

Abstract
Immunization with different adjuvants resulted in antithetic outcomes of infection with Chlamydia pneumoniae. Immunization with the outer major protein-2 from C. pneumoniae (OMP-2) emulsified in Freund's complete adjuvant (FCA) thus increased the susceptibility of mice to infection with the bacteria. The detrimental effect was not observed upon inoculation of irrelevant antigens or major outer membrane protein (MOMP) in FCA, but was also observed after immunization with FCA–chlamydial heat shock protein-60 (HSP-60). The harmful effect of FCA-OMP-2 depended on the presence of both CD4+ and CD8+ cells and was mediated by IL-10, as shown using gene-ablated mice. The increased susceptibility to infection caused by FCA-OMP-2 immunization was long-lasting and observed in mice infected 4 months after the last dose of immunogen. In contrast, partial protection against C. pneumoniae was observed when FCA was replaced with oligodeoxynucleotides containing immunostimulatory CpG motifs mixed with Freund's incomplete adjuvant (FIA-IS-CpG). These polar outcomes of infection related to the cytokine pattern: antigen-stimulated spleen cells from FCA-OMP-2-immunized mice showed higher IL-10/IFN-γ ratios than FIA-IS-CpG-OMP-2-immunized animals. In agreement, sera from FCA-OMP-2 showed higher anti-OMP-2 IgG1/IgG2a ratios than FIA-IS-CpG-OMP-2-immunized animals. Finally, OMP-2 also generated a protective response when delivered by a eukaryotic expression vector in tandem with CTLA4, a procedure that targeted OMP-2 to antigen-presenting cells.
Keywords: adjuvant, Chlamydia pneumoniae, interferon, IL-10, vaccine
Introduction
The successful elimination of pathogens following vaccination depends to large extent on the ability of the host's immune system to recognize and respond effectively, preferably with minimal injury to healthy tissue. In the design of effective non-replicating vaccines, immunological adjuvants serve as critical components that instruct and control the selective induction of appropriate type of antigen-specific immune responses. The most appropriate adjuvant for a given antigen will depend, to a large extent, on the type of immune response that is required for protective immunity. Thus, the type of adjuvant selected might orchestrate the type of immune response induced (Th1 or Th2), which in turn may have a significant impact on the protective efficacy of a vaccine. Consequently, induction of an inappropriate response type could increase susceptibility to infection.
Some adjuvants act as a signal 0, the so-called pathogen-recognition patterns. These molecules are the signatures of noxious substances recognized by pattern recognition receptors (PRR), a precondition for generation of signal 2. Among these adjuvants, bacterial DNA has been shown to possess unmethylated CpG motifs that allow discrimination of pathogen-derived DNA from self-DNA. CpG DNA, which are recognized by PRR [1,2], are the most potent adjuvants known for induction of Th1 response [3,4] and also trigger protective CD8+ T cell responses. Similar responses may be elicited by DNA vaccines (due to the CpG adjuvant effect of plasmid sequences) [5]. The mycobacterial components of Freund's complete adjuvant also represent a complex of different signal 0 substances.
Chlamydia pneumoniae, a Gram-negative intracellular bacteria, is responsible for a range of respiratory tract diseases in humans, and has also been reported to be associated to development of atherosclerosis [6]. A vaccine capable of protecting against chlamydial infections could provide considerable public health benefit. Such a vaccine should induce a Th1 response, and prime both CD4+ and CD8+ cells and IFN-γ-dependent and -independent effector mechanisms, components of the immune response shown to play a protective role in primary or secondary infections with different chlamydial species [7–12]. Relative success in protection have been met by some groups [13–16] but not by others [17] using DNA vaccines against chlamydia. Several chlamydial antigens have been scrutinized for their protective response. The major outer membrane protein (MOMP), a 60-kDa heat shock protein and an ATP/ADP translocase have all been shown to confer some degree of protection when delivered as a DNA vaccine [13–15]. The ability of protein vaccination to confer protection against chlamydial infection has also been indicated [18–20].
In this study we have focused upon the chlamydial cystein-rich outer membrane protein (OMP-2). OMP-2 is the second most abundant protein in C. trachomatis elementary bodies and homologues occur in all chlamydia species. Studies on C. pneumoniae and C. psittaci show that during the chlamydia replication cycle OMP-2 is expressed when reticulate bodies differentiate into elementary bodies [21]. Thus it is not surprising that this antigen elicits immune responses in vivo, and that OMP-2-specific memory cells can be demonstrated by challenge with elementary bodies in vitro [22–24]. Moreover, conservation of the sequence of OMP-2 among different C. trachomatis serovars may make it an attractive vaccine candidate.
We have used OMP-2 both as a component of a DNA vaccine and as a protein vaccine using FCA or CpG DNA as adjuvants. Surprisingly, FCA-OMP-2 immunization resulted in increased bacterial numbers in lungs, whereas immunization with FIA-IS-CpG-OMP-2 or a DNA vaccine containing the omp-2 gene gave a partial protection to infection. The opposite effect on the outcome of infection by using these adjuvants was linked to striking differences in the pattern of cytokines and antibody isotypes induced by immunization.
Material and methods
Recombinant HSP-60, OMP-2 and momp production
C. pneumoniae HSP-60, OMP-2 and momp were amplified from eukaryotic expression plasmids p-omp-2, p-hsp-60 and p-momp. The microbial signal sequence was deleted from the momp, hsp-60 and omp-2 genes which were then cloned into a prokaryotic expression vector ptrx-abp [25]. The DNA sequence was confirmed. ptrx-abp encodes a 26-kDa affinity fusion partner consisting of Escherichia coli thioredoxin (Trx) protein [26] and an albumin binding protein (ABP) derived from streptococcal protein G [27]. We have reported previously the construction of ptrx-abp-hsp-60 [15].
E. coli BL-21 (DE3) (Novagen, Inc., Madison, WI, USA) harbouring ptrx-abp-hsp-60, ptrx-abp-omp-2 and ptrx-abp-momp plasmids were grown overnight at 37°C in 100 ml Luria broth (LB) supplemented with 100 mg/l ampicillin. Thereafter, culture was diluted 1/100 in LB and grown 3–5 h. Expression of the recombinant fusion protein was induced by adding of 1 mm isopropyl-β-d-thiogalactosidase (IPTG). Protein production continued for 4–5 h at RT until bacteria reached an O.D.600 nm of 1·8–3. Cells were centrifuged and the pellets frozen at − 20°C, thawed, resuspended in 50 mm Tris-HCl, 0·2 mm NaCl, 0·05% Tween, pH 7·5 and sonicated. Sonicated cells were centrifuged, and the supernatant was applied on a human serum albumin–Sepharose column (Pharmacia, Stockholm, Sweden). Elution was performed using 0·5 m acetic acid pH 2·8. The size and purity of Trx-ABP-HSP-60, Trx-ABP-OMP-2 and Trx-ABP-MOMP was verified by SDS-PAGE and further confirmed by Western-blot.
Plasmid DNA constructions for immunization
P-omp-2
The omp-2 gene (accession no. X535511) from C. pneumoniae was amplified by PCR using a plasmid containing OMP-2 as a template, and cloned into the eukaryotic expression vector pCI (Promega, Madison, WI, USA) containing the murine IgG heavy chain signal sequence IgGSSeq. Fusion with IgG signal sequence improves secretion and immunogenicity of the different encoded antigens, including OMP-2 [28]. Primers used for PCR amplification for this gene were as follows:
Sense OMP-2: 5′ CCG AAT TCG AGC GGG GGT ATA GAG GCC GCC GCT GTA− 3′
Antisense OMP-2: 5′ GGC CGT CGACTT ATT ACA CGT GGG TAT TTT CTG TGT CTG A−3′
The sense primer introduced an EcoRI site (bold) and the antisense primer introduced a stop codon (underlined) and a SalI site (bold). The PCR product was digested with EcoRI and SalI and cloned into pCI in frame with the murine IgG leader to mediate secretion. Insertion was confirmed by sequencing (ABI systems, Perkin Elmer, Boston, MA, USA).
