

Abstract
Oral tolerance has been characterized as an immunological hyporesponsiveness to fed antigen. Previous studies have suggested that high-dose oral tolerance involves the preferential interaction of B7 with CTLA-4 on the T cell. To determine whether similar mechanisms are involved in the induction of low-dose oral tolerance, mice were treated with anti-CTLA-4 monoclonal antibody (MoAb), with or without IL-12, at the time of feeding. Results showed that anti-CTLA-4 MoAb alone failed to restore cellular proliferation, antibody titres and IFN-γ levels; however, IL-4 cytokine levels in OVA-fed mice were partially restored. In contrast, administration of IL-12 along with anti-CTLA-4 MoAb to mice during feeding completely prevented the suppression of Th1 immune responses, as shown by increased serum IgG2a titres, IFN-γ production and cell proliferation. These results suggest that blocking B7-CTLA-4 interactions in the presence of IL-12 prevents the induction of low-dose oral tolerance at the Th1 cell level.
Keywords: anti-CTLA-4, IL-12, oral, tolerance
INTRODUCTION
Oral administration of a specific soluble antigen induces systemic hyporesponsiveness upon antigenic challenge with the same specific antigen. Typically, feeding soluble antigen prior to immunization results in diminished CD4+ immune responses, characterized by decreased antibody secretion, lymphocyte proliferation and cytokine secretion [1–5]. The mechanism involved in oral tolerance induction is thought to vary, depending on the dose of antigen fed [6]. High doses of fed antigen are thought to lead to anergy or depletion of antigen-specific CD4+ cells [7–13] while low-dose oral tolerance most probably involves a combination of both active suppression (cytokines) and anergy [14–18].
Early in vitro studies using cloned cell lines found that primary stimulation of T cells in the absence of B7/CD28 interaction (the co-stimulatory signal) rendered cells anergic [19–22]. However, it has been suggested recently by Perez et al. that clonal anergy of Th1 cells arises not from the absence of a co-stimulatory signal, but from interaction of B7 with CLTA-4 rather than with CD28 on the T cell [23]. It is believed that binding of B7 to CTLA-4 results in the delivery of a negative or down-regulatory signal [24–26] manifested by inhibited IL-2 receptor expression [26] and restriction of normal cell cycle progression [27]. Studies have shown that suppressed immune responses induced either by UV-radiation [28], superantigen [29], i.p. injection of antigen [23] or oral administration of large doses of antigen [30] were prevented at least partially by in vivo administration of anti-CLTA-4 monoclonal antibody (MoAb). Further studies showed that suppression of IFN-γ levels in i.p. tolerized mice could be prevented if IL-12 was administered at the time of tolerance induction [31]. This result was not unexpected, given that IL-12 is a cytokine known to promote the differentiation of IFN-γ-producing Th1 cells [32].
Although it is generally accepted that anergy/depletion of antigen-specific CD4+ cells is the mechanism responsible for high-dose oral tolerance, it is possible that anergy plays a role in the induction of low dose oral tolerance as well. Previous results from our laboratory using a low-dose feeding regimen (1 mg/mouse for 3 consecutive days) have indicated that low doses of antigen administered orally can result in suppressed antibody, cellular proliferation and cytokine secretion [14]. In addition, others have shown that feeding low doses of chicken egg albumin (OVA) also leads to a reduction in these same types of immune responses [15,16,33]. Therefore, it was of profound interest to determine if this suppression was due, at least in part, to B7-CTLA-4 interaction and/or decreased levels of IL-12. Results from our present study indicate that administration of anti-CTLA-4 MoAb and IL-12 to mice at the time of low-dose oral tolerance induction prevents suppression of Th1 type immune responses, including IgG2a isotype antibody production, cellular proliferation and IFN-γ production.
MATERIALS AND METHODS
Mice
Female 6–8-week-old BALB/c mice were obtained from Harlan–Sprague Dawley (Indianapolis, IN, USA) and were housed in the animal facility at Thomas More College in accordance with guidelines outlined by the American Association for Laboratory Animal Care.
Antigen, antibodies and cytokines
OVA, Grade V, was obtained from Sigma Chemical Co. (St Louis, MO, USA). Rat IgG was purchased from Sigma Chemical Co. Anti-CTLA-4 MoAb was obtained from culture supernatants of hybridoma UC10-4F10-11 cells (ATCC, Rockville, MD, USA). Hybridomas were cultured in 1% Nutridoma-SP serum-free medium (Boehringer Mannheim, Indianapolis, IN, USA) and the secreted monoclonal antibodies were purified partially by ammonium sulphate precipitation. The antibody preparation was then dialysed against phosphate buffered saline (PBS). Sample purity and antibody concentration were determined by SDS-PAGE and Western blot analysis, using rat IgG (Sigma Chemical Co.) as a standard. Murine rIL-12 was obtained from R&D Systems (Minneapolis, MN, USA).
In vivo antibody and cytokine treatment of mice
Mice were injected i.p. with 75 µg/mouse of anti-CTLA-4 MoAb, 75 µg of rat IgG and/or 1 µg of IL-12. The concentration of antibodies and cytokines administered was based on those utilized in previously published reports [23,31,34]. Mice were treated on days 0, + 1 and + 2.
Induction of tolerance and immunization
Mice were tolerized orally by feeding 1 mg of OVA/mouse for 3 consecutive days; antigen was delivered in 0·5 ml of water by gastric intubation. Mice were fed on days 0, + 1 and + 2. Ten days after the last feeding (day + 12), all mice were immunized in the footpad and tail base with OVA (10 µg/mouse) emulsified in IFA.
