

Introduction
If the immune system can be considered the most highly evolved natural anti-infective therapeutic system, then it would seem to be a promising target for therapeutic manipulation. However, with the obvious exception of prophylactic vaccination (often described as the most successful of all medical public health measures), broadly applicable agents that make the immune system work better have yet to be discovered.
The term drug in its widest sense is generally used to include natural and synthetic therapeutic molecules from the very small (e.g. lithium salts) to the very large (e.g. insulin, interferon, monoclonal antibodies). However, the great majority of drugs conform to a much narrower definition encompassing small organic molecules with the great advantage of oral bioavailability, plus tractability in terms of large-scale synthesis, product quality-control, formulation and stability. In the field of immunology recent advances have identified large numbers of macromolecules that regulate host-defence, and many of these produce enhancement, amplification and/or diversion of immune responses in positive, therapeutically desirable directions. However, immunology has yet to provide small oral drugs that can be used to amplify and positively regulate immunity. A host of peptides and other bio-molecules have been tested in this context [1] and some have been exploited as components of investigational adjuvants for use locally with vaccines. However, none of these have found commercial application as licensed immunopotentiatory drugs.
New processes in drug discovery
The business of drug discovery has been transformed in the last 10 years by the industrialization of processes in chemistry, genetics and genomics and high-throughput biology. A broadly generic sequence of discovery activities has been adopted by the pharmaceutical industry through which the drugs of tomorrow will largely be generated [2]. This technology is designed principally to exploit new chemical entities (NCEs) but can also find applications relating to macromolecules. The process addresses unmet medical needs usually on a disease by disease basis and, in idealized form, proceeds as follows. First, a usable (tractable) biological target (usually a protein) is identified for the target disease. This is most easily achieved where there are precedented targets to choose from, i.e. targets already validated as established starting points for successful drugs. Non-precedented targets are usually chosen on the basis of scientific hypothesis which may subsequently turn out to be wrong, so carry a substantial additional risk of failure. Having selected a disease-specific target, many chemical compounds are then tested for binding activity, most frequently in simplified cell-free systems in a process in which leads are selected from ‘hits’ (positives for whatever reason in the binding assay). In the case of anti-infective drug discovery, it is of course the microbial genes and proteins that are the subjects of interrogation. Once a potential lead has been identified, chemical variants are generated, usually by substitution of chemical groups. These are tested for increased potency and other desirable features such as solubility. In this way structure-activity relationships are established in a process of lead optimization. Optimized leads are tested in cell and tissue contexts, and those continuing to show promise are moved on to testing in animal models where the desired target-specific behaviour is characterized together with pharmacokinetic, metabolic, and toxicological properties. At this point lead compounds are usually narrowed to a single candidate and tested for the first time in humans in small phase I clinical trials. Subjects are usually healthy volunteers, unless the nature of the disease target and the characteristics of the candidate preclude such testing.
The field of target identification has recently been massively expanded by the emergence of discovery genomics and genetics [2]. Genomics is focused on DNA sequence databases in order to identify gene and gene family sequences associated with specific diseases and therefore potentially encoding screenable targets. In this way new disease target genes can be identified on the basis of their sequence similarity with known disease-specific genes. The technology does not establish causal relevance between gene and disease but narrows the search to genes that may be involved in the disease pathway of interest. Discovery genetics aims to identify disease-related genes using information on patients genes, comparing them with healthy individuals. The search here is concentrated on known areas of sequence variability; and the frequency of alleles inherited by afflicted individuals focuses attention on potential genes of interest. Limitations to this approach include multiple genetic loci each contributing a small amount to total disease susceptibility, environmental factors with a greater influence than genetic ones, and complex inheritance patterns not conforming to simple Mendelian patterns. Despite these limitations discovery genetics provides an important complementary disease-relevance input to the large numbers of potential gene targets identified by discovery genomics. Both approaches feed into a third area of target validation termed functional genomics which examines the function of gene products. In this way, genes highlighted by genomic and genetic approaches can be tied to specific disease pathways and thus validated as drug targets. Only a proportion of the genes highlighted in this way will be suitable as tractable targets, i.e. genes that encode products amenable to high-throughput screening of chemicals in automated binding assays.