P-ctla4-omp-2
The extracellular domain of CTLA4 including its export signal sequence (accession no. X05719) was amplified from murine cDNA and cloned into the eukaryotic expression vector pCI. A synthetic 50 base pair hinge region, to mediate flexibility of the protein, was then cloned downstream the CTLA4 gene. The omp-2 gene was cloned downstream and in frame with the p-ctla4-hinge. The primers used for PCR amplification for each one of these genes were as follows:
Sense CTLA4: 5′ CCG GCT CGA GCC ACC ATG GCT TGT CTT GGA CTC CGG AGG 3′
Antisense CTLA4: 5′ CCA GAA CCA TGC CCG GAT TCT GAC GAA TTC GGA CGC GTG TTG CC 3′
Sense OMP-2: 5′ CCT TAC GCG TGA GCG GGG GTA TAG AGG CCG CTG TA 3′
Antisense OMP-2: 5′ GGC CGT CGA CTT ATT ACA CGT GGG TAT TTT CTG TGT CTG 3′
The sense primer of CTLA4 introduced an XhoI site (bold) and the antisense primer of CTLA4 introduced both an EcoRI and an MluI site (bold). The PCR product was digested with XhoI and MluI and cloned into the pCI vector. The hinge region, containing both an EcoRI and a MluI site (bold), was inserted into p-ctla4 vector downstream of the CTLA4 gene. The sense primer of OMP-2 introduced an MluI site (bold) and the antisense primer of OMP-2 introduced a stop codon (underlined) and a SalI site (bold). This PCR product was digested with MluI and SalI and cloned downstream and in frame with the p-ctla4-hinge to mediate secretion of the fusion antigen. In all cases insertion was confirmed by sequencing.
As control a p-ctla4 without omp-2 was constructed by introducing a stop codon downstream and in frame with p-ctla4-hinge.
E. coli strain XL1-blue (Stratagene, La Jolla, CA, USA) was used as bacterial host during plasmid construction and preparation. Plasmid DNA used for all cell transfections and immunizations was prepared from overnight cultures and purified using Qiagen Giga-Plasmid columns including an endotoxin removal step (Qiagen, Santa Clarita, CA, USA). DNA was dissolved in sterile H2O and stored at −20C°. Prior to injections, DNA was diluted in sterile PBS. Plasmid preparations contained less than 0·1 endotoxin units per µg of plasmid DNA (Limulus amoebocyte lysate assay (Bio Whittaker, Walkersville, MD, USA).
In vitro expression
Expression of proteins from plasmids was analysed initially in transiently transfected 293 cells. Cells were transfected using DOTAP-liposomes (Boehringer-Mannheim, Mannheim, Germany) according to the manufacturer's instructions. To analyse protein expression, cells were fixed with methanol and stained with sera from mice infected with C. pneumoniae. Alternatively, expression of transfected genes were analysed in cell supernatants and lysates by Western blot.
Binding assay
To analyse the CD80 (B7) binding capacity of the CTLA4-OMP-2 fusion protein 293 cells were transfected with p-ctla4-omp-2 using the lipofectamine (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. RMA cells expressing B7 (a kind gift from Hans-Gustaf Ljunggren, Stockholm, Sweden) and non-transfected controls were used; 2 × 105 B7-RMA or RMA cells were resuspended in 100 µl of supernatant containing the CTLA4-OMP-2 fusion protein and incubated on ice for 1 h. After incubation the cells were centrifuged and washed in PBS. The cells were then incubated for 1 h on ice, first with anti-OMP-2 antibodies (1/50), and then with FITC-labelled goat antimice immunoglobulins (1/30) in PBS 1% FCS. The samples were analysed using a fluorescence-activated cell sorter (FACS). As negative control supernatant from pCI transfected 293 cells was run in parallel.
Stimulatory oligodeoxynucleotides
Oligonucleotides (ODN) were purchased from Pharmacia Biotech, Uppsala, Sweden. The sequence of the CpG oligodeoxynucleotides (CpG) used was 5′ TTGGAACGTTCC TTTCCAACGTTGGTT TGGAACGTTCCTT 3′. The three CpG motifs are underlined. The non-CpG oligodeoxynucleotides (GpC) used was 5′ TTGGAAGCTTCCTTTCCAAGCTTGGTT TGGAAGCTTCCTT 3′. All the CpG dinucleotides are inverted to GpC.
Immunizations and challenges
C57Bl/6 mice (B6) 6–10 weeks of age were bred under specific pathogen-free conditions. Mutant mouse strains without CD4 showing a decreased helper cell activity [29], CD8 which is needed for development of cytotoxic T cells [30], recombination-activating gene-1 lacking both B and T cells (RAG-1) [31] and IL10 [32] were generated by homologous recombination in embryonic stem cells and back-crossed with C57Bl/6 mice. Mice deficient in both CD4 and CD8 [33] were also used.
All mice were anaesthetized with Metofane® (Mundelein, IL, USA) prior to immunization.
Mice were inoculated s.c. with 25 µg of OMP-2, HSP-60, MOMP, OVA or Trx-ABP emulsified in FCA or FIA, or with OMP-2 emulsified in FIA and mixed with 50 µg of immunostimulatory CpG or non-stimulatory GpC ODN. Control groups of mice received adjuvant alone (FCA or FIA-IS-CpG) or were left untreated.
Groups of eight to 10 mice received three times at 2-week intervals of 75 µg of either p-omp-2 or p-ctla4-omp-2 mixed with 75 µg empty control plasmid (pCI). Plasmids were diluted in 40 µl PBS and administered by intranasal inhalation (i.n.). Control groups of mice received 150 µg of p-ctla4, a mixture of p-ctla4 and p-omp-2 (75 µg each) or were left untreated.
Mycoplasma-free C. pneumoniae isolate Kajaani was propagated in HL cells. Infected cells were sonicated, cell remnants were removed by centrifugation and the bacteria were stored in small aliquots in sucrose–phosphate–glutamate (SPG) buffer at − 70°C until used. Infectivity as measured by inclusion-forming units (IFU) of bacterial preparation was determined in HL cell culture.
Mice were infected with 106 IFU of C. pneumoniae in 40 µl of PBS by i.n. (drops were applied to the nares under light anaesthesia with metophane).
Infectivity assay
Mice were sacrificed and right lungs were removed, minced and mechanically homogenized in 2 ml of SPG buffer. Homogenates were centrifuged for 10 min at 500 g to remove coarse tissue debris. Lysates were then diluted 10- and 100- fold in Dulbecco's MEM containing 5% FCS and streptomycin (DMEM 5% FCS). The infectious titre was assayed by culturing 500 µl of duplicate dilutions of the lysates on confluent HL cells grown on round 13-mm diameter coverslides in a shell vial. Inoculated cells were centrifuged for 1 h at 1600 r.p.m., supernatant removed and DMEM 5% FCS containing cycloheximide was added. Cells were incubated at 35°C for 72 h, fixed with methanol and stained with a FITC-conjugated Chlamydia genus-specific monoclonal antibody (Pathfinder Chlamydia Confirmation System, Kallestad Diagnostics, Chaska, MN, USA). Inclusion bodies were counted by fluorescence microscopy. Infectivity was expressed as IFU per lung.