Measurement of OVA-specific serum antibodies
Mice were bled from the retro-orbital plexus prior to sacrifice (day + 23) unless otherwise indicated. Sera from individual mice in each experimental group were stored at −70°C until time of assay. Enhanced binding 96-well ELISA plates were coated overnight at 4°C with 150 µl of a 2-µg/ml solution of OVA in 0·05 m carbonate buffer. After three washes with PBS/0·05% Tween 20, 150 µl aliquots of standard and serum in doubling dilutions were added to the plates and incubated for 1 h at 37°C. After three washes with PBS/0·05% Tween 20, 150 µl of either alkaline phosphatase (AP)-coupled goat antimurine antibody (H + L chain) (diluted 1 : 2000; Boehringer Mannheim), AP-coupled goat antimurine antibody IgG1 (diluted 1 : 1000; Zymed Laboratories, South San Francisco, CA, USA) or AP-coupled goat antimurine antibody IgG2a (diluted 1 : 1000; Zymed Laboratories) were added to the plates and incubated for an additional hour at 37°C. Plates were then washed again three times with PBS/0·05% Tween 20 and 100 µl/well of 4-nitrophenylphosphate (1 mg/ml) was added as a substrate. Absorbance was measured at a wavelength of 405 ηm using an automated microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). Sera from all mice (3–4 mice/group) were assayed individually.
Collection of tissue samples
After cervical dislocation of mice, the popliteal lymph nodes (PLN) were removed and single cell suspensions made. Unless otherwise stated, cells were cultured in supplemented RPMI 1640 (Gibco BRL) containing 5% heat inactivated FCS, 25 mm Hepes, 1 mm non-essential amino acids, 1 mm sodium pyruvate, 2 mm l-glutamine, 5 × 10−5m 2-mercaptoethanol, Pen/Step (100 units of penicillin and 100 mg of streptomycin) and 5 mg/ml gentomycin (cRPMI). Cultures were maintained at 37°C in humidified 5% CO2 atmosphere.
In vitro lymphocyte proliferative responses
Eleven days after footpad and tail base immunization, mice were sacrificed and single cell suspensions of PLN were prepared as described above. Cells were aliquoted at 5 × 105 cells/well in 96-well flat-bottom plates along with OVA at final concentrations of 1000 and 100 µg/ml. All cultures were then incubated at 37°C in humidified 5% CO2 for 48 h. Proliferative responses were assayed using the BrdU cell proliferation colourimetric kit (Boerhinger-Mannheim), as per the manufacturer's recommendations. Briefly, BrdU was added to cultures and the cells were incubated for 8 h at 37°C; BrdU was then removed by centrifugation. After air-drying for 1 h, fixdent solution was added to each well and cells were incubated for an additional 30 min at room temperature (RT). Fixdent was removed and enzyme-linked anti-BrdU MoAb was added; cells were incubated for 1 h at RT. Plates were then washed three times and substrate (TMB) was added for an additional 30 min. Absorbance was measured at a wavelength of 450 ηm using an automated microplate reader (BioTek Instruments, Inc.). Addition of 100 µg/ml of an unrelated antigen, bovine serum albumin (BSA), was added to control wells to assess non-specific proliferation.
Cytokine analysis
Single cell suspensions of PLN were obtained from mice as described above. Cells were aliquoted at 4 × 106cells/well in 24-well flat-bottom plates along with either cRPMI alone or 1000 µg/ml OVA (total volume of 2 ml). Cells used for TGF-β1 analysis were cultured in X-VIVO 20 serum-free media (BioWhittaker, Walkersville, MD, USA) + 1% Nutridoma SP (Roche Molecular Biochemicals, Indianapolis, IN, USA). Cells were then incubated at 37°C in humidified 5% CO2 for 48 h (for IL-4 and IFN-γ analysis) or 72 h (for TGF-β1 analysis). Supernatants were harvested and stored at −70°C until assayed. IL-4, IFNγ and TGF-β1 production was quantified using murine IL-4, IFNγ, and TGF-β1 ELISA sets, respectively (BD Biosciences, Pharmingen, San Diego, CA, USA). TGF-β1 was quantified using the human TGF-β1 immunoassay set which has 100% cross-reactivity with mouse TGF-β1. TGF-β1 samples were activated prior to assay by incubating samples with 1 N HCL at 1 : 25 dilution for 60 min; all samples were then neutralized by adding 1 N NaOH at 1: 25 dilution. All ELISAs were carried out as per the manufacturer's recommendation. Briefly, coating antibody was added to 96-well plates for 24 h at 4°C, then washed. Plates were blocked for 1 h at RT using BSA buffer. Next, standards and samples were added and allowed to incubate for 2 h at RT. After washing, biotinylated detecting antibody plus horseradish peroxidase conjugate was added for 1 h at RT. After washing, TMB substrate was added for 30 min and the reaction was terminated by the addition of 2 N sulphuric acid solution. Absorbance was measured on an ELISA reader (Bio Tek Instruments, Inc.) set at 450 ηm. The following ranges of concentrations were used to generate standard curves: IL-4 (7·8 pg/ml to 500 pg/ml), IFN-γ (31·3 pg/ml to 2000 pg/ml) and TGF-β1 (62·5–4000 pg/ml). The concentration of experimental samples was determined using the KC junior computer software program (Bio Tek Instruments, Inc.).
Statistical analysis
For proliferation and cytokine assays, pooled samples for each group were assayed in either quadruplicate (proliferation) or duplicate (cytokine) and expressed as O.D. ± standard deviation. For serum analysis experiments, serum from individual mice (3–4 mice/group) were run and the standard deviations determined. Statistical significance of differences among experimental values was determined using Student's t-test for multiple comparisons. Values of P ≤ 0·05 were considered significant, unless otherwise stated. All experiments were repeated at least once.
