

Abstract
Bronchiectasis is a common complication of primary antibody deficiency but the incidence of antibody deficiency as an underlying cause of bronchiectasis is largely undefined. In this study the humoral immune status of a cohort of bronchiectatic patients was investigated to detect the frequency of significant antibody deficiency and to determine the extent of immunological investigation which is appropriate for routine assessment of bronchiectasis patients. Fifty-six out-patients (with a mean age of 59·6 years) had serum immunoglobulins, IgG subclasses and specific antibodies to capsular polysaccharides of Haemophilus influenzae and Streptococcus pneumoniae measured. Where specific antibody levels were low, where possible, appropriate immunization with pneumococcal or conjugated Haemophilus polysaccharide vaccines was offered and the responses quantified. Three of 56 patients had low total serum IgG levels. Thirteen of 56 had deficiencies of either a single IgG subclass or combinations of two or more subclasses, with IgG4 being most frequently implicated (9/56). Twenty-nine of 56 had low basal specific polysaccharide antibody levels. Test immunization, where performed, produced satisfactory responses in all cases except one, where a specific defect of responsiveness to pneumococcal polysaccharide was identified. This study indicates that antibody deficiency is an uncommon aetiological/underlying factor in the causation of bronchiectasis beyond the fourth decade and that detailed investigation of humoral immune status as a routine in bronchiectasis patients, at least at this age, is not generally justified.
Keywords: antibody deficiency, bronchiectasis, Haemophilus influenzae, Streptococcus pneumoniae
INTRODUCTION
Bronchiectasis is a chronic debilitating condition characterized by abnormally and permanently dilated pulmonary airways [1,2]. The main symptoms are chronic cough with daily tenacious sputum production and recurrent respiratory infections. The exact aetiology of bronchiectasis is unknown, but the initiating event is thought to involve an infectious insult − usually a pneumonitis − with the subsequent establishment of a vicious circle of inflammation, microbial colonization (including organisms such as Haemophilus influenzae type b (Hib), untypeable Haemophilus strains, Streptococcus pneumoniae (pneumococcus) and Pseudomonas aeruginosa) and impaired mucociliary clearance [3]. The poor clearance of microorganisms and the ongoing inflammatory response cause irreparable damage to the bronchial wall, leading to permanent dilation of the bronchi. Predisposing factors to the development of bronchiectasis include bronchial obstruction, cystic fibrosis and congenital anatomical lung abnormalities. In addition, a variety of immunological abnormalities in patients with bronchiectasis have been described and include defects in neutrophil function [4,5] and deficiencies in humoral immunity. The latter can involve both the major immunoglobulin classes IgM, IgG and IgA [6–9] and the IgG subclasses IgG1, IgG2, IgG3 and IgG4 [6,8,9]. Impaired specific antibody responses to encapsulated pyogenic bacteria such as Hib and pneumococcus have also been reported [9,10].
Primary antibody deficiency diseases are genetically determined defects of the humoral arm of the immune system and involve an absence or reduced levels of one or more immunoglobulin classes or subclasses and/or defects of specific antibody formation [11]. The major, defined, more common forms of primary antibody deficiency, all of which have been linked with development of bronchiectasis [6–10], include X-linked agammaglobulinaemia, IgA deficiency, common variable immunodeficiency, IgG subclass deficiency and deficiency of antibody formation to specific pathogens. Many forms of primary antibody deficiency go undiagnosed or are diagnosed late, and untreated or suboptimally treated patients suffer unnecessarily from recurrent and often serious infections resulting in tissue damage. Bronchiectasis is a common long-term complication [12,13].
Early diagnosis of primary antibody deficiency and treatment with immunoglobulin (principally IgG) replacement therapy can enable affected individuals with panhypogammaglobulinaemia or significant IgG deficiency to live normal lives — the incidence of infections is considerably decreased and the development of bronchiectasis prevented or progression of established disease retarded [14,15]. The efficacy of current immunoglobulin replacement regimes is less certain for patients with isolated IgM deficiency or IgG subclass deficiency in which the capacity to make specific antibodies is intact and is unlikely to be of benefit in simple IgA deficiency.