In a similar way, the lead identification phase of drug discovery has been substantially impacted by recent advances in chemical synthesis such as combinatorial chemistry which exploits solid supports (e.g. silicon beads) to enormously accelerate the generation of representative chemical diversity libraries for automated screening [2]. Commensurate with these advances the screening process has likewise been adapted for high-throughput capacity by miniaturization, robotics, and the use of bead-based systems for both the storage and encoding of libraries and the screening itself. To overcome bottlenecks that this high volume activity inevitably produces downstream, predictive screening is being introduced for generally desirable characteristics of compounds such as tractable physicochemical properties, good absorption, distribution, metabolism and excretion (ADME) characteristics, and clean genetic and cytological toxicology. While the new wave of technologies clearly relies much less on scientific insight than previously, judgements nevertheless need to be applied to every output, together with accumulated wisdom on the known tractability of pathways and families of targets and chemical entities.
From this brief account of the generic new technologies of drug discovery, it is apparent that many of the approaches are not directly relevant to the discovery of immunopotentiatory drugs. Instead they are biased towards disease-specific targets which may not be helpful in identifying immunopotentiatory targets (except the genes responsible for primary immunodeficiencies of which more than 30 are already known [3]). Despite this, the power and scale of generic NCE screening is likely to prove useful for immunomodulation as more and more receptor families are characterized and intracellular pathways expanded through identification of key molecules in signalling cascades, many of which will be essential to immune function. The nature of targets that might prove valuable in the discovery of immunopotentiatory NCEs is considered later.
Current candidates for immunopotentiatory drugs
It takes around 10 years to take a drug from preclinical safety testing to licensure as a medicine, and the period from initial discovery of a biological activity to preclinical development is also several years [2]. This means that the compounds we have today as candidates for broadly applicable immunopotentiatory drugs were first identified long before the revolution in drug discovery technology. Imiquimod, licensed as Aldara cream, serves as a good example of the serendipity and complexity involved in nonidealized drug discovery and development [4–10]. Despite a long history in the development phase, imiquimod is now successful as an immunopotentiatory drug, although licensed only for a single indication (ano-genital warts). Imiquimod also has additional value in illustrating how research on an established drug can tell us a good deal about how to apply the new technologies to discovering the next generation of immunopotentiatory drugs.
Imiquimod was first noted in a programme screening for antiherpes virus activity in the 1980s [4]. The slight toxicity of the drug series produced a slight reduction in herpes cytopathology in Vero cell cultures sufficient for compounds to be tested further in a guinea-pig model. Here, complete protection against herpetic lesions was observed. In fact all the antiviral effects of imiquimod were subsequently shown to be a consequence of its ability to induce pro-inflammatory cytokines driving Th1 immune responses [4–8]. Although a small molecule, imiquimod has limited oral bioavailability and is effectively restricted to topical application. In vitro analyses of imiquimod's effects on immune cells show that it induces the synthesis of IFN-α, TNF-α and other pro-inflammatory cytokines. It also induces antigen nonspecific B-cell proliferation through a mechanism that may be unrelated to cytokine induction. In vitro effects seem to depend on the activation of transcription factors such as NFκB that bind to promoter regions of IFN-α and other cytokines including IL-1, IL-1RA, IL-6, IL-8 and IL-12p40, although IFN-α seems to be induced at lower concentrations of drug. In vivo studies of topical application in hairless mice have shown that imiquimod induces increased IFN-α mRNA and increased IFN-α and TNF-α proteins in treated skin. These effects occur within 1–4 h. Accompanying changes include enlargement, morphological activation and increased migration of Langerhans cells from treated sites to regional lymph nodes; effects likely to facilitate the presentation of antigens present at the treatment site. The ultimate effect of imquimod is enhancement of Th1 type immune responses through downstream effects, i.e. T-cell cytokines such as IL-2 and IFN-γ are enhanced indirectly through the mediation of IFN-α and IL-12. Imiquimod also down-regulates Th2 cytokines such as IL-5 in mouse and human systems in a manner consistent with its positive effects on Th1 cytokines.
Clinical studies confirm the immunopotentiatory effects of imiquimod at the sites of genital warts [5]. Biopsies of treated wart tissue subjected to RT-PCR analysis revealed drug-induced increases in IFN-α, and TNF-α mRNAs, and these changes correlated with reductions in HPV DNA & RNA, and the resolution of warts. Th-1 T-cell-derived cytokines were also increased in many subjects while decreases in CD1a mRNA suggested increased migration of Langerhans cells to regional nodes, consistent with animal observations. These immunopotentiatory effects are believed to underlie the approximately 50% success rate of imiquimod in resolving outbreaks of HPV lesions (ano-genital warts) within a 16-week treatment period (although up to a third of patients may interrupt the thrice weekly treatment course to allow local erythema to subside). No systemic effects are expected as only very small amounts of topical drug become systemic. As a general point of caution, it should be noted that imiquimod was ineffective in both a human-reconstituted SCID mouse model and a rabbit infection model of the disease for which it was eventually licensed [4].