RESULTS
Effect of anti-CTLA-4 MoAb treatment on cellular responses
Initial experiments were performed to assess the role of CTLA-4 in the suppressed proliferation response of mice fed low doses of OVA. As expected, control (rat IgG-treated) mice fed low doses of OVA exhibited decreased levels of proliferation compared to water-fed mice; treatment with anti-CTLA-4 MoAb during feeding of antigen failed to prevent this suppression (Fig. 1). Suppression indices (SI) for PLN from rat-IgG-treated, OVA-fed mice stimulated with 1000 and 100 µg/ml of OVA were 0·44 and 0·50, respectively, while those for anti-CTLA-4-treated, OVA-fed mice were 0·43 and 0·28, respectively. (Suppression indices were calculated by dividing values of OVA-fed mice by those of the corresponding water-fed mice.)
Fig. 1.
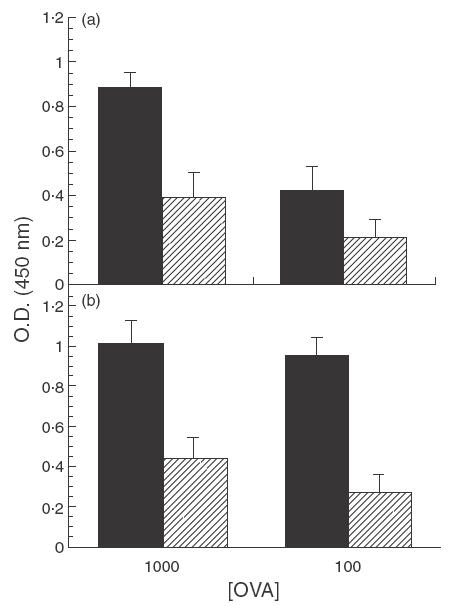
Effect of in vivo anti-CTLA-4 MoAb administration on cell proliferation in orally tolerized mice. Mice were treated i.p. with either (a) rat IgG (75 µg/mouse) or (b) anti-CTLA-4 MoAb (75 µg/mouse) on days 0, + 1 and + 2 and fed OVA (1 mg/mouse) on days 0, + 1 and + 2. Ten days after feeding, mice were injected with 10 µg of OVA/IFA in the footpad and tail base and sacrificed 11 days later. Popliteal lymph node (PLN) cells were incubated in vitro with 1000 and 100 µg of OVA and proliferation was assessed using a BrdU cell proliferation colourimetric kit (see Materials and Methods). The error bars represent standard deviations calculated from quadruplicate samples. (a) ▪, Rat IgG/H2O;  , rat IgG/OVA. (b) ▪, Anti-CTLA-4/H2O; , anti-CTLA-4/OVA.
, rat IgG/OVA. (b) ▪, Anti-CTLA-4/H2O; , anti-CTLA-4/OVA.
Previous studies have shown that in vivo injection of anti-CTLA-4 MoAb prevented suppressed cytokine levels in mice tolerized by feeding high doses of antigen (IL-4 and IFN-γ) [30] but not by i.p./i.v. administration of antigen (IFN-γ) [31]. Using a low-dose feeding regimen our results showed that, as expected, PLN from rat IgG, OVA-fed mice exhibited suppressed IL-4 and IFN-γ levels compared to water-fed control mice (Table 1). When treated with anti-CTLA-4 MoAb, IFN-γ levels continued to remain suppressed in OVA-fed mice compared to water-fed controls, while IL-4 levels were restored (Table 1).
Table 1.
Effect of anti-CTLA-4 on IL-4, IFN-γ and TGF-β1 secretion in the PLN1
| Treatment | Fed | In vitro stimulus | [IL-4] (pg/ml) | [IFN-γ] (pg/ml) | [TGF-β1] (pg/ml) |
|---|---|---|---|---|---|
| Rat IgG | H20 | 1000 µg OVA | 6·69 ± 0·34 | 13780 ± 187 | 68 ± 65 |
| Rat IgG | OVA | 1000 µg OVA | 0·09 ± 0·12*2 | 293 ± 13* | 312 ± 36* |
| Rat IgG | H20 | Medium | 0·00 ± 0·00 | 0 ± 0 | 10 ± 14 |
| Rat IgG | OVA | Medium | 0·00 ± 0·00 | 0 ± 0 | 151 ± 76 |
| Anti-CTLA-4 | H20 | 1000 µg OVA | 46·38 ± 0·67 | 21188 ± 1987 | 824 ± 18 |
| Anti-CTLA-4 | OVA | 1000 µg OVA | 51·37 ± 1·14 | 1682 ± 161* | 477 ± 79* |
| Anti-CTLA-4 | H20 | Medium | 30·93 ± 0·60 | 2169 ± 65 | 355 ± 6 |
| Anti-CTLA-4 | OVA | Medium | 10·87 ± 0·07* | 121 ± 7* | 287 ± 68* |
Mice were treated i.p. with either rat IgG or anti-CTLA-4 (75 µg/mouse) on days 0, + 1 and + 2 and fed OVA (1 mg) on days 0, + 1 and + 2. Ten days after feeding, mice were injected with 10 µg of OVA/IFA in the footpad and tail base and sacrificed 11 days later. PLN cells were incubated in vitro with and without 1000 µg of OVA and cytokine levels were assessed using IL-4, IFN-γ and TGF-β1-specific ELISA kits (see Materials and Methods section).
Asterisks indicate Student's t-test values that differ significantly (P ≤ 0·01) from the corresponding value for the water-fed control. The experiment was repeated twice.
In addition to IL-4 and IFN-γ, TGF-β1 levels were assessed, due to the putative role that this cytokine plays in oral tolerance induction [17,18]. As shown in Table 1, significantly higher levels of TGF-β1 were secreted from antigen-stimulated PLN cells in tolerized mice treated with rat IgG. In contrast, when mice were simultaneously fed low doses of OVA and treated with anti-CTLA-4 MoAb, the level of TGF-β1 from cells was significantly lower than that of anti-CTLA-4 MoAb-treated, water-fed mice. It should be noted that treatment with anti-CTLA-4 MoAb appeared to result in an overall increase in cytokine production.