The purpose of this study was to determine the detailed humoral immune status of patients with bronchiectasis attending a local chest clinic. The long-term objective is to improve patient management by determining the degree of immunological screening which is appropriate in the routine assessment of patients with bronchiectasis on presentation, and to identify those patients who may benefit from specific treatment of antibody deficiency. The parameters examined included measurement of total immunoglobulin, IgG subclasses and specific antibody levels to both Hib and pneumococcus in the patient group and the ability of those patients with low specific antibody levels to respond to appropriate immunization. Previous groups [6–9] have studied bronchiectatic cohorts with similar objectives although the spectrum of laboratory parameters examined by each differed from those of the current study.
MATERIALS AND METHODS
Patients
The study group consisted of 56 patients (33 male, 23 female, age range 37–84 years, mean age 59·6 years) with bronchiectasis all of whom were regularly attending an outpatient chest clinic. Bronchiectasis had been confirmed by high resolution computerized tomography and/or bronchography in all cases. The group had not previously been investigated for possible immune deficiency but other secondary causes of bronchiectasis not related to antibody deficiency, such as cystic fibrosis, allergic bronchopulmonary aspergillosis and α-1 antitrypsin deficiency, had already been excluded. Forty-seven of the group were lifelong non-smokers, seven had stopped smoking cigarettes between 11 and 37 years prior to the study and two were active smokers at the time of investigation. A detailed history of recent respiratory infections requiring antibiotics in the group in the 12 months prior to the study revealed a spectrum of organisms isolated included Candida, coliforms, Hib, P. aeruginosa, Moraxella catarrhalis, Staphylococcus aureus and pneumococcus. No patients were known to have been vaccinated previously against Hib or pneumococcus.
Patients involved in the study were bled at a routine clinic visit and the sera obtained were then tested immediately according to the study protocol or frozen at − 70°C for batch testing. A humoral immune status screen was performed on each sample and consisted of assessment of total immunoglobulin levels, IgG subclasses and levels of specific antibody against the surface capsular polysaccharides of S. pneumoniae and Hib.
Immunoglobulin quantification
Total serum immunoglobulin levels (IgM, IgG and IgA) were quantified by rate nephelometery using standard sera (Dade Behring Ltd, Milton Keynes, UK) traceable to international reference preparations for IgM, IgG and IgA. The presence of any paraprotein bands was excluded by agarose gel electrophoresis (REP Ultra SP Procedure Kit, Helena Laboratories, Gateshead, Tyne & Wear, UK).
IgG subclass quantification
The IgG subclasses IgG1–IgG4 were quantified by a commercial single radial immunodiffusion method (The Binding Site Ltd, Birmingham, UK) employing stabilized, pooled, normal human serum calibrators and pre-poured agarose gel plates containing mono-subclass specific sheep antihuman polyclonal antisera. The lower limit of sensitivity for each subclass assay was 1·4 mg/dL (IgG1), 0·8 mg/dL (IgG2), 6 mg/dL (IgG3) and 2·5 mg/dL (IgG4).
Specific antibody quantification
Serum IgG to Hib capsular polysaccharide and pneumococcal capsular polysaccharide (PCP) were measured using commercial ELISA assays (Binding Site Ltd) employed with minor modifications from the methods recommended by the manufacturer. For Hib capsular polysaccharide antibody assays patient sera were diluted 1 : 10, 1 : 50 or 1 : 100 according to measurable antibody present with levels calculated using the appropriate dilution factor. The assay employs six prediluted, stabilized human sera calibrated against the Food and Drug Administration human anti-Hib capsular polysaccharide reference serum. The assay was employed principally in this study with a lower limit of measurement of 0·11 µg/ml.
For anti-PCP measurement sera were again assayed at dilutions of 1 : 10, 1 : 50 or 1 : 100 as appropriate and antibodies to non-specific cell wall polysaccharide (c-Ps) [16] were removed by absorption of patient sera and calibrators with c-Ps. The assay was adjusted for maximal sensitivity in the lower ranges, such that the detection range was 3·3 µg/ml−84 µg/ml.
Immunization and repeat testing schedule
Patients whose sera contained low levels of specific antibody against Hib polysaccharide or PCP were immunized with, as appropriate, single intramuscular doses of HibTITER® (Wyeth Laboratories, Maidenhead, UK) or Act-Hib® (Pasteur Merieux MSD Ltd, Maidenhead, UK) and/or Pneumovax®II (Pasteur Merieux). One month after immunization a venous blood sample was taken and the serum assayed for levels of polysaccharide antibodies (Hib and/or PCP). Post-immunization samples were assayed at dilutions of up to 1 : 1000 to ensure that O.D. values fell within the range of the calibration curve(s). Pre- and post-immunization samples from each patient were processed routinely within the same assay for both Hib- and PCP-specific antibodies.