As a small-molecule topical immunopotentiator promoting Th1 responses imiquimod is a first-in-class drug that would seem to offer benefit in a range of applications wherever skin lesions are an important manifestation of infection. This would include genital herpes (HSV-2) and Molluscum contagiosum (a skin infection caused by a member of the pox virus family), and might also be extendable to nonviral cutaneous diseases such as basal cell carcinoma, squamous cell carcinoma and cutaneous Leishmaniasis [4,8–10]. Such applications will require extensive clinical trials to demonstrate efficacy. It is not yet clear if imiquimod can impact atopic skin disorders with a Th2 pathology. Imiquimod has already proved fruitful as a chemical starting point from which the more potent analogue, resiquimod, has been developed. Resquimod is active at tenfold lower concentrations in inducing pro-inflammatory cytokines, is effective by a number of local routes in controlling experimental HSV-2 infection, and has promising characteristics in terms of oral bioavailability [5]. As such it is an interesting candidate as a systemic immunopotentiator for viral infection. Resiquimod's mechanism of action, primarily working through the induction of IFN-α, means it is logical to test resiquimod in clinical indications where systemic IFN-α is effective. Hepatitis C infection is therefore the most interesting testing ground for oral resiquimod and trials are underway.
Perhaps the greatest importance of imiquimod and its analogues for present purposes is as a lesson on where to focus the new high-throughput generic technologies of drug discovery. Investigation of the physiological target for imiquimod and resiquimod, has recently shown that these imidazoquinolines activate immune cells through a member of the toll-like receptor family, TLR-7 [11]. Some redundancy may exist it this family in that human TLR-8 may also mediate the response to resiquimod [12]. This provides an important and intellectually satisfying link with the long history of immunological adjuvant research in which conserved structural components of pathogens have been tested and exploited as local agents to amplify immune responses to vaccines. These pathogen associated molecular patterns (PAMPs), first exploited by the use of whole mycobacteria in Freunds complete adjuvant, now include molecules constituting the structure of gram negative bacterial lipopolysaccharides (LPS), gram positive bacterial teichoic acids, unmethylated CpG motifs, viral double stranded RNA, yeast mannans and other conserved structures, not present in eukaryotic cells, that signal the presence of a pathogen [13–15]. The link with the imidazoquinolines comes with the fact that antigen presenting cells (APC) of the immune system respond to these immunopotentiatory PAMPs through toll-like receptors whose generic specificities are currently being mapped [16–20]. Thus, TLR-2 appears important in sensing peptidoglycans, lipoproteins, and zymosan, TLR-3 in sensing viral RNA, TLR-4 in sensing LPS, TLR-5 in sensing bacterial flagellin, and TLR-9 in sensing unmethylated CpG DNA. It therefore makes very good sense to find that a small xenobiotic molecule that enhances Th-1 immunity so effectively does so through the same mechanism as the pathogen-derived danger signals that allow the immune system to distinguish between innocuous and pathogenic molecular encounters (Fig. 1).
Fig. 1.
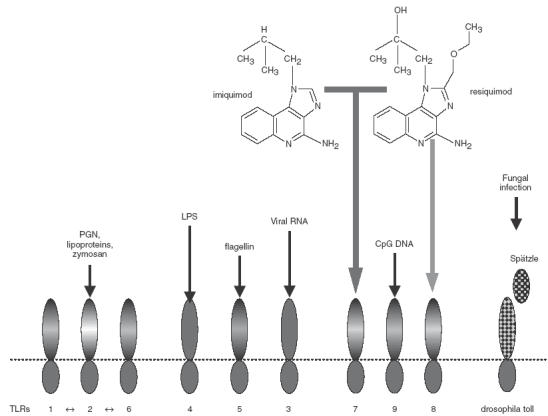
Human toll-like receptors 1–9, expressed on dendritic cells and other APC, mediate the detection of pathogen associated molecular patterns and the up-regulation of costimulatory molecules and cytokines in response to these pathogen-associated signals. The low-molecular-weight immunopotentiatory drugs imiquimod and resiquimod signal via TLR-7 (while resiquimod can also signal via human, but not murine, TLR-8 at higher drug concentrations).