Effect of anti-CTLA-4 MoAb treatment on the humoral responses
To determine whether suppression of humoral responses involved CTLA-4, tolerized mice were treated with anti-CTLA-4 MoAb and antibody titres were assessed. Similar to rat IgG control mice, total Ig serum antibody remained significantly suppressed in anti-CTLA-4 MoAb-treated OVA-fed mice compared to water-fed control (Fig. 2a). Furthermore, analysis of IgG1 and IgG2a isotypes revealed that these subclasses of antibody also remained suppressed in anti-CTLA-4-treated OVA-fed mice (Fig. 2b,c). It is interesting to note that anti-CTLA-4 MoAb treatment appeared to enhance suppression of the IgG1 isotype.
Fig. 2.
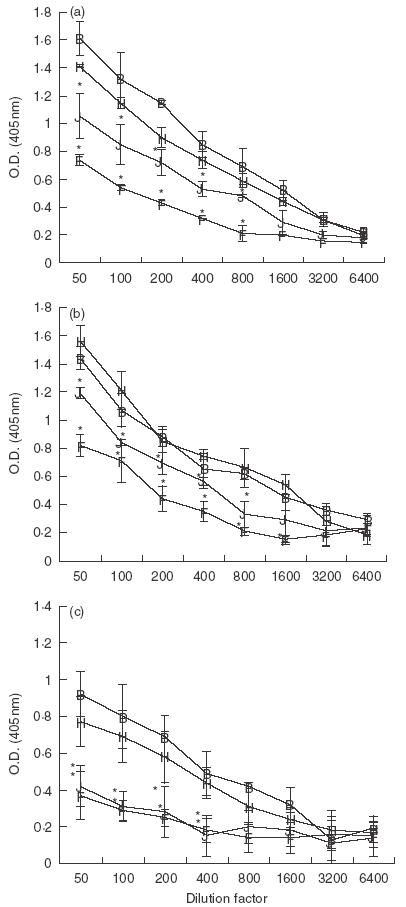
Effect of in vivo anti-CTLA-4 MoAb administration on serum antibody production in orally tolerized mice. Mice were treated i.p. with either rat IgG (75 µg/mouse) or anti-CTLA-4 MoAb (75 µg/mouse) on days 0, + 1 and + 2 and fed OVA (1 mg) on days 0, + 1 and + 2. Ten days after feeding, mice were injected with 10 µg of OVA/IFA in the footpad and tail base and bled 10 days later. Sera from mice were stored at −70°C. Sera from individual mice (3–4 mice/group) were measured for total Ig (a) IgG1 (b) or IgG2a (c) using ELISA (see Materials and Methods). Asterisks indicate Student's t-test values that differ significantly (P ≤ 0·05) from the corresponding value for the water-fed control. B, Rat IgG/H2O; J, rat IgG/OVA; H, anti-CTLA-4/H2O; F, anti-CTLA-4/OVA.
To confirm the results of serum antibody levels measured on day 10 after immunization, antibody titres in experimental mice over time were assessed. To accomplish this, mice were fed low doses of OVA while simultaneously treated with either anti-CTLA-4 MoAb or rat IgG. Eight days after the last feeding, mice were immunized i.p. with OVA/IFA and bled at 1-week intervals for 4 weeks. Results from this experiment demonstrated that total IgG serum antibody, as well as isotypes (IgG1 and IgG2a) remained suppressed on days 21 and 28 after immunization (data not shown).
Effect of anti-CTLA-4 MoAb and IL-12 treatment on cellular responses
Our results indicated that anti-CTLA-4 MoAb treatment alone could not prevent suppressed immune responses in mice fed low doses of OVA; therefore, it was of interest to determine if IL-12, in addition to anti-CTLA-4 MoAb, could mediate abrogation. As seen previously, cells from OVA-fed mice treated with rat IgG or anti-CLTA-4 MoAb alone exhibited suppressed proliferation (Fig. 3a). In contrast, PLN cells from OVA-fed mice treated with both anti-CTLA-4 MoAb and IL-12 were no longer suppressed when compared to water-fed controls (Fig. 3b). Interestingly, treatment with IL-12 alone appeared to partially inhibit suppressed proliferative responses as well, since no significant suppression was observed in this group when cells were stimulated in vitro with 1000 µg/ml of OVA (Fig. 3b). The SI for PLN from rat-IgG-treated, OVA-fed mice stimulated with 1000 and 100 µg/ml of OVA were 0·57 and 0·48, respectively, those for anti-CTLA-4-treated, OVA-fed mice were 0·53 and 0·43, respectively, those for IL-12-treated, OVA-fed mice were 0·88 and 0·13, respectively, and those for anti-CTLA-4/IL-12-treated, OVA-fed mice were 1·4 and 1·1, respectively. It should be noted that administration of IL-12 appeared to down-regulate overall proliferation responses; therefore evidence of suppression was limited to comparison with the water-fed control of the corresponding group.
Fig. 3.