Normal ranges
The locally derived reference ranges (geometric mean ± 2 s.d.G) in routine usage for IgM, IgG and IgA were, respectively, 0·5–1·5 g/l, 8·0–14·0 g/l and 0·9–3·5 g/l. IgG subclass adult reference ranges were constructed using sera from 60 (30 female, 30 male, age range 40–79 years, mean age 57·5 years) age-matched normal adult controls. Identical methodology to that of the test sera was used to give 95th percentile ranges of 318–1020 mg/dL (IgG1), 125–665 mg/dL (IgG2), 20–192 mg/dL (IgG3) and 6·1–134 mg/dL (IgG4). For Hib-specific antibody levels sera from 168 normal unimmunized adults (80 male, 88 female, age range 20–80 years, mean 47·8 years) were assayed with a resulting range (geometric mean 1·25 µg/ml ± 2 s.d.G) of 0·12–13·28 µg/ml. Patient samples were considered to have below normal levels if no Hib antibodies were detected (<0·11 µg/ml) when serum was assayed at 1:10. This is broadly in agreement with previous studies [17,18] which have shown that, for a non-vaccinated population, a lower limit of 0·15 µg/ml gives a good inverse correlation with the incidence of H. influenzae type b infection and that anti-Hib levels maintained above 0·15 µg/ml reflects an ability to respond to specific capsular polysaccharide and to produce a protective response to natural infection. A maintained level of 1·0 µg/ml has been suggested as a correlate of the level of specific antibody which offers optimal post-immunization protection [17,18], although whether this may also apply in naturally acquired immunity is unproven. In the normal adult group used to establish the reference range 4·8% of individuals had naturally acquired Hib antibody levels below 0·15 µg/ml and 41·1% were found to have levels below 1·0 µg/ml. To derive a normal range for the PCP assay, 240 sera (120 female, 120 male, age range 16–80 years, mean 45·8 years) were obtained from the local unimmunized normal adult population producing a normal range lower limit (95th centiles) of 15·4 µg/ml.
RESULTS
Total serum immunoglobulins
Three patients had serum total IgG levels below the local normal reference range (see Table 1). The three sera assayed at 4·0, 4·7 and 7·8 g/l. None of the 56 patients had any evidence of IgM or of partial/complete IgA deficiency. One patient showed IgM levels above the reference range (5·4 g/l), and seven and 19 patients had elevated levels of IgG (ranging from 14·8 to 28·6 g/l) and IgA (3·6–8·0 g/l), respectively (Table 2).
Table 1.
Numbers of bronchiectasis patients with serum immunoglobulin concentrations below the normal range lower limits
| IgM | IgG | IgG1 | IgG2 | IgG3 | IgG4 | IgA |
|---|---|---|---|---|---|---|
| 0 | 3 | 3 | 3 | 1 | 9 | 0 |
| 0%a | 5·4% | 5·4% | 5·4% | 1·8% | 16·1% | 0% |
Numbers of patients expressed as a percentage of the total study group (n = 56).
Table 2.
Numbers of bronchiectasis patients with serum immunoglobulin concentrations above the normal range upper limits
| IgM | IgG | IgG1 | IgG2 | IgG3 | IgG4 | IgA |
|---|---|---|---|---|---|---|
| 1 | 7 | 6 | 3 | 4 | 1 | 19 |
| 1·8%a | 12·5% | 10·7% | 5·4% | 7·1% | 1·8% | 33·9% |
Numbers of patients expressed as a percentage of the total study group (n = 56)
IgG subclasses
The incidence of low levels of the four subclasses is shown in Table 1. Thirteen patients had subnormal IgG subclass levels either as isolated low levels of a single subclass or as combinations of two or more subclasses. Three patients had low serum IgG1 levels (202, 296 and 316 mg/dL), with two of these also having low total IgG levels. Three patients also had low IgG2 (68, 96 and 106 mg/dL) levels. Only one patient was identified as having a low IgG3 level (19 mg/dL). Eight patients with measurable IgG4 had levels below the lower limit of normal (ranging from 2·6 to 5·4 mg/dL), and for one serum sample IgG4 was undetectable using this radial immunodiffusion method. In only two cases were more than one subclass deficiency combined within the same subject, one with low levels of IgG2 (68 mg/dL) and IgG4 (4·0 mg/dL) and the other with low levels of IgG1 (202 mg/dL), IgG2 (96 mg/dL) and IgG4 (2·6 mg/dL). Re-assessment of IgG subclass levels in these patients with low measured levels was undertaken in samples drawn between 4 and 7 months after primary sampling for the study. An identical profile of measured deficiencies as were found in the primary samples resulted for this group demonstrating that, at least in the short term, the IgG subclass abnormalities detected were not transient.