Competitive PCR assay
Accumulation of IFN-γ and β-actin mRNA in freshly extracted lungs from vaccinated and infected mice was measured by competitive PCR assays [34]. Competitor fragments with a different length but using the same primers as the target DNA were constructed using composite primers [35] and an exogenous DNA fragment, or by subcloning of mutated (deleted or ligated) cytokine cDNA. Competitors were amplified by purified PCR (Qiagen, Studio City, CA, USA) and quantified in a spectrophotometer.
The primer sequences for the amplification of the cDNA were:
Sense IFN-γ: 5′ AAC GCT ACA CAC TGC ATC TTG G 3′
Anti-sense IFN-γ: 5′ GAC TTC AAA GAG TCT GAG G 3′
Sense β-actin: 5′ GTG GGC CGC TCT AGG CAC CAA 3′
Anti-sense β-actin: 5′ CTC TTT GAT GTC ACG CAC GAT TTC 3′.
Ten- or threefold serial dilutions of the competitor were amplified in the presence of a constant amount of cDNA. Reactions were carried out for 28–45 cycles in a thermal cycler (Perkin-Elmer Cetus, CT, USA) using an annealing step at 60°C.
Each experiment was performed two or three times and a representative set of results is presented.
Specific antibody determinations
Anti-OMP-2 antibodies in the sera from immunized mice were measured by ELISA [15]. Plates were coated overnight with 0·7 µg per ml of Trx-ABP-OMP-2 fusion protein. After blocking, sera from individual mice were then added at 1 : 1000 or 1 : 20000 dilutions (for total IgG) and 1 : 50 or 1 : 200 (for IgG2a) and 1 : 500 or 1 : 2000 (for IgG1). Plates were developed with alkaline phosphatase-conjugated goat antimouse IgG, IgG2a or IgG1 (γ-chain-specific) (Jackson, MO). The assay was standardized between plates by including titration of a pool of positive sera from OMP-2-immunized mice.
Cytokine determination in supernatants from antigen-stimulated spleen cells
Spleen cells (105 in proliferation assays or 3 × 105 cells for IFN-γ-tests) from individual mice were suspended in RPMI containing 10% FCS and distributed in round-bottomed microtitre plates. Triplicate wells were cultured for 3 days at 37°C in 5% CO2 atmosphere in the presence of different concentrations of Trx-ABP-OMP-2. A capture ELISA was used to measure the presence of IFN-γ in culture supernatants obtained 48 h after co-incubation with recombinant protein. Antimurine IFN-γ MoAb R-4-A-62 (ImmunoKontakt, Switzerland) was used as capture antibody (10 µg/ml), using biotinylated XMG.1 antimouse IFN-γ (4 µg/ml) (ImmunoKontakt) for detection. Quantification of IFN-γ in supernatants was calculated using parallel titrations of recombinant murine IFN-γ (Pharmingen, San Diego, CA, USA).
Pathology
Left lungs from FCA-HSP-60, FCA-OMP-2 or FCA administered and C. pneumoniae-infected mice were fixed in 4% formalin and processed for conventional histopathological examination after paraffin embedding. Three non-consecutive sagital sections were cut at 2 µm, deparaffinized and stained with haematoxylin–eosin. A single-blind microscopic evaluation of two sets of serial sections from each organ was performed on precoded slides. Histocytometry was performed with the aid of an ‘integrationsplatte’ eyepiece (Zeiss) with 100 hits. Presence or absence of pneumoniae for every hit was scored. All the area of the lung section was analysed.
Results
We studied first the effect of immunization using FCA as adjuvant on the outcome of infection with C. pneumoniae. Surprisingly, lungs from mice immunized s.c. with OMP-2 or HSP-60 showed a significant increase of bacterial load after infection (Fig. 1a). Groups receiving the C. pneumoniae MOMP, irrelevant antigen OVA, Trx-ABP protein, FCA or left untreated before challenge with C. pneumoniae showed no increased susceptibility to C. pneumoniae (Fig. 1a). These increases in susceptibility were noted at different times after infection (Fig. 1b). The enhanced bacterial load in FCA-OMP-2- and FCA-HSP-60-immunized mice was linked to a dramatically increased pathological outcome of infection (Fig. 2a–d). Whereas lungs from FCA-treated mice demonstrated perivascular and peribronchiolar mononuclear cell infiltrates and almost completely normal or slightly affected lung architecture (Fig. 2a), those from FCA-OMP-2- (Fig. 2b) and FCA-HSP-60- (not shown) immunized mice showed large areas of lung consolidation, larger mononuclear infiltrates and bronchi often filled with inflammatory exudates containing polymorphs.
Fig. 1.
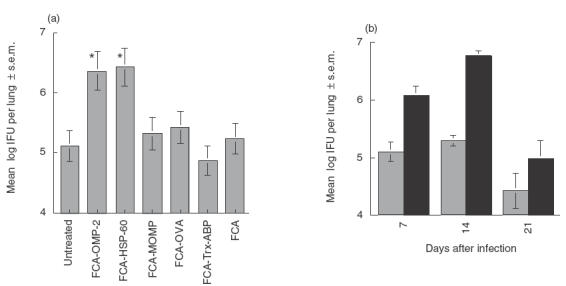
Immunization with FCA-OMP-2 diminishes resistance of mice to C. pneumoniae infection. (a) Groups of B6 mice (eight to 24 animals per group) were immunized s.c. with FCA-OMP-2, FCA-HSP-60, FCA-MOMP, FCA-OVA or FCA-Trx-ABP on three occasions at 0, 2 and 4 weeks or left untreated. Mice were infected intranasally with 106 C. pneumoniae 2 weeks after the last immunization dose. Mice were sacrificed 14 days after infection and the amounts of IFU per lung measured. Cumulative data from three independent experiments are shown. Differences vs. Trx-ABP, OVA, MOMP, FCA-treated and infected mice as well as untreated and infected mice are significant (P < 0·05 Student's t-test). (b) Groups of B6 mice (eight to 16 individuals per group) were immunized s.c. with FCA-OMP-2 on three occasions at 0, 2 and 4 weeks. Fourteen days after the last immunization, mice were challenged intranasally with 106 C. pneumoniae. Mice were sacrificed 7, 14 and 21 days after challenge and individual IFU determined in lung homogenates. The means of the log10-transformed IFU per lung ± s.e.m. are depicted. Cumulative data from two independent experiments are shown. (b)  , Untreated; ▪, FCA + OMP-2.
, Untreated; ▪, FCA + OMP-2.
Fig. 2.