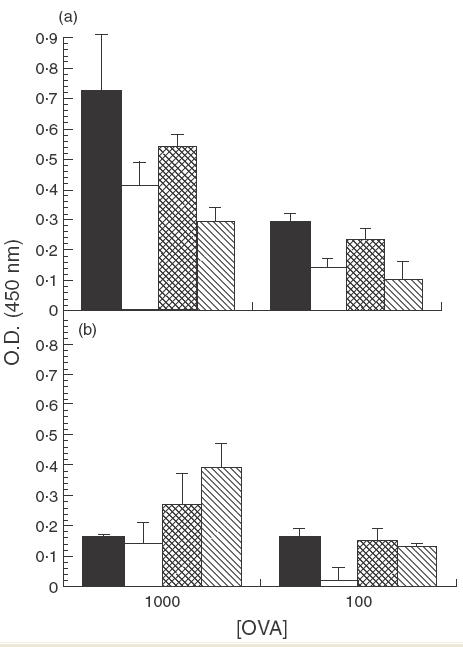
Effect of in vivo anti-CTLA-4 MoAb and IL-12 administration on cell proliferation in orally tolerized mice. Mice were treated i.p. with either rat IgG (75 µg/mouse) or anti-CTLA-4 MoAb (75 µg/mouse) (a) or with IL-12 (1 µg/mouse) or anti-CTLA-4/IL-12 (b) on days 0, + 1 and + 2. Mice were fed OVA (1 mg/mouse) on days 0, + 1 and + 2. Ten days after feeding, mice were injected with 10 µg of OVA/IFA in the footpad and tail base and sacrificed 11 days later. PLN cells were removed and incubated in vitro with 1000 µg/ml or 100 µg/ml of OVA and proliferation was assessed using a BrdU cell proliferation colourimetric kit (see Materials and Methods section). The error bars represent stadard deviations calculated from quadruplicate samples. In (a): ▪, Rat IgG/H2O; □, rat IgG/OVA;  , anti-CTLA-4/H2O;
, anti-CTLA-4/H2O; , anti-CTLA-4/OVA. In (b): ▪, IL-12/H2O; □, IL-12/OVA; , IL-12/anti-CTLA-4/H2O; , IL-12/anti-CTLA-4/OVA.
, anti-CTLA-4/OVA. In (b): ▪, IL-12/H2O; □, IL-12/OVA; , IL-12/anti-CTLA-4/H2O; , IL-12/anti-CTLA-4/OVA.
The effect of anti-CTLA-4 MoAb and IL-12 treatment on suppressed cytokine levels in tolerized mice was also assessed. Results showed that rat IgG, OVA-fed mice exhibited reduced IFN-γ levels compared to water-fed mice and treatment with anti-CTLA-4 MoAb or IL-12 alone had little or no effect on this suppression (Table 2). In contrast, OVA-fed mice treated with both anti-CTLA-4 MoAb and IL-12 failed to exhibit significantly suppressed levels of IFN-γ compared to water-fed controls. As expected, levels of IL-4 were suppressed in control, orally tolerized mice (Table 2). Tolerized mice treated with anti-CTLA-4 MoAb alone continued to exhibit significantly suppressed levels of IL-4, although to a much lesser degree than that of rat IgG control mice. When IL-12 was co-administered with anti-CTLA-4 MoAb, levels of IL-4 continued to remain profoundly suppressed in OVA-fed mice (Table 2). Treatment with IL-12 alone resulted in minimal or undetectable levels of IL-4.
Table 2.
Effect of anti-CTLA-4 and IL-12 administration on IL-4 and IFN-γ secretion in the PLN1
| Treatment | Fed | In vitro stimulus | [IL-4] (pg/ml) | [IFN-γ] (pg/ml) |
|---|---|---|---|---|
| Rat IgG | H20 | 1000 µg OVA | 26·92 ± 6·13 | 5533 ± 745 |
| Rat IgG | OVA | 1000 µg OVA | 0·04 ± 0·07*2 | 2680 ± 79* |
| Rat IgG | H20 | Medium | 0·34 ± 0·60 | 289 ± 21 |
| Rat IgG | OA | Medium | 0·65 ± 1·12 | 177 ± 11* |
| Anti-CTLA-4 | H20 | 1000 µg OVA | 34·83 ± 5·50 | 11971 ± 1267 |
| Anti-CTLA-4 | OVA | 1000 µg OVA | 22·95 ± 3·4* | 3149 ± 275* |
| Anti-CTLA-4 | H20 | Medium | 11·01 ± 3·51 | 814 ± 43 |
| Anti-CTLA-4 | OA | Medium | 11·97 ± 5·38 | 233 ± 26* |
| IL-12 | H20 | 1000 µg OVA | 2·40 ± 3·59 | 3496 ± 324 |
| IL-12 | OVA | 1000 µg OVA | 0·00 ± 0·00 | 1123 ± 81* |
| IL-12 | H20 | Medium | 0·00 ± 0·00 | 200 ± 28 |
| IL-12 | OA | Medium | 0·34 ± 0·60 | 187 ± 15* |
| IL-12/anti-CTLA-4 | H20 | 1000 µg OVA | 75·93 ± 16·06 | 4825 ± 117 |
| IL-12/anti-CTLA-4 | OVA | 1000 µg OVA | 7·01 ± 5·02* | 4253 ± 231 |
| IL-12/anti-CTLA-4 | H20 | Medium | 14·22 ± 1·98 | 1027 ± 37 |
| IL-12/anti-CTLA-4 | OVA | Medium | 0·00 ± 0·0 | 415 ± 14* |
Mice were treated i.p. with either rat IgG (75 µg/mouse), anti-CTLA-4 MoAb (75 µg/mouse), IL-12 (1 µg/mouse) or anti-CTLA-4 (75 µg/mouse) and IL-12 on days 0, + 1 and + 2 and fed OVA (1 mg/mouse) on days 0, + 1 and + 2. Ten days after feeding, mice were injected with 10 µg of OVA/IFA in the footpad and tail base and sacrificed 11 days later. PLN cells were incubated in vitro with and without 1000 µg of OVA and cytokine levels were assessed using IL-4 and IFN-γ-specific ELISA kits (see Materials and Methods section).
Asterisks indicate Student's t-test values that differ significantly (P ≤ 0·01) from the corresponding value for the water-fed control. The experiment was repeated twice.
Effect of anti-CTLA-4 MoAb and IL-12 treatment on humoral response
Because previous attempts to prevent humoral suppression in OVA-fed mice by administration of anti-CTLA-4 MoAb failed (Fig. 2), it was of interest to determine whether IL-12 could affect this response. When mice were fed low doses of OVA, total Ig, IgG1 and IgG2a titres were significantly suppressed in rat IgG control mice (Fig. 4a,e,i) as well as in mice treated with anti-CTLA-4 MoAb alone (Fig. 4b,f,j) or IL-12 alone (Fig. 4c,g,k). In contrast, when tolerized mice were treated with anti-CTLA-4 MoAb and IL-12, they no longer exhibited significantly suppressed IgG2a titres when compared to water-fed controls (Fig. 4l). Serum antibody titres for total Ig and IgG1 in anti-CTLA-4/IL-12-treated mice remained significantly suppressed (Fig. 4d,h).