A second candidate immunopotentiatory drug currently in clinical trials and with an equally long history of development, may also have informative connections with pathogen-associated danger signals, though in a very different way. In the late eighties, studies on the interaction between T-cells and APC implicated transient covalent bond formation between specialized cell-surface groups as important in optimal induction of specific T-cell activation [21–23]. This mechanism provided a physiological explanation for the experimental phenomenon of oxidative mitogenesis and identified chemical ligands as potential targets for immunomodulation. The Schiff base-forming cell-surface ligands defined in these studies (specialized carbonyls and amines) were subsequently targeted in programmes looking at the generation of such groups and their chemical modification [24]. Small Schiff base-forming chemicals were found to mimic the effects of cell-surface ligands promoting antigen-specific T-cell activation and were therefore investigated as candidate immunopotentiatory drugs. Although there were few precedents for drugs working through covalent reactivity, attempts to modify sickle cell haemoglobin through Schiff base formation, were encouraging to this approach [25]. In the event it was a lead candidate from the sickle-cell programme that was chosen for development as an immunopotentiatory drug (effects on immunity occur at much lower doses than those modifying haemoglobin) [26]. Tucaresol, the Schiff base-forming substituted benzaldehyde selected for development, was subsequently shown to provide a costimulatory signal to CD4+ T-cells, rapidly activating Na+ and K+ transport [26], converging with T-cell receptor (TCR) signalling at the level of the MAP kinase ERK-2 [27], and priming for increased intensity of calcium signalling [28]. The latter effect may underlie the Th1 deviation induced by tucaresol [29,30]. Human studies in vitro and animal studies in vivo have shown that Schiff base formation by tucaresol on T-cell-surface amines enhances TCR-dependent IL-2 and IFN-γ production, promoting CD4 and CD8 T-cell responses to antigen and exerting favourable therapeutic effects in animal models of tumour growth [26], mycobacterial infection (Orme, unpublished observation), viral infection [26] and protozoal infection [31]. Unlike imiquimod, tucaresol is orally bioavailable and best used as an oral drug. In pilot clinical studies tucaresol proved active as an oral immunopotentiatory drug promoting Th1 cytokine production [32]. Subsequent clinical studies have concentrated on finding suitable doses (tucaresol has a bell-shaped dose–response curve) which may be indication-specific (Ferrari et al. unpublished observation), and investigating combinations with antiviral drugs to prevent gross symptoms that may be associated with immune hyper-reactivity [33,34]. The therapeutic utility of tucaresol as an oral immunopotentiatory drug has therefore yet to be determined. Like imiquimod, tucaresol has already provided a starting point for analogues such as isotucaresol reported to be active as immunological adjuvants [35].
In another parallel with imiquimod, an intriguing link appears to exist between tucaresol and potential pathophysiological danger signals, though not in terms of macromolecular receptors (tucaresol's primary macromolecular targets are not yet identified). Instead, convergence has occurred between studies of small molecules produced by activated neutrophils and studies of small Schiff base-forming immunopotentiators. Activated neutrophils have been shown to employ the myeloperoxidase, H202–Cl− dependent pathway to convert l-tyrosine to p-hydroxyphenylacetaldehyde in near quantitative yield [36]. This small Schiff base-forming product partitions preferentially to the plasma membrane where it is highly reactive with ɛ-amino groups in vivo [37]. By chance, p-hydroxyphenylacetaldehyde was also the first Schiff base-forming compound to be studied as an immunopotentiator where it was comparably to tucaresol in amplifying Th1 immune responses in vitro and in vivo [26]. Putting these two sets of observations together strongly suggests a picture in which small Schiff base forming products of activated neutrophils act directly to costimulate Th-cells, thereby functioning as danger signals linking acute first-line defences of the innate immune system with the subsequent adaptive immune response (Fig. 2).
Fig. 2.
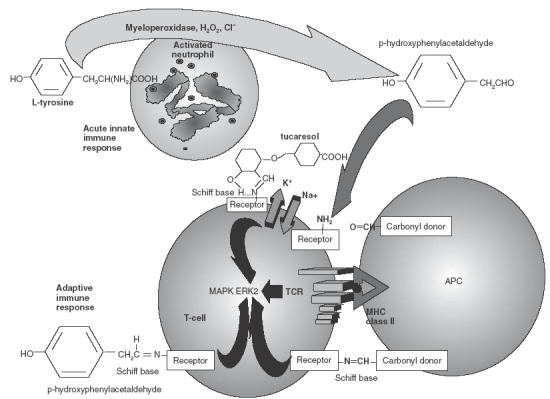
Activated neutrophils, which mediate the acute phase of innate immune responses, use the myeloperoxidase, hydrogen peroxide, chloride pathway to convert l-tyrosine to p-hydroxyphenylacetaldehyde which partitions to plasma membranes to forms Schiff bases with ɛ-amino groups. In separate studies the latter process has been shown to costimulate T-cells and amplify Th1 responses in the same way as the immunopotentiatory drug tucaresol.