Table 2 shows numbers of cases with IgG subclass levels above the normal ranges. Eleven sera had high levels, six of IgG1 (ranging from 1030 to 2270 mg/dL), three of IgG2 levels (702–780 mg/dL), four of IgG3 levels (210–461 mg/dL) and one with a high IgG4 level (304 mg/dL). Elevated levels of IgG subclasses occurred as isolated findings except in two cases, one with combined elevated levels of IgG1 (1740 mg/dL), IgG2 (760 mg/dL) and IgG3 (210 mg/dL) and the other of IgG2 (780 mg/dL) and IgG3 (240 mg/dL).
Specific (Hib and PCP) antibodies
Twenty-three patients showed below normal levels of anti-Hib antibodies, three patients had below normal levels of anti-PCP antibodies and three patients had combined subnormal levels of antibodies against both Hib and PCP (Table 3). For the remaining patients, Hib antibody levels ranged between 0·19 and 4·20 µg/ml (mean 0·59 µg/ml) (Fig. 1) and PCP antibody levels ranged from 19·1 µg/ml to 72·1 µg/ml in 11 patients and were>84 µg/ml in the remaining 39 (Fig. 2). Antibody levels could not be correlated to any degree with organisms identified during infective episodes. Eight of these patients had chronic pseudomonas carriage with the remainder all experiencing a range of infections with staphylococci, Moraxella and coliforms requiring antibiotic therapy in the 12 months prior to the start of the study.
Table 3.
Numbers of bronchiectasis patients with below normal levels of IgG to Hib and PCP
Numbers of patients expressed as a percentage of the total study group (n = 56).
Specific Hib IgG antibody undetectable (<0·11 µg/ml).
Fig. 1.
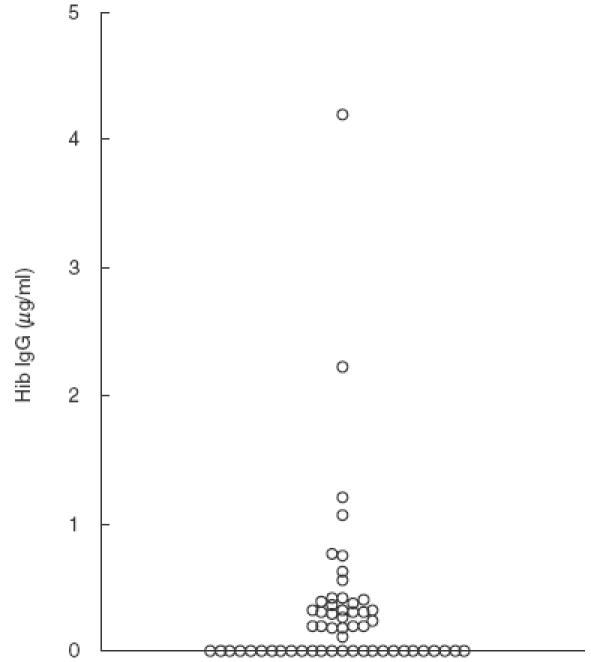
Serum IgG antibodies to Hib in 56 patients with bronchiectasis. The antigen preparation consisted of Hib oligosaccharide conjugated to human serum albumin. Antibody levels <0·11 µg/ml were denoted as being low.
Fig. 2.