Fig. 4.
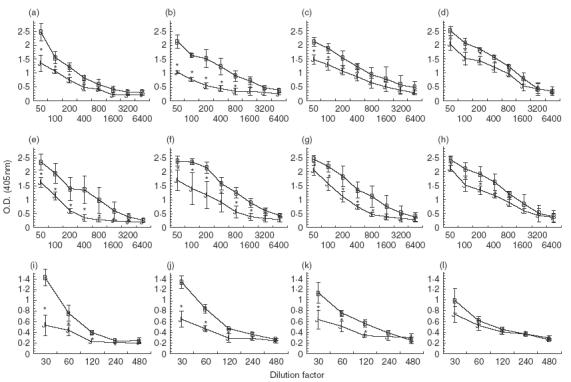
Effect of in vivo anti-CTLA-4 MoAb and IL-12 administration on serum antibody production in orally tolerized mice. Mice were treated i. p. with either rat IgG (75 µ g /mouse) (a,e,i), anti-CTLA-4 MoAb (75 µg/mouse) (b,f,j), IL-12 (1 µg/mouse) (c,g,k) or anti-CTLA-4 and IL-12 (d,h,l) on days 0, + 1 and + 2 and fed OVA (1 mg/mouse) on days 0, + 1 and + 2. Ten days after feeding, mice were injected with 10 µg of OVA/IFA in the footpad and tail base and bled 10 days later. Sera were stored at −70°C. Sera from individual mice (3–4 mice/group) were measured for total Ig (a–d), IgG1 (e–h) or IgG2a (I–l) using ELISA (see Materials and Methods). Asterisks indicate Student's t-test values that differ significantly (P ≤ 0·05) from the corresponding value for the water-fed control. (a, e,i) B, Rat IgG/H2O; J, rat IgG/OVA. (b, f, j) B, Anti-CTLA-4/H2O; J, anti-CTLA-4/OVA. (c, g, k) B, IL-12/H2O; J, IL-12/OVA. (d, h, l) B, IL-12/anti-CTLA-4/H2O; J, IL-12/anti-CTLA-4/OVA.
DISCUSSION
While low-dose oral tolerance has been observed and studied for years, the mechanism responsible for inducing this suppressed immune state remains to be fully elucidated. Previously, others have shown that administration of anti-CTLA-4 MoAb inhibited suppressed proliferative responses in mice tolerized by either i.p. injection of antigen or high-dose antigen feeding [30,31]. Therefore, it was of interest to determine whether CTLA-4 was also involved in the induction of low-dose oral tolerance. In our system, administration of anti-CTLA-4 MoAb did not result in restoration of the humoral or proliferative response in mice fed low doses of OVA. Discrepancies in our results compared to those of others may be due to the fact that i.p./i.v.-induced tolerance and high-dose oral tolerance are mediated exclusively via anergy [6,16,35], while mechanisms other than anergy alone are thought to be responsible for inducing low-dose oral tolerance.
With respect to cytokine profiles, our results are in agreement with those of Van Parjis et al., who showed that anti-CTLA-4 MoAb treatment alone could not restore IFN-γ levels in i.p./i.v.-tolerized mice [31]. Interestingly, administration of anti-CTLA-4 MoAb during low-dose feeding appeared to reverse IL-4 suppression at least partially and is thus similar to results observed when high-dose feeding regimens were utilized [30]. Results from previous studies have suggested that the signalling pathways for cytokine production and proliferation may differ, indicating that various immune responses in ‘anergic’ T cells may be independently regulated [36,37]. Thus it is possible that B7-CTLA-4 interaction, while not sufficient to induce suppressed proliferative responses and IFN-γ production in mice fed low doses of OVA, could act as a negative regulator of IL-4. However, it should be noted that the level of IL-4 in these experiments was very low; therefore, despite the consistency with which we observed this response, this data should be interpreted with caution.
In agreement with others [17,18,38–41], our low-dose feeding regimen resulted in elevated level of TGF-β1. Previous studies have found that cross-linking of CTLA-4 induces TGF-β1 production from CD4+ T cells and results in the inhibition of proliferation as well as IFN-γ and IL-4 production [42,43]. In our hands, treatment of tolerized mice with anti-CTLA-4 MoAb appeared to decrease the ability of mice to produce TGF-β1 compared to the corresponding water-fed control mice; however, decreased levels of TGF-β1 did not correspond to an increase in either cellular proliferation or IFN-γ production. Furthermore, as seen with IL-4 and IFN-γ, anti-CTLA-4 MoAb treatment appeared to result in an overall increase in TGF-β1 production. Such a result is at odds with the hypothesis that simple stimulation of CTLA-4 is necessary for TGF-β1 production and indicates that additional factors must be involved in induction of low-dose oral tolerance. The ability of anti-CTLA-4 MoAb treatment to stimulate an overall increase in the production of cytokines may be due the critical role CTLA-4 plays as a general negative regulator of T cell activation [44,45]. Alternatively, it is possible that anti-CTLA-4 MoAb functions as an agonist in vivo.