Other small-molecule drugs are sometimes termed immunomodulatory and include inosine pranobex, ribavirin and thalidomide. However, in all three cases the mechanism of action is unclear and the drugs are pleiotropic. This means they are unlikely to be useful as guides in target identification. Inosine pranobex (isoprinosine) is licensed in Europe for treatment of H. simplex infections, subacute sclerosing panencephalitis, acute viral encephalitis caused by H. simplex, and treatment of EBV and measles in immunosuppressed patients [1]. The drug can augment the production of IFN-α and was developed as an antiviral agent [38]. Recent success in delaying the progression of AIDS may be through interference with folate synthesis essential for the replication of P. carinii [39]. Ribavirin is licensed for use with pegylated IFN-α in chronic HCV infection [40] While it has been reported to be immunomodulatory, favouring Th1 responses [41], its primary mechanism appears to be through interference, as a rapidly phosphorylated molecule, with the biosynthesis of gaunosine triphosphate essential in replication of DNA and RNA viral genomes [42]. Recently, interest has grown in the possibility of lethal mutagenesis induced by ribavirin in RNA virus genomes [43,44]. Thalidomide, synthesized as a potential antihistaminic, then widely exploited as a sedative until withdrawn in 1962 because of teratogenic properties, is now approved for use in patients with erythema-nodosum-leprosum where its beneficial effects were discovered by chance [45,46]. Its immunomodulatory effects seem to be due to suppression of TNF-α production through promoting the degradation of TNF-α mRNA. It is also antiangiogenic and beneficial in the management of multiple myeloma through antiproliferative effects and modulation of adhesion molecules [47]. Analogues with higher potency against TNF-α have been synthesized [45].
Potential targets in the immune system for novel immunopotentiatory drugs
Advances in understanding the molecular basis of immunity suggest a host of targets that might attract the application of new technologies aimed at discovering novel immunopotentiatory drugs. As with valuable precedented targets for conventional drugs, the few presently existing immunopotentiatory compounds, though not ideal, may be useful for narrowing this wealth of targets to tractable front-runners. At first pass, and without applying this filter, several categories of target suggest themselves. These include pro-inflammatory cytokines and their receptors [48,49], costimulatory macromolecules and their receptors [50], ion-channels, seven trans-membrane receptors, and other target families falling out of large scale screening programmes and also present on T-cells and other leucocytes, and key signalling proteins in the pathways utilized by such receptors. Although it might seem easier to discovery inhibitors through NCE screening, interrogation of down-regulatory pathways may identify inhibitors that up-regulate immune responses. It may also prove possible to generate small molecule mimetics of immunopotentiatory proteins. As a key upstream mediator, IFN-α might prove an attractive target while interleukin targets downstream might permit finer manipulation of immune deviation. Key intracellular signalling proteins may make tractable targets providing that sufficient specificity for the immune element of function can be conferred [51–54]. It is here that existing immunopotentiatory drugs may provide some focus on an otherwise large and diverse collection of opportunities. Identification of the macromolecular targets of tucaresol (and, by inference, natural receptors for acute mediators from activated neutrophils), may provide targets validated by a compound known to be active as an oral Th1 immunopotentiator. More immediately, the fact that imiquimod, the first licensed small molecule Th1 potentiator, works through toll-like receptors clearly indicates the potential value of the TLR family as targets for a new generation of immunopotentiatory compounds [11,12]. Since these receptors appear to utilize a common signalling pathway, their intracellular signalling elements may also repay investigation [11].
In paying attention to this target, can we be sure that the imidazoquinolines are directly ligating TLR receptor binding domains? Studies thus far have utilized cells and cell lines positive and negative for TLR 7 & 8 in in vitro reporter systems for intracellular signalling or cytokine production as well as demonstrating in vivo effects in TLR receptor positive and negative TG mice. In this way the action of imidazoquinolines has been firmly associated with specific TLR expression (along with the intracellular signalling pathway utilized by these receptors). Thus, TRL 7 is both necessary and sufficient to mediate imidazoquinoline signalling in simplified systems in which only drug and cultured cells are present [11], and the same is true for TLR 8 [12]. Direct ligation of receptors is therefore the simplest explanation for these effects. However, it is possible that serum components participate in this ligation or that an intermediate ligand might be induced. The simpler the system the less likely is the latter possibility, and the fact that a positive response is seen in HEK 293 cells transiently transfected with TLR7 and a luciferase reporter system makes it unlikely that an intermediate endogenous ligand is being generated. Nevertheless, direct binding of imidazoquinolines to TLR domains has not yet been investigated.