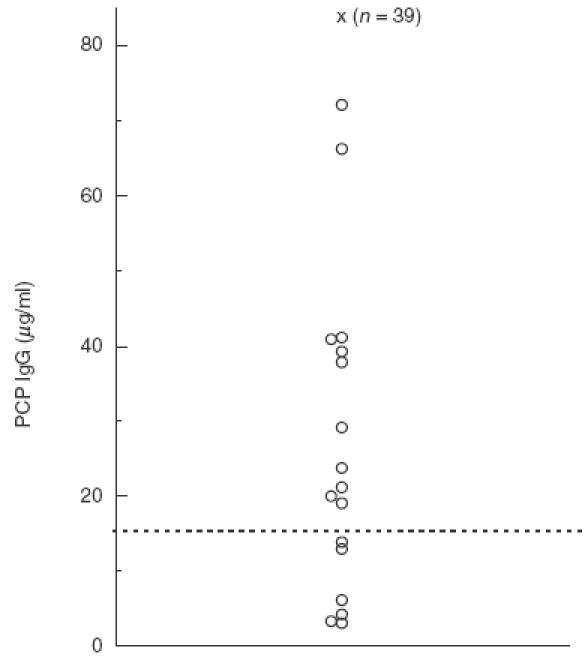
Serum IgG antibodies to pneumococcal capsular polysaccharide (PCP) in 56 patients with bronchiectasis. The antigen preparation contained PCP purified from 23 common serotypes of S. pneumoniae. The horizontal line (–––) indicates the lower limit of the normal range (15·4 µg/ml). The patient samples indicated by ‘x’ had PCP IgG levels of>84 µg/ml.
Immunization of patients with subnormal levels of Hib and/or PCP antibodies
Immunization with conjugated Hib vaccine and/or Pneumovax® II was offered as appropriate to patients with low Hib and/or PCP antibody levels. In 13 cases of subnormal anti-Hib levels this was not taken up either because patients refused vaccination (10 patients) or had died in the interim (three patients). One of the three patients with subnormal levels of anti-PCP refused immunization with Pneumovax®II. Immunization of two of the three patients with combined subnormal levels of Hib and PCP antibodies was not performed because of death in one case and refusal in the other. Consequently, appropriate immunization was possible in only 10 patients with low anti-Hib levels, two patients with low anti-PCP levels and a single patient with combined subnormal anti-Hib and anti-PCP levels. More detailed information on these 13 patients, including post-immunization antibody levels, is given in Table 4. The mean percentage FEV1/predicted FEV1 was 62·2% with an average of three infective episodes requiring antibiotics (range 0–11) in the 12 months prior to immunization.
Table 4.
IgG associated humoral immune values for 13 vaccinated bronchiectatic patients
| Post-immunization serology | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient no. | Total IgG (g/l) | IgG1 (mg/dL) | IgG2 (mg/dL) | IgG3 (mg/dL) | IgG4 (mg/dL) | Anti-Hib (µg/ml) | Anti-PCP (µg/ml) | Vaccination Hib and/or Pneumovax | Hib (µg/ml) and/or PCP (µg/ml) | |
| 3 | 8·4 | 636 | 424 | 144 | 8·5 | <0·11* | >84 | Hib | 45 | |
| 5 | 13·2 | 797 | 521 | 178 | 18·3 | <0·11* | >84 | Hib | 66 | |
| 13 | 10·4 | 631 | 240 | 90 | 46·0 | <0·11* | 21·1 | Hib | 56 | |
| 17 | 15·5* | 1330* | 67·6* | 42 | 4·0* | <0·11* | 8·3* | Hib + Pneumovax | 10 | 5.4* |
| 28 | 8·9 | 537 | 136 | 101 | 5·4* | <0·11* | >84 | Hib | 9 | |
| 29 | 14·7* | 972 | 363 | 77 | 70·9 | 0·2 | 4·2* | Pneumovax | 150 | |
| 33 | 13·3 | 769 | 349 | 86 | 22·3 | <0·11* | >84 | Hib | 16 | |
| 44 | 11·6 | 797 | 318 | 82 | 20·0 | <0·11* | >84 | Hib | 275 | |
| 45 | 10·5 | 513 | 424 | 83 | 29·8 | <0·11* | >84 | Hib | 11 | |
| 46 | 4·7* | 316* | 147 | 20 | 12·7 | <0·11* | >84 | Hib | 22 | |
| 48 | 7·8* | 561 | 293 | 89 | 16·3 | <0·11* | >84 | Hib | 39 | |
| 49 | 13·4 | 797 | 408 | 74 | 60·0 | <0·11* | >84 | Hib | 12 | |
| 55 | 8·9 | 513 | 147 | 110 | (u)* | 0·19 | 13·9* | Pneumovax | 562 | |
Result outside relevant normal range. (u) = undetectable by assay employed.