Our studies indicated that the co-administration of IL-12 and CTLA-4 MoAb was able to prevent suppression of cellular proliferation and IFN-γ production in OVA-fed mice, while IL-4 levels remained suppressed. The inability of this treatment to prevent IL-4 suppression was not unexpected, given that IL-12 is a known Th1 cell differentiating factor and would thus favour the development of Th1 cells over IL-4-secreting Th2 cells. It is interesting to note that the addition of IL-12 abrogates the enhanced IFN-γ production observed in mice treated with anti-CLTA-4 MoAb alone; the reason for this is unknown and is currently under investigation. With respect to proliferation, recent studies have presented evidence that IL-12 may up-regulate IL-2Rα on T cells, thus permitting the formation of the high-affinity IL-12R [46]. Given this finding, it is possible that IL-12 may aid in the promotion of proliferation (via the IL-2–IL-2R autocrine response) in addition to promoting the differentiation of IFN-γ-secreting Th1 cells. Our results are consistent with those of others who found that tolerance to fed antigen is associated with decreased levels of IL-12 in lymphoid tissue [47], while anti-IL-12 MoAb administration at the time of antigen feeding moderately enhanced suppressed immune responses [48]. The ability to generate both low- and high-dose oral tolerance in IL-12 knockout mice [33] is also consistent with the hypothesis that decreased IL-12 levels are not necessary and in fact may be required for the induction of Th1 cell oral tolerance.
Loss of significant IgG2a isotype suppression observed in orally tolerized mice treated with anti-CTLA-4 and IL-12 may be due to enhanced IFN-γ production by Th1 cells (IFN-γ is required by B cells to produce the IgG2a isotype). Consistent with the low IL-4 levels observed in these same mice, total Ig and IgG1 isotypes remained significantly suppressed.
Results from this study indicate that while B7-CTLA-4 interaction is important in establishing low-dose oral tolerance, other factors are required to see full suppression of immune responses. The administration of IL-12 along with anti-CTLA-4 MoAb to mice during feeding was able to prevent the suppression of Th1 immune responses, perhaps by allowing for the differentiation of Th1 cells. It is expected that the knowledge gained from this study will help elucidate the mechanism involved in oral tolerance induction. Such information is crucial for development of treatments for autoimmune diseases such as multiple sclerosis, rheumatoid arthritis and uveitis.
Acknowledgments
This study was supported by NIH grant no. R15/AI/DE/45517–01.
REFERENCES
- 1.Saklayen MG, Pesce AJ, Pollak VE, Michael JG. Kinetics of oral tolerance: study of variables affecting tolerance induced by oral administration of antigen. Int Archs Allergy Appl Immun. 1984;73:5–9. doi: 10.1159/000233428. [DOI] [PubMed] [Google Scholar]
- 2.Mowat A, Strobel S, Drummond HE, Ferguson A. Immunological responses to fed protein antigens in mice. I. Reversal of oral tolerance to ovalbumin by cyclophosphamide. Immunol. 1982;45:105–13. [PMC free article] [PubMed] [Google Scholar]
- 3.Challacombe SJ, Tomasi TB. Systemic tolerance and secretory immunity after oral immunization. J Exp Med. 1980;152:1459–72. doi: 10.1084/jem.152.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domen PL, Muckerheide A, Michael JG. Cationization of protein antigens. III. Abrogation of oral tolerance. J Immunol. 1987;139:3195–8. [PubMed] [Google Scholar]
- 5.Titus RG, Chiller JM. Orally induced tolerance: definition at the cellular level. Int Archs Allergy Appl Immun. 1981;65:323–38. [PubMed] [Google Scholar]
- 6.Freidman A, Weiner HL. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc Natl Acad Sci USA. 1994;91:6688–92. doi: 10.1073/pnas.91.14.6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanson DG, Miller SD. Inhibition of specific immune responses by feeding protein antigens. V. Induction of the tolerant state in the absence of specific suppresser T cells. J Immunol. 1982;128:2378–81. [PubMed] [Google Scholar]
- 8.Whitacre C, Gienapp I, Orosz CG, Bitar DM. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J Immunol. 1991;147:2155–63. [PubMed] [Google Scholar]
- 9.Whitacre C, Gienapp I, Cox K, et al. Oral tolerance in experimental autoimmune encephalomyelitis (EAE): T cell anergy. J Immunol. 1993;150:245A. [PubMed] [Google Scholar]
- 10.Melamed D, Friedman A. Direct evidence for anergy in T lymphocytes tolerized by oral administration of ovalbumin. Eur J Immunol. 1993;23:935–42. doi: 10.1002/eji.1830230426. [DOI] [PubMed] [Google Scholar]
- 11.Melamed D, Friedman A. In vivo tolerization of Th1 lymphocytes following a single feeding with ovalbumin: anergy in the absence of suppression. Eur J Immunol. 1994;24:1974–81. doi: 10.1002/eji.1830240906. [DOI] [PubMed] [Google Scholar]
- 12.Barone KS, Jain SL, Michael JG. Effect of in vivo depletion of CD4+ and CD8+ cells on the induction and maintenance of oral tolerance. Cell Immunol. 1995;163:19–29. doi: 10.1006/cimm.1995.1094. [DOI] [PubMed] [Google Scholar]
- 13.Shi HN, Grusby MJ, Nagler-Anderson C. Orally induced peripheral nonresponsiveness is maintained in the absence of functional Th1 or Th2 cells. J Immunol. 1999;162:5143–8. [PubMed] [Google Scholar]
- 14.Barone KS, Tolarova DD, Ormsby I, Doetschman T, Michael JG. Induction of oral tolerance in TGFβ1 null mice. J Immunol. 1998;161:154–60. [PubMed] [Google Scholar]
- 15.Garside P, Mowat AM. Mechanisms of oral tolerance. Crit Rev Immunol. 1997;17:119–37. doi: 10.1615/critrevimmunol.v17.i2.10. [DOI] [PubMed] [Google Scholar]