Despite this the current evidence may be judged sufficiently strong to justify TLR-based compound screening approaches to identify second generation candidate immunopotentiatory drugs. This principal example serves to illustrate a general process in which, as more immune enhancing compounds are identified, more targets will be validated in an iterative refinement of targets and chemical ligands that will eventually provide the immunopotentiatory drugs of the future.
REFERENCES
- 1.Masihi KN. Immunomodulatory agents for the prophylaxis and therapy of infections. Int J Antimicrob Agents. 2000;14:181–91. doi: 10.1016/s0924-8579(99)00161-2. [DOI] [PubMed] [Google Scholar]
- 2.Sykes RB. Rock Carling Fellowhip of the Nuffield Trust. London: The Stationary Office; 2000. New medicines, the practice of medicine and public policy. [Google Scholar]
- 3.Buckley RH. Primary Immunodeficiency Diseases. In: Paul W, editor. Fundamental Immunology. 4. New York: Lippincot-Raven; 1999. pp. 1427–53. [Google Scholar]
- 4.Miller RL, Gerster JF, Owens ML, Slade HB, Tomami MA. Imiquimod applied topically. a novel class of immune response modifier and new class of drug. Int J Immunopharm. 1999;21:1–14. doi: 10.1016/s0192-0561(98)00068-x. [DOI] [PubMed] [Google Scholar]
- 5.Dockrell DH, Kinghorn GR. Imiquimod and Resiquimod as novel immunomodulators. J Antimicrob Chemother. 2001;48:751–5. doi: 10.1093/jac/48.6.751. [DOI] [PubMed] [Google Scholar]
- 6.Testerman TL, Gerster JF, Imbertson LM, Reiter MJ, Miller RL, Gibson SJ, Wagner TL, Tomai MA. Cytokine induction by the immunomodulators imiquimod and S-27609. J Leukocyte Biol. 1995;58:365–72. doi: 10.1002/jlb.58.3.365. [DOI] [PubMed] [Google Scholar]
- 7.Weeks CE, Gibson SJ. Induction of interferon and other cytokines by imiquimod and its hydroxylated metabolite R-842 in human blood cells in vitro. J Interferon Res. 1994;14:81–5. doi: 10.1089/jir.1994.14.81. [DOI] [PubMed] [Google Scholar]
- 8.Harrison CJ, Miller RL, Bernstein DI. Post-therapy suppression of genital herpes simplex virus (HSV) recurrences and enhancement of HSV-specific T-cell memory by imiquimod inguinea pigs. Antimicrob Agents Chemother. 1994;38:2059–64. doi: 10.1128/aac.38.9.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein DI, Harrison CJ, Tomai MA, Miller RL. Daily or weekly therapy with resiquimod (R-848) reduces genital recurrences in herpes simplex virus-infected guinea pigs during and after treatment. J Infect Dis. 2001;183:44–849. doi: 10.1086/319262. [DOI] [PubMed] [Google Scholar]
- 10.Arevelo I, Ward B, Miller R, Meng T-C, Najar E, Alvarez E, Matlashewski G, Lianos-Cuentas A. Successful treatment of drug-resistant cutaneous leishmaniasis in humans by use of imiquimod, an immunomodulator. Clin Infect Dis. 2001;33:1874–51. doi: 10.1086/324161. [DOI] [PubMed] [Google Scholar]
- 11.Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent pathway. Nature Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 12.Jurk M, Heil F, Vollmer J, Schetter C, Krieg M, Wagner H, Lipford G, Bauer S. Human TLR-7 or TLR-8 independently confer responsiveness to the antiviral compound R-848. Nature Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 13.Medzhitov R, Janeway CA. Innate immune response recognition and control of adaptive immune responses. Sem Immunol. 1998;10:351–3. doi: 10.1006/smim.1998.0136. [DOI] [PubMed] [Google Scholar]
- 14.Medzhitov R, Janeway CA. How does the immune system distinguish self from nonself? Sem Immunol. 2000;12:185–8. doi: 10.1006/smim.2000.0230. [DOI] [PubMed] [Google Scholar]
- 15.Medzhitov R, Janeway CA. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 16.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 17.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-Iike receptor 5. Nature. 2001;410:1099–103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 19.Yang RB, Mark MR, Gray A, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signaling. Nature. 1998;395:284–8. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]