In all cases, except one, immunization produced satisfactory specific antibody responses. In the case of immunization with H. influenzae type b-conjugated polysaccharide vaccine an adequate response was deemed to be one which produced optimal protective antibody levels above 1·0 µg/ml [17] with an arbitrarily defined relative incremental rise in measurable antibody levels (minimum 10-fold). The 11 patients immunized produced minimal relative rises between 90- and 2750-fold above baseline levels. In the case of pneumococcal immunization, in the absence of a defined and accepted correlate of protective immunity, an adequate response was again taken to be an increase in the level of measured specific antibody to within or above the normal range involving a minimum 10-fold rise between pre- and post-immunization samples. The single case (no.17) which produced an inadequate response was a 49-year-old female who had high levels of total IgG and IgG1 but subnormal levels of IgG2 and IgG4. Her response to Hib immunization was satisfactory, but no measurable antibody response to PCP was induced following Pneumovax®II immunization. Repeat test immunization with Pneumovax®II was not possible in this patient.
DISCUSSION
Bronchiectasis is found frequently in patients with proven antibody deficiency, often occurring as a result of late diagnosis or suboptimal treatment [13]. In contrast the results of this study show that tissue damage caused by recurrent infection due to underlying antibody deficiency is only rarely an aetiological factor in the causation of bronchiectasis, at least in patients within the age range studied here. The results also suggest that expensive and detailed immunological investigation of bronchiecta-sis patients in this age group on a routine basis is generally unwarranted.
The reported incidence of an association between bronchiectasis and antibody deficiency appears to vary considerably between different groups of bronchiectatic patients. In two recent reports by Hill et al. [19] and De Gracia et al. [9], in which IgG subclass deficiency was principally considered, the frequencies of detection were 6% and 48%, respectively. In the study described in this paper, the incidence of IgG subclass deficiency was 23% with the relative contribution of subnormal IgG4 levels being similar to that found by Hill and colleagues. However, the findings differ from those of De Gracia and coworkers, both in the overall incidence of immunoglobulin deficiency (23% compared to 48%), and the subclass involved (principally IgG4 in this study and IgG2 in that of De Gracia et al.). The predominance of subnormal IgG4 levels in bronchiectasis patients in this study may not be surprising, as studies of IgG subclass levels in the normal population have reported that between 10% [20] and 25% [21] of adults have undetectable serum IgG4. The sensitivity of IgG4 subclass measurement is method-dependent and care should therefore be exercised in definitively interpreting isolated subnormal or absent IgG4 levels as being indicative of humoral immune deficiency especially where relatively insensitive methods such as radial immunodiffusion are used for quantification.
The elevated levels of IgA, IgG and the G1 and G3 subclasses in bronchiectasis patients described here are in agreement with previous studies [6,9,19] and are reflective of reactive responses to airway inflammation in bronchiectasis. The predominance of IgA in these cases is consistent with repeated antigenic challenge at the bronchial mucosal surface.
It is known that normal or elevated serum levels of IgG can co-exist with and mask an IgG subclass deficiency [22]. However, deficiencies of the major immunoglobulin classes or IgG subclasses did not appear to have a frequent role in the development of bronchiectasis in the patients investigated in this study. Therefore, it was surmised that the low incidence of detected antibody defects may be concealing a more subtle humoral defect, such as an inability to produce specific antibodies against the capsular polysaccharides of S. pneumoniae or H. influenzae. To investigate this further, basal and, where possible, post-immunization serum levels of Hib and pneumococcal-specific antibodies were measured in these patients. The findings would suggest that, as with the major immunoglobulin classes and IgG subclasses, defects of specific antibody production in bronchiectasis are relatively infrequent with only a single case (no. 17) with absent PCP reactivity being identified. It should be noted, however, that the approach taken in this study of measuring the total pneumococcal IgG level to all 23 polysaccharide serotypes employed in the ELISA may mask deficient natural or immunization responses to one or more individual serotypes [16]. The reasons for the relatively high incidences of subnormal basal specific antibody levels, particularly those against Hib, in the study cohort in comparison to the normal population are unknown. The study does not exclude the possibility that this group of patients may respond to natural infection or to immunization in a normal quantitative manner initially, but have a poor duration of post-exposure protective or measurable antibody levels.