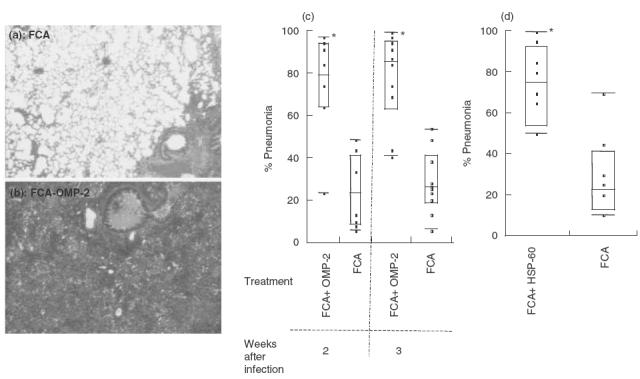
Increased severity of pneumonia in FCA-OMP-2 and FCA-HSP-60 immunized and C. pneumoniae-infected mice. Haematoxylin–eosin stainings of lungs from mice s.c. administered with FCA (a) or FCA-OMP-2 (b) 14 days after infection with C. pneumoniae. Note the large consolidated area in the lung of FCA-0MP-2-immunized mice, whereas a nearly normal lung morphology is preserved in control mice (a = 60×). The individual percentage of pneumonia in lung tissue sections from FCA-OMP-2 immunized mice 14 and 21 days after infection (c) or FCA-HSP-60 immunized mice and 14 days after infection (d). The median pneumonia per group is shown as a line in the middle of each box, and the 25th and 75th quantiles are the ends. The external bars indicate maximum and minimum values. *Differences between FCA-OMP-2 and FCA-HSP-60 versus FCA-treated groups are significant (P < 0·01, Mann–Whitney–Wilcoxon U-test).
A similar increase in bacterial load was observed when infection was delayed until 4 months after the last FCA-OMP-2 immunization dose. Mice (eight animals per group) immunized with FCA-OMP 24 months before infection showed 9·95 ± 1≈23 × 105 (mean ± s.e.m.) IFU per lung while FCA-inoculated controls had 1·04 ± 0·21 × 105 IFU (P < 0·05, t-test of log10 transformed data). This suggests that immunization with FCA-OMP-2 induced long-lasting memory immune responses that facilitate C. pneumoniae survival or growth.
We next explored the relevance of B and T cells for the observed enhancement of infectivity. B and T cell-deficient mice inoculated with FCA-OMP-2 were compared to controls immunized with adjuvant alone. Both CD4+ and CD8+ cells were required to be present in order for the increased susceptibility to infection to occur, because CD4−, CD8− or CD4−/CD8− FCA-OMP-2-immunized mice all showed similar susceptibility to infection as mice treated with adjuvant only (Fig. 3a–e).
Fig. 3.
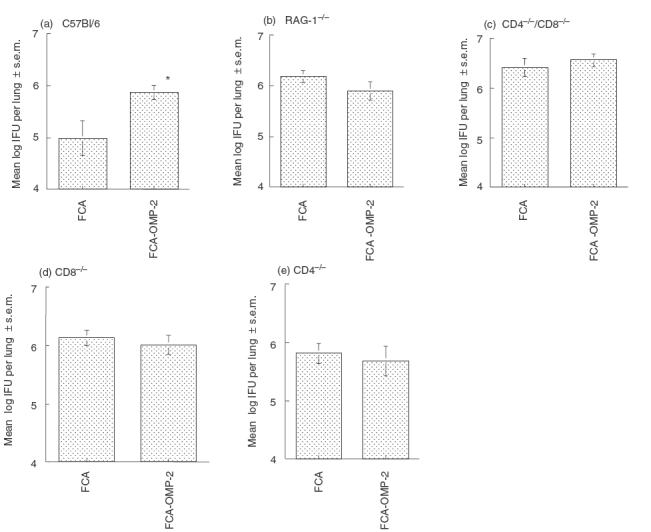
CD4+ and CD8+ T cells are required simultaneously for the harmful effect induced by FCA-OMP-2 immunization. B6 (a), RAG-1−/− [31] (b); CD4−/−/CD8−/− [33] (c); CD8−/−[30] (D); and CD4−/− [29] (e) mice (eight to 16 individuals per group) were immunized with FCA-OMP-2 or FCA as described in legend to Fig. 1. Fourteen days after the last immunization mice were infected with C. pneumoniae. Mice were sacrificed 14 days after challenge and individual IFU determined in lung homogenates. The mean log10 IFU per lung ± s.e.m. are depicted. Cumulative data from two independent experiments are shown. Differences versus FCA-treated and infected mice are significant (P < 0·05, Student's t-test).
The role of the adjuvant in the adverse reaction was then analysed. For this purpose the effect of using FCA with that of FIA containing immunostimulatory CpG (FIA-IS-CpG) motifs or non-stimulatory GpC oligonucleotides (FIA-NIS-GpC) as adjuvants for OMP-2 immunization was compared. Mice immunized with FIA-NIS-GpC-OMP-2 showed no increased susceptibility to infection with C. pneumoniae (Fig. 4). Thus, mycobacterium components from FCA are necessary to induce the detrimental effect of OMP-2 immunization. Moreover, immunization with FIA-IS-CpG-OMP-2 resulted in protection, as measured by a lower bacterial load in lungs (Fig. 4).
Fig. 4.
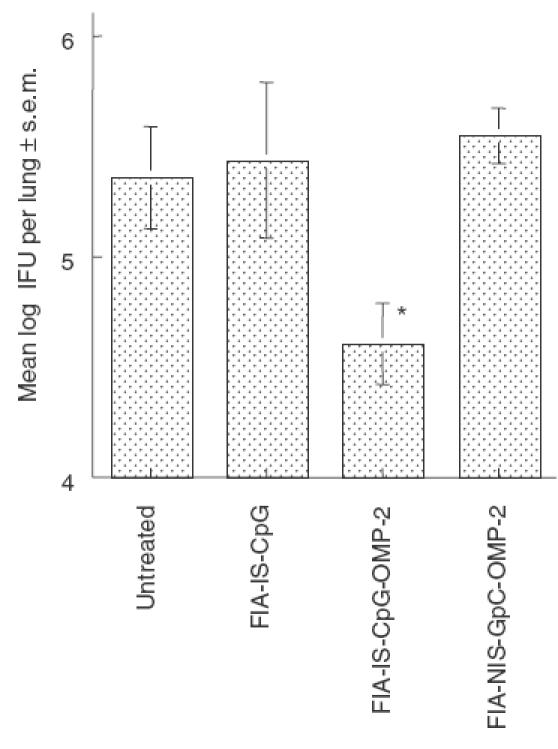
Immunization with OMP-2 using FIA-IS-CpG as adjuvant results in a partial protection against infection with C. pneumoniae. Mice (eight animals per group except FIA-IS-CpG-OMP-2, which contains 15 animals) were immunized with OMP-2 in the presence of immunostimulatory CpG or non-stimulatory GpC oligonucleotides. Control animals were administered with FIA-IS-CpG or left untreated. The mean of the log10 IFU per lung ± s.e.m. is depicted. Cumulative data from two independent experiments are shown. Differences versus FIA-IS-CpG, FIA-NIS-GpC-OMP-2 and untreated, infected mice are significant (P < 0·05, Student's t-test).
The latter findings led us to test the efficacy of DNA vaccines for OMP-2 containing immunostimulatory CpG motifs. In addition we included a DNA vaccine coding for the extracellular domain of CTLA-4 in fusion with OMP-2 (p-ctla4-omp-2). This fusion protein secreted from transfected cells bound B7-1 on the surface of transfected RMA cells (Fig. 5a). Mice immunized with p-ctla4-omp-2 showed 10-fold higher anti-OMP-2 IgG titres than those immunized with p-omp-2, suggesting an enhanced immunogenicity of the fusion protein (Fig. 5b). DNA vaccination with the fusion construct showed a 10-fold reduction of bacterial load in lungs (Fig. 5c). The non-protective effect of inoculation with p-ctla4 alone or of co-inoculation with p-ctla4 and p-omp-2 suggests that targeting OMP-2 protein to B7+ cells is likely to be of importance to achieve protection.
Fig. 5.