- 20.Seiling PA, Modlin RL. Toll-like receptors. mammalian ‘taste receptors’ for a smorgasbord of microbial invaders. Current Opinion Microbiol. 2002;5:70–5. doi: 10.1016/s1369-5274(02)00288-6. [DOI] [PubMed] [Google Scholar]
- 21.Rhodes J. Evidence for an intercellular covalent reaction essential in antigen specific T cell activation. J Immunol. 1989;143:1482–9. [PubMed] [Google Scholar]
- 22.Gao X-M, Rhodes J. An essential role for constitutive Schiff base-forming ligands in antigen presentation to murine T cell clones. J Immunol. 1990;144:2883–90. [PubMed] [Google Scholar]
- 23.Rhodes J. E-Rosettes provide an analogue for Schiff base formation in specific T cell activation. J Immunol. 1990;145:463–9. [PubMed] [Google Scholar]
- 24.Zheng B, Brett S, Tite JP, Lifely MR, Brodie TA, Rhodes J. Galactose oxidation in the design of immunogenic vaccines. Science. 1992;256:1560–3. doi: 10.1126/science.1598588. [DOI] [PubMed] [Google Scholar]
- 25.Rolan PE, Parker JE, Gray SJ, Weatherley BC, Ingram J, Leavens W, Wootton R, Posner J. The pharmacokinetics, tolerability and pharmacodynamics of tucaresol (589C80; 4[2-formyl-3-hydroxyphenoxymethyl] benzoic acid), a potential anti-sickling agent, following oral administration to healthy subjects. Brit J Clin Pharmacol. 1993;35:419–25. doi: 10.1111/j.1365-2125.1993.tb04160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhodes J, Chen H, Hall SR, Beesley JE, Jenkins DC, Collins P, Zheng B. Therapeutic potentiation of the immune system by costimulatory Schiff base-forming drugs. Nature. 1995;377:71–5. doi: 10.1038/377071a0. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Hall S, Heffernan B, Thompson NT, Rogers MV, Rhodes J. Convergence of Schiff base costimulatory signaling and TCR signaling at the level of mitogen activated protein kinase ERK2. J Immunol. 1997;159:2274–81. [PubMed] [Google Scholar]
- 28.Hall SR, Rhodes J. Schiff base-mediated co-stimulation primes the T-cell-receptor-dependent calcium signalling pathway in CD4 T-cells. Immunology. 2001;104:50–7. doi: 10.1046/j.0019-2805.2001.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noble A, Truman JP, Vyas B, Vukamanovic-Stejic M, Hirst WJ, Kemeny DM. The balance of protein kinase C and calcium signaling directs T cell subset development. J Immunol. 2000;164:1807–13. doi: 10.4049/jimmunol.164.4.1807. [DOI] [PubMed] [Google Scholar]
- 30.Noble A, Thomas MJ, Kemeny DM. Early Th1/Th2 cell polarisation in the absence of IL-4 and Il-12: T cell receptor signaling regulates the response to cytokines in CD4 and CD8 T-cells. Euro J Immunol. 2001;31:2227–35. doi: 10.1002/1521-4141(200107)31:7<2227::aid-immu2227>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 31.Smith AC, Yardley V, Rhodes J, Croft SL. Activity of the novel immunomodulatory compound tucaresol against experimental visceral leishmaniasis. Antimicrob Agents Chemother. 2000;44:1494–8. doi: 10.1128/aac.44.6.1494-1498.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirkwood JM, Schucter LM, Donelly S, et al. A novel immunopotentiatory agent, tucaresol: results from a multicenter, pilot study in patients with metastatic melanoma. Proc Am Assoc Cancer Res. 1997;38:402. [Google Scholar]
- 33.Clerici M, Bosis S, Rizzardi G, Comombo F, Rhodes J, Bray D, Piconi S. In vitro immunomdulatory properties of tucaresol in HIV infection. Clin Immunol. 2000;97:211–20. doi: 10.1006/clim.2000.4937. [DOI] [PubMed] [Google Scholar]
- 34.Gori A, Trabattoni D, Bandera A, et al. Safety and immunomodulatory properties of tucaresol HIV-Infected Patients. In: Gatelli J, Casabona J, editors. XIVth International AIDS Confererce. Barcelona: 2002. Poster no: MoPeA3032. [Google Scholar]
- 35.Johnson D. Novel amphipathic aldehydes and their use as adjuvants and immunoeffectors. International Patent (PCT) Application No. WO 01/70663 A2. 2001 [Google Scholar]