- 16.Inada S, Yoshino S, Azizul M, Ogata Y, Kohashi O. Clonal anergy is a potent mechanism of oral tolerance in suppression of acute antigen-induced arthritis in rats by oral administration of the inducing antigen. Cell Immunol. 1997;175:67–75. doi: 10.1006/cimm.1996.1049. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance. suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–40. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Inobe J, Kuchroo VK, Baron JL, Janeway CA, Weiner HL. Oral tolerance in myelin basic protein T-cell receptor transgenic mice. Suppression of autoimmune encephalomyelitis and dose-dependent induction of regulatory cells. Proc Natl Acad Sci USA. 1996;93:388–91. doi: 10.1073/pnas.93.1.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042–9. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–56. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 21.Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a co-stimulatory signaling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:444–80. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- 22.Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signaling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–9. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 23.Perez VL, Van Parjis L, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–7. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- 24.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 26.Walunas TL, Bakker CY, Bluestone JA. CLTA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541–50. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krummel M, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–40. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarz A, Beissert S, Grosse-Heitmeyer K, et al. Evidence for functional relevance of CTLA-4 in ultraviolet-radiation-induced tolerance. J Immunol. 2000;165:1824–31. doi: 10.4049/jimmunol.165.4.1824. [DOI] [PubMed] [Google Scholar]
- 29.Walunas TL, Bluestone JA. CTLA-4 regulates tolerance induction and T cell differentiation in vivo. J Immunol. 1998;160:3855–60. [PubMed] [Google Scholar]
- 30.Samoilova EB, Horton JL, Zhang H, Khoury J, Weiner HL, Chen Y. CTLA-4 is required for the induction of high dose oral tolerance. Int Immunol. 1998;10:491–8. doi: 10.1093/intimm/10.4.491. [DOI] [PubMed] [Google Scholar]
- 31.Van Parijs VL, Perez L, Biuckians A, Maki RG, London CA, Abbas AK. Role of interleukin 12 and costimulators in T cell anergy in vivo. J Exp Med. 1997;186:1119–28. doi: 10.1084/jem.186.7.1119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Magram J, Connaughton SE, Warrier RR, et al. IL-12 deficient mice are defective in IFN-γ production and type 1 cytokine response. Immunity. 1996;4:471–81. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 33.Mowat AM, Steel M, Leishman AJ, Garside P. Normal induction of oral tolerance in the absence of a functional IL-12-dependent IFN-γ signaling pathway. J Immunol. 1999;163:4728–36. [PubMed] [Google Scholar]
- 34.Claessen AM, Von Blomberg BME, De Groot J, Wolvers DA, Kraal G, Scheper RJ. Reversal of mucosal tolerance by subcutaneous administration of interleukin-12 at the site of attempted sensitization. Immunol. 1996;88:363–7. doi: 10.1046/j.1365-2567.1996.d01-659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garside P, Steel M, Worthey EA, et al. Th2 cells are subject to high dose oral tolerance and are not essential for its induction. J Immunol. 1995;154:5649–55. [PubMed] [Google Scholar]
- 36.Sloan-Lancaster J, Evavold BD, Allan PM. Th2 cell clonal anergy as a consequence of partial activation. J Exp Med. 1994;180:1195–205. doi: 10.1084/jem.180.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karpus WJ, Peterson JD, Miller SD. Anergy in vivo: down-regulation of antigen-specific CD4+ Th1 but not Th2 cytokine responses. Int Immunol. 1994;6:721–30. doi: 10.1093/intimm/6.5.721. [DOI] [PubMed] [Google Scholar]
- 38.Miller A, Lider O, Roberts AB, Sporn M, Weiner HL. Suppresser T cells generated by oral tolerization to myelin basic proteins suppress both in vitro and in vivo responses by the release of transforming growth factor β after antigen-specific triggering. Proc Natl Acad Sci USA. 1992;8:421–5. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khoury SJ, Hancock WW, Weiner HL. Oral tolerance to myelin basic protein and natural recovery from experimental autoimmune encephalomyelitis are associated with downregulation of inflammatory cytokines and differential upregulation of transforming growth factor β, Interleukin 4 and prostaglandin E expression in the brain. J Exp Med. 1992;176:1355–64. doi: 10.1084/jem.176.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y, Inobe J, Weiner HL. Induction of oral tolerance to myelin basic protein in CD8-depleted mice: both CD4+ and CD8+ cells mediate active suppression. J Immunol. 1995;155:910–6. [PubMed] [Google Scholar]
- 41.Caspi RR, Stiff LR, Morawetz R, et al. Cytokine-dependent modulation of oral tolerance in murine model of autoimmune uveitis. Ann NY Acad Sci. 1996;778:315–24. doi: 10.1111/j.1749-6632.1996.tb21139.x. [DOI] [PubMed] [Google Scholar]
- 42.Chen W, Jin W, Wahl SM. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production in muringe CD4+ T cells. J Exp Med. 1998;188:1849–57. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomes NA, Gattass CR, Barreto-de-Souza V, Wilson ME, DosReis GA. TGF-β mediates CTLA-4 suppression of cellular immunity in murine kalaazar. J Immunol. 2000;16:2001–8. doi: 10.4049/jimmunol.164.4.2001. [DOI] [PubMed] [Google Scholar]
- 44.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 45.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 46.Min B, Legge KL, Bell JJ, et al. Neonatal exposure to antigen induces a defective CD40 ligand expression that undermines both IL-12 production by APC and IL-2 receptor up-regulation on splenic T cells and perpetuates IFN-γ-dependent T cell anergy. J Immunol. 2001;166:5594–603. doi: 10.4049/jimmunol.166.9.5594. [DOI] [PubMed] [Google Scholar]
- 47.Karpus WJ, Kennedy KJ, Kunkel SL, Lukacs NW. Monocyte chemotactic protein 1 regulates oral tolerance induction by inhibition of T helper cell-related cytokines. J Exp Med. 1998;187:733–41. doi: 10.1084/jem.187.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marth T, Strober W, Kelsall BL. High dose oral tolerance in ovalbumin TCR-transgenic mice. systemic neutralization of IL-12 augments TGFβ secretion and T cell apoptosis. J Immunol. 1996;157:2348–57. [PubMed] [Google Scholar]