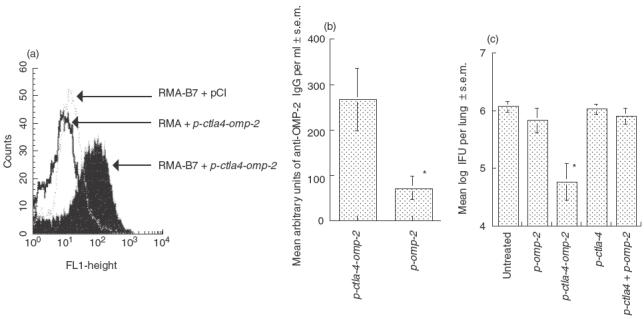
Protective effect of i.n. p-ctla4-omp-2 immunization against C. pneumoniae. (a) CTLA4-OMP-2-secreted protein binds to both B7 and to C. pneumoniae specific antibodies. Supernatants were obtained from p-ctla4-omp-2 or empty plasmid transfected L-293 cells. Supernatants were co-incubated with B7-transfected RMA cells or non-transfected controls. The complex was then stained using anti-C. pneumoniae mouse polyclonal antibodies followed by a FITC-labelled rabbit antimouse secondary antibody. Binding of CTLA4-OMP-2 to the B7 transfected RMA cells was measured by FACS. (b) Titres of anti-OMP-2 IgG were determined in sera from individual immunized mice (eight per group) with p-ctla4-omp-2, p-omp-2 and non-immunized mice 14 days after the last immunization dose. The relative arbitrary units were obtained by comparison with the titration of a positive pool serum. The mean log10 arbitrary units of anti-OMP-2 IgG per ml of sera ± s.e.m. are depicted. *Differences versus p-omp-2-immunized or untreated mice mice are significant (P < 0·05. Student's t-test). (c) Groups of B6 mice (eight to16 individuals per group) were immunized i.n. with p-ctla4-omp-2 on three occasions, at 0, 2 and 4 weeks. Fourteen days after the last immunization, mice were challenged with 106 IFU of C. pneumoniae. Mice were sacrificed 7 days after challenge and IFU determined in lung homogenates from individual mice. Data are pooled from two independent experiments. The means of the log10-transformed IFU per lung ± ss.e.m. are depicted. Differences versus p-omp-2, p-ctla4, p-ctla4 + p-omp-2 and untreated and infected mice are significant (P < 0·05, Student's t-test).
We next analysed how the various outcomes of infection after immunization with OMP-2 with the different types of adjuvants related to the type of immune response elicited. IFN-γ mRNA levels in lungs from FIA-IS-CpG-OMP-2-immunized mice were higher than those from FCA-OMP-2-immunized animals (Fig. 6a). Also, IFN-γ levels in supernatants from OMP-2-stimulated spleen cells from FIA-IS-CpG-OMP-2-immunized mice were higher than those from FCA-OMP-2-immunized animals (Fig. 6b). On the contrary, OMP-2-stimulated spleen cells from FCA-OMP-2-immunized mice showed higher levels of IL-10 than those immunized with FIA-IS-CpG-OMP-2 (Fig. 6c). IL-10 probably mediated the harmful effect induced by FCA-OMP-2 immunization, as FCA-OMP-2-immunized IL-10−/− mice showed similar susceptibility to C. pneumoniae as FCA-treated controls (Fig. 6d).
Fig. 6.
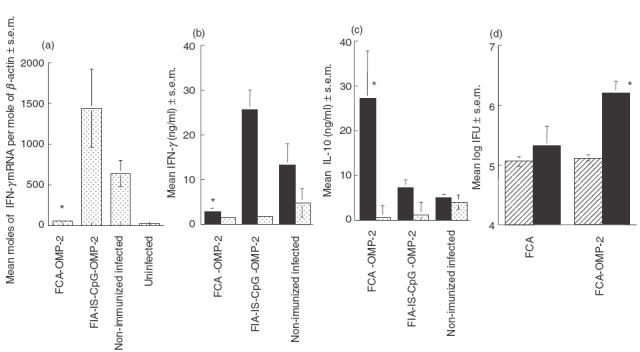
Higher IFN-γ and lower IL-10 levels are present in supernatants from spleen cells of mice immunized with FIA-IS-CpG-OMP-2 and infected with C. pneumoniae compared to those immunized with FCA-OMP-2. (a) Mice were immunized with FCA-OMP-2 and FIA-IS-CpG-OMP-2 before infection with C. pneumoniae. Infected non-immunized and untreated controls wee also used. RNA was extracted from the left lungs of at least four mice per group and reverse transcribed. IFN-γ and β-actin were measured by a competitive PCR. Differences versus FIA-IS-CpG-OMP-2-immunized or non-immunized infected mice are significant (P < 0·05, Student's t-test). (b,c) IFN-γ (b) and IL-10 (c) levels were measured in supernatants from FCA-OMP-2, FIA-IS-CpG-OMP-2-immunized or non-immunized controls 14 days after infection with C. pneumoniae. Spleen cells from individual mice (2 × 106 cells per ml) were co-incubated with recombinant OMP-2 for 48 h at 37°C, and the concentration of IFN-γ and IL-10 in supernatants from at least 6 mice per group were measured by ELISA. Differences in cytokine levels versus FIA-IS-CpG-OMP-2-immunized or non-immunized and infected, OMP-2-stimulated cultures are significant (P < 0·05, Student's t-test). Differences in IFN-γ levels in OMP-2-stimulated spleen cell cultures from FIA-IS-CpG-OMP-2 and untreated infected mice are significant (P < 0·05, Student's t-test). (d) IL-10−/−[32] and WT mice were immunized s.c. three times with FCA-OMP-2 or FCA as described above and challenged with C. pneumoniae 14 days after the last immunization dose. Mice were sacrificed 14 days after C. pneumoniae infection and individual IFU determined in lung homogenates. The mean log10 IFU per lung ± s.e.m. are depicted. Differences versus FCA-treated, infected mice are significant (P < 0·05, Student's t-test). (b) ▪, OMP-2-stimulated;  , medium. (d)
, medium. (d)  , IL-10−/−; ▪, WT.
, IL-10−/−; ▪, WT.
The shift in the cytokine pattern in mice immunized with OMP-2 with different adjuvants also related to that of the isotype pattern of specific antibodies in the sera. Similar levels of anti-OMP-2 IgG antibodies were detected in sera from mice immunized with FIA-IS-CpG-OMP-2 or FCA-OMP-2 immunization (Fig. 7a). However, anti-OMP-2 IgG2a titres were higher and IgG1 were lower in FIA-IS-CpG-OMP-2 than in FCA-OMP-2-immunized mice (Fig. 7b,c). The switch was due to the presence of CpG: mice immunized with FIA-NIS-GpC-OMP-2 showed lower IgG2a and higher IgG1 anti-OMP-2 levels compared to those immunized with stimulatory FIA-IS-CpG-OMP-2 (data not shown).
Fig. 7.