- 36.Hazen SL, Hsu FF, Heinecke JW. p-hydrophenylacetaldehyde is the major product of l-tyrosine oxidation by activated human phactocytes: a chloride dependent mechanism for the conversion of free amino acids into reactive aldehydes by myeloperoxidase. J Biol Chem. 1996;271:1861–7. doi: 10.1074/jbc.271.4.1861. [DOI] [PubMed] [Google Scholar]
- 37.Hazen SL, Guat JP, Hsu FF, Crowley JR, d’Avignon A, Heinecke JW. p-hyroxyphenylacetaldehye, the major product of l-tyrosine oxidation by the myeloperoxidase-H2-O2-Chloride system of phagocytes, covalently modifies ɛ-amino groups of protein lysine residues. J Biol Chem. 1997;272:16990–8. doi: 10.1074/jbc.272.27.16990. [DOI] [PubMed] [Google Scholar]
- 38.Campoli-Richards DM, Sorkin EM, Heel RC. Inosine pranobex. A preliminary review of its pharmacodynamics and pharmacokinetic properties, and therapeutic efficacy. Drugs. 1986;32:383–424. doi: 10.2165/00003495-198632050-00001. [DOI] [PubMed] [Google Scholar]
- 39.Kovacs JA, Powell F, Voeller D, Allegra CJ. Inhibition of pneumocystis-carinii dihydropteroate synthetase by para-acetoamidobenzioc acid – possible mechansims of action of isoprinosine in human immunodeficiency virus infection. Antimicrob Agents Chemother. 1993;37:1227–31. doi: 10.1128/aac.37.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snell NJ. Ribavirin. current status of a broad spectrum anti-viral agent. Expert Opin Pharmacother. 2001;2:1317–24. doi: 10.1517/14656566.2.8.1317. [DOI] [PubMed] [Google Scholar]
- 41.Souvignet C, Zarski JP. Combination treatment for chronic hepatitis C. What is the role of ribavirin. Fundamental Clin Pharmacol. 2000;14:321–5. doi: 10.1111/j.1472-8206.2000.tb00412.x. [DOI] [PubMed] [Google Scholar]
- 42.Eriksson B, Helgstrand E, Johansson NG, et al. Inhibition of influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob Agents Chemother. 1977;11:946–51. doi: 10.1128/aac.11.6.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crotty S, Maag D, Arnold JJ, Zhong W, Lau JYN, Hong Zhi Andino R, Cameron CE. The broad spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nature Med. 2000;6:1375–9. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 44.Cameron CE, Castro C. The mechanism of action of ribavirin: lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase. Curr Opinion Infect Dis. 2001;14:757–64. doi: 10.1097/00001432-200112000-00015. [DOI] [PubMed] [Google Scholar]
- 45.Eriksson T, Bjorkman S, Höglund P. Clinical pharmacology of thalidomide. Euro J Clin Pharm. 2001;57:365–76. doi: 10.1007/s002280100320. [DOI] [PubMed] [Google Scholar]
- 46.Meierhofer C, Dunzendorfer S, Wiedermann CJ. Theoretical basis for the activity of thalidomide. Biodrugs. 2001;15:681–703. doi: 10.2165/00063030-200115100-00005. [DOI] [PubMed] [Google Scholar]
- 47.Hussein MA, Juturi JV, Lieberman I. Multitple myeloma. present and future. Curr Opin Oncol. 2002;14:31–5. doi: 10.1097/00001622-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 48.Leonard WJ, Type I. cytokines and interferons and their receptors. In: Paul W, editor. Fundamental Immunology. 4. New York: Lippincot-Raven; 1999. pp. 741–74. [Google Scholar]
- 49.Krakauer T, Vilcek J, Oppenheim JJ. Proinflammatory cytokines. TNFα and IL-1 families, TGF-β, and others. In: Paul W, editor. Fundamental Immunology. 4. New York: Lippincot-Raven; 1999. pp. 775–811. [Google Scholar]
- 50.Bluestone JA, Khattri R, van Seventer GA. Accessory molecules. In: Paul W, editor. Fundamental Immunology. 4. New York: Lippincot-Raven; 1999. pp. 449–78. [Google Scholar]
- 51.Majetis R, Weiss A. Regulatory mechanisms for receptor protein tyrosine phosphatases. Chem Rev. 2001;101:2441–8. doi: 10.1021/cr000085m. [DOI] [PubMed] [Google Scholar]
- 52.Cantrell DA. T-cell antigen receptor signal transduction. Immunology. 2002;105:369–74. doi: 10.1046/j.1365-2567.2002.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong C, Flavell RA. MAP kinases in the immune response. Ann Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 54.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulatator of IL-12R expression in naïve CD4+ T-cells. Nature Immunol. 2002;3:449–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]