaio MP, et al. αβT cell regulation and CD40 ligand dependence in murine systemic autoimmunity. J Immunol. 1997;158:2464–70. [PubMed] [Google Scholar]
- 49.Daikh DI, Finck BK, Linsley PS, Hollenbaugh D, Wofsy D. Long-term inhibition of murine lupus by brief simultaneous blockade of B7/CD28 and CD40/gp39 co-stimulation pathways. J Immunol. 1997;159:3104–8. [PubMed] [Google Scholar]