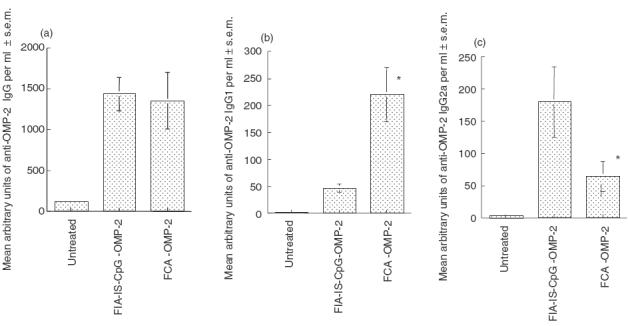
The isotype pattern of anti-OMP-2 antibodies depends on the adjuvant used for OMP-2 immunization. Specific antibody levels production after s.c. protein immunization against C. pneumoniae. The mean titres of anti OMP-2 IgG (a), IgG1 (b) and IgG2a (c) antibodies were determined in individual serum samples from mice (six per group) immunized with FCA-OMP-2 or CpG OMP-2 or non-treated controls. Differences versus FIA-IS-CpG-OMP-2-immunized or untreated infected mice are significant (P < 0·05, Student's t-test).
Discussion
One challenge in vaccine development is the selection of an adjuvant that favours the generation of protective immune mechanisms. The type of adjuvant used can direct the type of Th response generated to an administered antigen [36–38]. The importance of adjuvant in the outcome of experimental leishmaniasis and tuberculosis infections has been shown [39,40]. In this report we have added C. pneumoniae to the list of pathogens where adjuvant choice is essential in determining the outcome of infection.
The effect of FCA-OMP-2 (or HSP-60) immunization observed was antigen-specific, because immunization with MOMP or with non-relevant antigens with FCA did not change susceptibility to C. pneumoniae. Moreover, the requirement for the simultaneous presence of CD4+ and CD8+ cells and the ability to induce a harmful response, even several months after the last immunization dose, all suggest that a specific adaptive immune response to FCA-OMP-2 accounts for the detrimental effect. OMP-2 and HSP-60 are both highly immunogenic, and the immune responses elicited by both of them have been associated with a pathological outcome of infection [41–45]. However, a direct pathogenic effect of FCA-OMP-2 in the absence of infection was not observed in hearts or lungs, in contrast to the reports using C. trachomatis and C. pneumoniae OMP-2-derived peptides in BALB/c animals [45]. In fact, using the same peptides as used by these authors [1] we failed to reproduce their results even when using BALB/c mice (not shown).
We have shown here that FIA-IS-CpG-OMP-2 and p-ctla4-omp-2 protected against infection with C. pneumoniae. Similarly, DNA vaccines containing hsp-60 have been shown to protect mice against infection with C. pneumoniae [15,16]. Fusion in tandem of omp-2 with ctla4 increased the immunogenicity of the DNA vaccine, as also shown with other immunogens [46]. p-ctla4 omp-2 fusion also provided increased protection compared to p-omp-2. Increased immunogenicity of the correct type and protection are due probably to targeting of the expressed fusion protein to antigen-presenting cells, as no effect was achieved by co-inoculation of p-omp-2 and p-ctla4. Alternatively, fusion to CTLA-4 could increase the stability of OMP-2.
Results from ours and other laboratories have demonstrated the importance of IFN-γ in the resistance against primary infection with C. pneumoniae [7,8,47]. In this report we show that a shift towards a Th1 immune response by including immunostimulatory CpG in adjuvants was linked to increased protection. Confirming previous data, FIA-IS-CpG proved to be a better Th1-inducing adjuvant than FCA [4]. Indeed, although FCA has generally been shown to be a Th1 cytokine inducer (as exemplified in [48]), reports in which FCA induces a Th2 or a non-polarized response are not sporadic [48–55]. Th2 responses after immunization with antigen in the presence of FCA have been shown to depend on the nature of the antigen [52], on the antigen dose, on the genetic background [56] and on bystander immune responses to irrelevant antigens or infectious agents [52,54], which will alter the cytokine milieu of the host.The detrimental effect measured after FCA-OMP-2 immunization was associated with high IL-10 and low IFN-γ levels. Similarly, IL-10−/− mice were resistant to FCA-OMP-2-induced detrimental effects, indicating that IL-10 accounts for increased susceptibility to infection after FCA-OMP-2 immunization. However, IL-10 did not hamper a protective response as IL-10−/− FCA-OMP-2-immunized mice were still not protected against challenge with C. pneumoniae.
Previous observations suggest that inadequate immunity induced commonly against Chlamydia is at due least partially to negative immunoregulation by IL-10. High IL-10-producer mouse strains are more susceptible to infections [57]. Also, increased IL-10 levels in the endocervical secretion of women with chlamydial infection could predispose to enhanced HIV-1 transmission [58]. Finally, IL-10 has been indicated to mediate susceptibility against primary or secondary chlamydial infections [57–60].
The shift in cytokine pattern after immunization with different adjuvants was reflected in the isotype pattern of specific anti-OMP-2 antibodies. However, the antibody response to OMP-2 may be of limited utility, as OMP-2 in elementary bodies is confined to the inner surface of the outer membrane [61].
In summary, our data suggest that HSP-60 and OMP-2, but not MOMP, could act as Trojan horses, enhancing infectivity if administered with Th2-inducing adjuvants. On the other hand, OMP-2 inoculated with Th1 preferentially inducing adjuvants confer partial protection against C. pneumoniae. MOMP is the only protein of the three tested expressed in the outer surface of C. pneumoniae and accessible to humoral neutralizing antibodies [44]. We will therefore test if FCA-MOMP immunization in B cell-deficient mice will paradoxically enhance susceptibility to infection. In line with our results, inoculation of DNA vaccines containing MOMP or of liposome-MOMP suspension have been shown to induce Th1 responses and protect mice against infection with C. trachomatis and C. pneumoniae [13,14,18,62], while Th2 immune responses were recorded after immunization with dendritic cells pulsed with recombinant MOMP [63].
No sterilizing protection against C. pneumoniae was observed in this as well as in previous reports. However, those reports and data presented herein all indicate that vaccine development for chlamydia seems more possible now than in the past. The present data indicate that proper selection of the adjuvant will be crucial in such development.
Acknowledgments
This work was supported by grants from the Swedish Medical Research Council, the Swedish Cancer Society, the Karolinska Institute, the Swedish Society of Medicine and Amgen. We thank Ms Berit Olsson for excellent technical assistance.