In the case of patient 17, the results would indicate a defect of antibody production against at least one thymus-independent antigen (PCP), although the response to peptide conjugated Hib vaccine does not rule out a more widespread defect of natural responsiveness to unconjugated polysaccharide antigens. Detailed review of this case has shown that she has Sjögren's syndrome, with which her immunoglobulin profile is compatible, but little evidence of problems with recurrent infections with encapsulated bacteria (one episode of coliform infection in the 12 months prior to the study), no evidence of low grade pulmonary inflammation and no evidence, currently, of progression of her bronchiectasis. Therefore, on clinical grounds, her defined immunological abnormality does not presently merit active immunological intervention with immunoglobulin replacement. This serves to reaffirm the integrated and overlapping nature of the effector defences of the immune system where deficiencies in one area may be compensated for by defence mechanisms in other areas. In addition, in considering the immune competence of individual patients the role of factors other than those discussed here should not be forgotten. These include antibody avidity and idiotype, individual isotype responses to infection or immunization, the duration of effective antibody responses, antibody responses to non-polysaccharide components of bacteria and the potential importance of local pulmonary, rather than systemic, antibody production [19].
The findings of this study suggest that detailed investigation of humoral immune status, including assessing basal levels of specific antibody, in most cases of bronchiectasis is difficult to justify, although it should be acknowledged that the age of the patient cohort investigated here differed from previous studies [6–9,19]. Routine measurement of the major serum immunoglobulin isotypes may be justifiable in terms of ready availability, ease of measurement and relative inexpense but a different, more intensive general approach to investigation may be required in younger patients [9]. A definitive policy for clinical units and laboratories would be based optimally upon formal analysis of the costs of screening all patients against the benefits, financial and otherwise, of detecting the occasional case who may benefit from active management of clinically significant antibody deficiency. Consideration of the costs should not only take into account the direct and indirect costs of antibody replacement but should weigh this against the global costs and quality of life issues of missed diagnosis which may include frequent admissions to hospital or outpatient clinics, antibiotic costs and frequent or prolonged periods off work. For patients with recognized indicators of antibody deficiency such as recurrent, severe or persistent infections, infection with unusual pathogens or with other indicators such as a family history of immune problems, unexplained joint symptoms, excessive or deficient lymphoid tissues, unexplained hepatosplenomegaly or failure to thrive in infancy [13] an alternative approach is indicated. In such patients more detailed investigation is appropriate and should include methods such as those described in this study as well as specialist immunological advice and input. The latter may be useful in ensuring that clinical investigation protocols optimally reflect current scientific knowledge and of relevance in relation to the design of this study are current and growing indications that determination of IgG subclass levels are of limited, possibly very limited, value in the assessment of patients with suspected immunodeficiency [11].
Acknowledgments
This study was supported by a grant from the University of Aberdeen. The authors are grateful for the co-operation of the Thoracic Physicians Drs J. A. R. Friend, D. Godden, J. S. Legge and S. Watt for the provision of sera and data on bronchiectasis patients. We thank the Clinical Biochemistry and Immunopathology laboratories of Aberdeen Royal Infirmary for performing the quantification of total immunoglobulins and IgG subclasses, respectively. We are grateful to Dr D. A. Stead for his constructive advice throughout the project.