REFERENCES
- 1.Schnare M, Holt AC, Takeda K, et al. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. 2000;10:1139–42. doi: 10.1016/s0960-9822(00)00700-4. [DOI] [PubMed] [Google Scholar]
- 2.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 3.Krieg AM, Yi AK, Schorr J, et al. The role of CpG dinucleotides in DNA vaccines. Trends Microbiol. 1998;6:23–7. doi: 10.1016/S0966-842X(97)01145-1. [DOI] [PubMed] [Google Scholar]
- 4.Sun S, Kishimoto H, Sprent J. DNA as an adjuvant: capacity of insect DNA and synthetic oligodeoxynucleotides to augment T cell responses to specific antigen. J Exp Med. 1998;187:1145–50. doi: 10.1084/jem.187.7.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donnelly JJ, Ulmer JB, Shiver JW, et al. DNA Vaccines Annu Rev Immunol. 1997;15:617–48. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 6.Kuo CC, Jackson LA, Campbell LA, et al. Chlamydia pneumoniae (TWAR) Clin Microbiol Rev. 1995;8:451–61. doi: 10.1128/cmr.8.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry LL, Feilzer K, Caldwell HD. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J Immunol. 1997;158:3344–52. [PubMed] [Google Scholar]
- 8.Rottenberg M, Gigliotti Rothfuchs AC, Gigliotti D, et al. J Immunol. 1999. Role of innate and adaptive immunity in the outcome of primary infection with Chlamydia pneumoniae, as analyzed in genetically modified mice; pp. 2829–36. [PubMed] [Google Scholar]
- 9.Williams DM, Grubbs BG, Pack E, et al. Humoral and cellular immunity in secondary infection due to murine Chlamydia trachomatis. Infect Immun. 1997;65:2876–82. doi: 10.1128/iai.65.7.2876-2882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson M, Schon K, Ward M, et al. Genital tract infection with Chlamydia trachomatis fails to induce protective immunity in gamma interferon receptor-deficient mice despite a strong local immunoglobulin A response. Infect Immun. 1997;65:1032–44. doi: 10.1128/iai.65.3.1032-1044.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Starnbach MN, Bevan MJ, Lampe MF. Protective cytotoxic T lymphocytes are induced during murine infection with Chlamydia trachomatis. J Immunol. 1994;153:5183–9. [PubMed] [Google Scholar]
- 12.Igietseme JU, Black CM, Caldwell HD. Chlamydia vaccines: strategies and status. Biodrugs. 2002;16:19–35. doi: 10.2165/00063030-200216010-00003. [DOI] [PubMed] [Google Scholar]
- 13.Zhang D, Yang X, Berry J, et al. DNA vaccination with the major outer-membrane protein gene induces acquired immunity to Chlamydia trachomatis (mouse pneumonitis) infection. J Infect Dis. 1997;176:1035–40. doi: 10.1086/516545. [DOI] [PubMed] [Google Scholar]
- 14.Murdin AD, Dunn P, Sodoyer R, et al. Use of a mouse lung challenge model to identify antigens protective against Chlamydia pneumoniae lung infection. J Infect Dis. 2000;181(Suppl. 3):S544–51. doi: 10.1086/315605. [DOI] [PubMed] [Google Scholar]
- 15.Svanholm C, Bandholtz L, Castanos-Velez E, et al. Protective DNA immunization against Chlamydia pneumoniae. Scand J Immunol. 2000;51:345–53. doi: 10.1046/j.1365-3083.2000.00684.x. [DOI] [PubMed] [Google Scholar]
- 16.Penttila T, Vuola JM, Puurula V, et al. Immunity to Chlamydia pneumoniae induced by vaccination with DNA vectors expressing a cytoplasmic protein (Hsp60) or outer membrane proteins (MOMP and Omp2) Vaccine. 2000;19:1256–65. doi: 10.1016/s0264-410x(00)00237-1. [DOI] [PubMed] [Google Scholar]
- 17.Pal S, Barnhart KM, Wei Q, et al. Vaccination of mice with DNA plasmids coding for the Chlamydia trachomatis major outer membrane protein elicits an immune response but fails to protect against a genital challenge. Vaccine. 1999;17:459–65. doi: 10.1016/s0264-410x(98)00219-9. [DOI] [PubMed] [Google Scholar]
- 18.Igietseme JU, Murdin A. Induction of protective immunity against Chlamydia trachomatis genital infection by a vaccine based on major outer membrane protein-lipophilic immune response-stimulating complexes. Infect Immun. 2000;68:6798–806. doi: 10.1128/iai.68.12.6798-6806.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal S, Theodor I, Peterson EM, et al. Immunization with an acellular vaccine consisting of the outer membrane complex of Chlamydia trachomatis induces protection against a genital challenge. Infect Immun. 1997;65:3361–9. doi: 10.1128/iai.65.8.3361-3369.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su H, Messer R, Whitmire W, et al. Vaccination against chlamydial genital tract infection after immunization with dendritic cells pulsed ex vivo with nonviable Chlamydiae. J Exp Med. 1998;188:809–18. doi: 10.1084/jem.188.5.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson MW, Lambden PR, Everson JS, et al. Immunoreactivity of the 60 kDa cysteine-rich proteins of Chlamydia trachomatis, Chlamydia psittaci and Chlamydia pneumoniae expressed in Escherichia coli. Microbiology. 1994;140:2003–11. doi: 10.1099/13500872-140-8-2003. [DOI] [PubMed] [Google Scholar]
- 22.Mygind P, Christiansen G, Persson K, et al. Analysis of the humoral immune response to chlamydia outer membrane protein 2. Clin Diagn Lab Immunol. 1998;5:313–8. doi: 10.1128/cdli.5.3.313-318.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pal S, Fielder TJ, Peterson EM, et al. Analysis of the immune response in mice following intrauterine infection with the Chlamydia trachomatis mouse pneumonitis biovar. Infect Immun. 1993;61:772–6. doi: 10.1128/iai.61.2.772-776.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beatty PR, Stephens RS. Identification of Chlamydia trachomatis antigens by use of murine T cell lines. Infect Immun. 1992;60:4598–603. doi: 10.1128/iai.60.11.4598-4603.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaVallie ER, Lu Z, Diblasio-Smith EA, et al. Thioredoxin as a fusion partner for production of soluble recombinant proteins in Escherichia coli. Meth Enzymol. 2000;326:322–40. doi: 10.1016/s0076-6879(00)26063-1. [DOI] [PubMed] [Google Scholar]
- 26.LaVallie ER, DiBlasio EA, Kovacic S, et al. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (NY) 1993;11:187–93. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- 27.Nygren P, Eliasson M, Abrahamsen L, et al. Analysis and use of serum albumin binding domains of streptococcal protein G. J Mol Recognit. 1998;1:69–76. doi: 10.1002/jmr.300010204. [DOI] [PubMed] [Google Scholar]
- 28.Svanholm C, Bandholtz L, Lobell A, et al. Enhnacement of antibody responses by DNA immunization using expression vectors mediating efficient antigen presentation. J Immunol Meth. 1999;228:121–30. doi: 10.1016/s0022-1759(99)00086-1. [DOI] [PubMed] [Google Scholar]
- 29.Rahemtulla A, Fung-Leung WP, Schilham MW, et al. Normal development and function of CD8+ cells but markedly decreased helper activity in mice lacking CD4. Nature. 1991;353:180–4. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- 30.Fung-Leung WP, Schilham MW, Rahemtulla A, et al. CD8 is needed for the development of cytotoxic T cells but not helper T cells. Cell. 1991;65:443–9. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- 31.Mombaerts P, Iacomini J, Johnson R, et al. RAG-1 deficient mice have no mature T and B lymphocytes. Cell. 1992;68:869–77. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 33.Schilham MW, Fung-Leung WP, Rahemtulla A, et al. Alloreactive cyotoxic T cells can develop in mice lacking both CD4 and CD8. Eur J Immunol. 1993;23:1299–304. doi: 10.1002/eji.1830230617. [DOI] [PubMed] [Google Scholar]
- 34.Rottenberg M, Castaños-Velez E, Mesquita R, et al. Intracellular colocalization of inducible nitric oxide synthase and Trypanosoma cruzi: evidence for a dual pathway of iNOS induction. Eur J Immunol. 1996;26:3203–13. doi: 10.1002/eji.1830261254. [DOI] [PubMed] [Google Scholar]
- 35.Siebert PD, Larrick JW. Competitive PCR. Nature. 1992;359:557–9. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- 36.Lipford GB, Bauer M, Blank C, et al. CpG-containing synthetic oligonucleotides promote B and cytotoxic T cell responses to protein antigen: a new class of vaccine adjuvants. Eur J Immunol. 1997;27:2340–4. doi: 10.1002/eji.1830270931. [DOI] [PubMed] [Google Scholar]
- 37.Davis HL, Weeranta R, Waldschmidt TJ, et al. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J Immunol. 1998;160:870–6. [PubMed] [Google Scholar]
- 38.Roman M, Martin-Orozco E, Goodman JS, et al. Immunostimulatory DNA sequences function as T helper-1-promoting adjuvants. Nat Med. 1997;3:849–54. doi: 10.1038/nm0897-849. [DOI] [PubMed] [Google Scholar]
- 39.Stacey KJ, Blackwell JM. Immunostimulatory DNA as an adjuvant in vaccination against Leishmania major. Infect Immun. 1999;67:3719–26. doi: 10.1128/iai.67.8.3719-3726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindblad EB, Elhay MJ, Silva R, et al. Adjuvant modulation of immune responses to tuberculosis subunit vaccines. Infect Immun. 1997;65:623–9. doi: 10.1128/iai.65.2.623-629.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrison RP, Lyng K, Caldwell HD. Chlamydial disease pathogenesis. Ocular hypersensitivity elicited by a genus-specific 57-kD protein. J Exp Med. 1989;169:663–75. doi: 10.1084/jem.169.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodall JC, Beacock-Sharp H, Deane KH, et al. Recognition of the 60 kilodalton cysteine-rich outer membrane protein OMP2 by CD4 (+) T cells from humans infected with Chlamydia trachomatis. Clin Exp Immunol. 2001;126:488–93. doi: 10.1046/j.1365-2249.2001.01709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinnunen A, Paavonen J, Surcel HM. Heat shock protein 60 specificT cell response in chlamydial infections. Scand J Immunol. 2001;54:76–81. doi: 10.1046/j.1365-3083.2001.00940.x. [DOI] [PubMed] [Google Scholar]
- 44.Wolf K, Fischer E, Mead D, et al. Chlamydia pneumoniae major outer membrane protein is a surface-exposed antigen that elicits antibodies primarily directed against conformation-dependent determinants. Infect Immun. 2001;69:3082–91. doi: 10.1128/IAI.69.5.3082-3091.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bachmaier K, Neu N, de la Maza LM, et al. Chlamydia infections and heart disease linked through antigenic mimicry. Science. 1999;283:1335–9. doi: 10.1126/science.283.5406.1335. [DOI] [PubMed] [Google Scholar]
- 46.Boyle JS, Brady JL, Lew AM. Enhanced responses to a DNA vaccine encoding a fusion antigen that is directed to sites of immune induction. Nature. 1998;392:408–11. doi: 10.1038/32932. [DOI] [PubMed] [Google Scholar]
- 47.Rottenberg ME, Gigliotti Rothfuchs A, Gigliotti D, et al. Regulation and role of IFN-gamma in the innate resistance to infection with Chlamydia pneumoniae. J Immunol. 2000;164:4812–8. doi: 10.4049/jimmunol.164.9.4812. [DOI] [PubMed] [Google Scholar]
- 48.Yip HC, Karulin AY, Tary-Lehmann M, et al. Adjuvant-guided type-1 and type-2 immunity: infectious/noninfectious dichotomy defines the class of response. J Immunol. 1999;162:3942–9. [PubMed] [Google Scholar]
- 49.Victoratos P, Yiangou M, Avramidis N, et al. Regulation of cytokine gene expression by adjuvants in vivo. Clin Exp Immunol. 1997;109:569–78. doi: 10.1046/j.1365-2249.1997.4631361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moureau C, Vidal PL, Bennasser Y, et al. Characterization of humoral and cellular immune responses in mice induced by immunization with HIV-1 Nef regulatory protein encapsulated in poly (DL-lactide-co-glycolide) microparticles. Mol Immunol. 2002;38:607–18. doi: 10.1016/s0161-5890(01)00096-7. [DOI] [PubMed] [Google Scholar]
- 51.Maron R, Hancock WW, Slavin A, et al. Genetic susceptibility or resistance to autoimmune encephalomyelitis in MHC congenic mice is associated with differential production of pro- and anti-inflammatory cytokines. Int Immunol. 1999;11:1573–80. doi: 10.1093/intimm/11.9.1573. [DOI] [PubMed] [Google Scholar]
- 52.Holland MJ, Harcus YM, Riches PL, et al. Proteins secreted by the parasitic nematode Nippostrongylus brasiliensis act as adjuvants for Th2 responses. Eur J Immunol. 2000;30:1977–87. doi: 10.1002/1521-4141(200007)30:7<1977::AID-IMMU1977>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 53.Comoy EE, Capron A, Thyphronitis G. Adjuvant is the major parameter influencing the isotype profiles generated during immunization with a protein antigen, the Schistosoma mansoni Sm28-GST. Scand J Immunol. 1998;47:444–52. doi: 10.1046/j.1365-3083.1998.00330.x. [DOI] [PubMed] [Google Scholar]
- 54.Comoy EE, Pestel J, Duez C, et al. The house dust mite allergen, Dermatophagoides pteronyssinus, promotes type 2 responses by modulating the balance between IL-4 and IFN-gamma. J Immunol. 1998;160:2456–62. [PubMed] [Google Scholar]
- 55.Brewer JM, Conacher M, Satoskar A, et al. In interleukin-4-deficient mice, alum not only generates T helper 1 responses equivalent to freund's complete adjuvant, but continues to induce T helper 2 cytokine production. Eur J Immunol. 1996;26:2062–6. doi: 10.1002/eji.1830260915. [DOI] [PubMed] [Google Scholar]
- 56.Locksley R, Reiner S, Hatam F, et al. Helper T cells without CD4. Control of Leishmaniasis in CD4 deficient mice. Science. 1993;261:1448–50. doi: 10.1126/science.8367726. [DOI] [PubMed] [Google Scholar]
- 57.Yang X, HayGlass KT, Brunham RC. Genetically determined differences in IL-10 and IFN-gamma responses correlate with clearance of Chlamydia trachomatis mouse pneumonitis infection. J Immunol. 1996;156:4338–44. [PubMed] [Google Scholar]
- 58.Cohen CR, Plummer FA, Mugo N, et al. Increased interleukin-10 in the the endocervical secretions of women with non-ulcerative sexually transmitted diseases: a mechanism for enhanced HIV-1 transmission? Aids. 1999;13:327–32. doi: 10.1097/00002030-199902250-00004. [DOI] [PubMed] [Google Scholar]
- 59.Yang X, Gartner J, Zhu L, et al. IL-10 gene knockout mice show enhanced Th1-like protective immunity and absent granuloma formation following Chlamydia trachomatis lung infection. J Immunol. 1999;162:1010–7. [PubMed] [Google Scholar]
- 60.Igietseme JU, Ananaba GA, Bolier J, et al. Suppression of endogenous IL-10 gene expression in dendritic cells enhances antigen presentation for specific Th1 induction: potential for cellular vaccine development. J Immunol. 2000;164:4212–9. doi: 10.4049/jimmunol.164.8.4212. [DOI] [PubMed] [Google Scholar]
- 61.Mygind P, Christiansen G, Birkelund S. Topological analysis of Chlamydia trachomatis L2 outer membrane protein 2. J Bacteriol. 1998;180:5784–7. doi: 10.1128/jb.180.21.5784-5787.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang DJ, Yang X, Shen C, et al. Characterization of immune responses following intramuscular DNA immunization with the MOMP gene of Chlamydia trachomatis mouse pneumonitis strain. Immunology. 1999;96:314–21. doi: 10.1046/j.1365-2567.1999.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shaw J, Grund V, Durling L, et al. Dendritic cells pulsed with a recombinant chlamydial major outer membrane protein antigen elicit a CD4 (+) type 2 rather than type 1 immune response that is not protective. Infect Immun. 2002;70:1097–105. doi: 10.1128/IAI.70.3.1097-1105.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]