REFERENCES
- 1.Nicotra MB. Bronchiectasis. Semin Respir Med. 1994;9:31–40. [PubMed] [Google Scholar]
- 2.Barker AF. Bronchiectasis. Semin Thorac Cardiovas Surg. 1995;7:112–8. [PubMed] [Google Scholar]
- 3.Cole PJ. Inflammation. A two-edged sword – the model of bronchiectasis. Eur J Respir Dis. 1986;69:6–15. [PubMed] [Google Scholar]
- 4.Cazzola G, Valletta EA, Ciaffoni S, Roata C, Mastella G. Neutrophil function and humoral immunity in children with recurrent infections of the lower respiratory tract and chronic bronchial suppuration. Ann Allergy. 1989;63:213–8. [PubMed] [Google Scholar]
- 5.Nikolaizik WH, Warner JO. Aetiology of chronic suppurative lung disease. Arch Dis Child. 1994;70:141–2. doi: 10.1136/adc.70.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanely PJ, Corbo G, Cole PJ. Serum IgG subclasses in chronic and recurrent respiratory infections. Clin Exp Immunol. 1984;58:703–8. [PMC free article] [PubMed] [Google Scholar]
- 7.Barker AF, Craig S, Bardana EJ. Humoral immunity in bronchiectasis. Ann Allergy. 1987;59:179–82. [PubMed] [Google Scholar]
- 8.Feldman C, Weltman M, Wadee A, Sussman G, Smith C, Zwi S. A study of immunoglobulin G subclass levels in black and white patients with various forms of obstructive lung disease. S Afr Med J. 1992;83:9–12. [PubMed] [Google Scholar]
- 9.De Gracia J, Rodrigo MJ, Morell F, Vendrell M, Miravitlles M, Cruz MJ, Codina M, Bofill JM. IgG subclass deficiencies associated with bronchiectasis. Am J Respir Crit Care Med. 1996;153:650–5. doi: 10.1164/ajrccm.153.2.8564113. [DOI] [PubMed] [Google Scholar]
- 10.Ambrosino DM, Siber GR, Chilmonczyk BA, Jernberg JA, Finberg RW. An immunodeficiency characterised by impaired antibody responses to polysaccharides. N Engl J Med. 1987;316:790–3. doi: 10.1056/NEJM198703263161306. [DOI] [PubMed] [Google Scholar]
- 11.IUIS Scientific Group. Primary immunodeficiency diseases. Report of an IUIS Scientific Group. Clin Exp Immunol. 1999;118(Suppl. 1):1–34. doi: 10.1046/j.1365-2249.1999.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spickett GP, Misbah SA, Chapel HM. Primary antibody deficiency in adults. Lancet. 1991;337:281–4. doi: 10.1016/0140-6736(91)90882-p. [DOI] [PubMed] [Google Scholar]
- 13.Chapel H. Consensus on diagnosis and management of primary antibody deficiencies. Br Med J. 1994;308:581–5. doi: 10.1136/bmj.308.6928.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haeney M. Intravenous immune globulin in primary immunodeficiency. Clin Exp Immunol. 1994;97(Suppl. 1):11–15. [Review] [PMC free article] [PubMed] [Google Scholar]
- 15.Skull S, Kemp A. Treatment of hypogammaglobulinaemia with intravenous immunoglobulin. Arch Dis Child. 1996;74:527–30. doi: 10.1136/adc.74.6.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musher DM, Luchi MJ, Watson DA, Hamilton R, Baughn RE. Pneumococcal polysaccharide vaccine in young adults and older bronchitics: determination of IgG responses by ELISA and the effect of absorption of serum with non-type specific cell wall polysaccharide. J Infect Dis. 1990;161:728–35. doi: 10.1093/infdis/161.4.728. [DOI] [PubMed] [Google Scholar]
- 17.Käyhty H, Peltda H, Karanko V, Mäkelä PH. The protective level of serum antibodies to the capsular polysaccharide of Haemophilus influenzae type b. J Infect Dis. 1983;147:1100. doi: 10.1093/infdis/147.6.1100. [DOI] [PubMed] [Google Scholar]
- 18.Anderson P. The protective level of serum antibodies to the capsular polysaccharide of Haemophilus influenzae type b. J Infect Dis. 1984;149:1034. doi: 10.1093/infdis/149.6.1034. [DOI] [PubMed] [Google Scholar]
- 19.Hill SL, Mitchell JL, Burnett D, Stockley RA. IgG subclasses in the serum and sputum from patients with bronchiectasis. Thorax. 1998;53:463–8. doi: 10.1136/thx.53.6.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.French MAH, Harrison G. Serum IgG subclasses concentrations in healthy adults: a study using monoclonal antibodies. Clin Exp Immunol. 1984;56:473–5. [PMC free article] [PubMed] [Google Scholar]
- 21.Oxelius V. Quantitative and qualitative investigation of serum IgG subclasses in immunodeficiency diseases. Clin Exp Immunol. 1979;36:112–16. [PMC free article] [PubMed] [Google Scholar]
- 22.Shield JPH, Strobel S, Levinsky RJ, Morgan G. Immunodeficiency presenting as hypergammaglobulinaemia with IgG2 subclass deficiency. Lancet. 1992;449:448–50. doi: 10.1016/0140-6736(92)91769-5. [DOI] [PubMed] [Google Scholar]