

Abstract
The human peripheral cannabinoid receptor (CB2) was expressed as a fusion with the maltose-binding protein (at the N-terminus), thioredoxin A (at the C-terminus) and two small affinity tags (a Strep-tag and a polyhistidine tag). Expression levels of the recombinant receptor in Escherichia coli BL21(DE3) cells were dependent on location and type of tags in the expression construct, and were as high as 1-2 mg per liter of bacterial culture. The recombinant receptor was ligand binding-competent, and activated cognate G-proteins in an in vitro coupled assay. The fusion CB2-125 protein was purified by immobilized metal affinity chromatography on a Ni-NTA resin. Maltose-binding protein, thioredoxin and a decahistidine tag were removed from the fusion by treatment with Tobacco etch virus (Tev) protease. Purification to over 90% homogeneity of the resulting CB2, containing an N-terminal Strep-tag was achieved by affinity chromatography on a StrepTactin resin. Circular dichroism spectroscopy indicated an α-helical content of the purified recombinant protein of ∼54%. The expression and purification protocol allows for production of large (milligram) quantities of functional peripheral cannabinoid receptor, suitable for subsequent structural characterization. Preliminary results of reconstitution experiments indicate that the CB2 has retained its ligand-binding properties.
Keywords: Peripheral cannabinoid receptor, CB2, Affinity tags, Chromatographic purification, Reconstitution, Functional, Heterologous expression
The peripheral type cannabinoid receptor, CB2, belongs to the class A, rhodopsin-like G protein-coupled receptors (GPCR)1 with a trans-membrane domain of seven helices. GPCRs are involved in a large array of physiological activities. While the human genome encodes several hundred different GPCRs, potentially making them one of the most important group of drug targets for pharmaceutical industry [1], the structure of only one of these receptors, bovine rhodopsin, is currently available at atomic resolution. Unlike rhodopsin, which is conveniently obtained from bovine rod outer segment discs in large quantities at high purity, the natural expression levels of most GPCRs in tissues are quite low, preventing their large-scale purification as well as the application of most experimental methods to study their structure. In particular NMR and crystallography require heterologous expression of structurally unperturbed, functionally active GPCR. Heterologous expression of membrane proteins as a fusion with various expression partners in Escherichia coli cells is a commonly used technique that potentially yields milligram quantities of these polypeptides. N-terminal fusion with the maltose-binding protein (MBP) has been particularly beneficial for expression of recombinant membrane proteins in E. coli. It appears that the periplasmically localized MBP facilitates both the insertion of the hydrophobic polypeptide into the cytoplasmic membrane of E. coli and its proper folding. For example, both the full-length MBP sequence as well as a truncated version of MBP, consisting solely of the 36 N-terminal amino acid residues (26 amino acids of the signal sequence and the first 10 amino acids of the mature MBP) yielded expression of the GPCR 5-HT1A [2].
Earlier we reported a method for the heterologous expression of functional peripheral cannabinoid receptor as a fusion with maltose-binding protein and thioredoxin A (TrxA) in cells of E. coli [3] which was ligand binding-competent. We further demonstrated activation of cognate G proteins by the recombinant CB2 in response to binding of cannabinoid agonist CP55,940. The recombinant CB2 was solubilized from E. coli cells and purified to a high degree of homogeneity. While the fusion with the MBP and TrxA resulted in significantly increased expression levels, these expression tags also increased the total molecular weight of the recombinant protein to over 100 kDa, which is detrimental to most NMR structural studies. In this paper, we describe an efficient procedure for removing these expression partners by the action of specific Tev protease, and for purification of the cleaved CB2 via small affinity tags. This approach allows for the production of milligram quantities of purified CB2 and its functional reconstitution into lipid bilayers.
The previously expressed fusion CB2 had two Tev protease cleavage sites, one between MBP and CB2, and the second between CB2 and TrxA, as well as a decahistidine tag at the C-terminus of the fusion. Expression levels of the CB2-fusion were up to 2 mg per liter of bacterial culture [3,4]. Despite the significant advantages offered by this combination of expression tags, it also had certain drawbacks. In particular, the two Tev protease cleavage sites had different protease affinity which complicated the determination of optimal conditions for cleavage. Incomplete cleavage resulted in additional protein loss. Therefore, in this study we compare expression levels and ligand-binding characteristics of several new constructs harboring different combinations of MBP, TrxA and affinity tags fused to the CB2 sequence.
Since the N-terminal maltose-binding protein has a beneficial effect on expression levels of functional CB2, it was intriguing to investigate whether the signal sequence of MBP (first 26 amino acid residues), fused to the N-terminus of the GPCR, would be sufficient to ensure expression, correct folding, and insertion of the recombinant CB2 into cytoplasmic membranes of E. coli. Therefore, in the two constructs described in this work the signal sequence of MBP (S, Fig. 1) is fused to StrepTag II followed by the CB2 sequence. We provide evidence that the presence of both full-length MBP and TrxA in the fusion construct leads to higher concentrations of functional receptor in plasma membranes of E-coli.
Fig. 1.
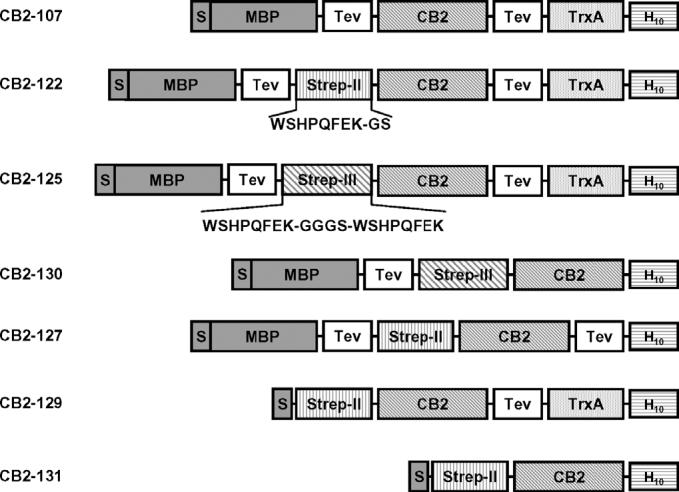
CB2-fusion constructs for expression in E. coli.
The presence of a polyhistidine tag at the C-terminus of the fusion CB2 was instrumental for affinity purification of the recombinant protein on a Ni-NTA resin, yielding 400-700-fold enrichment of the CB2-fusion to over 80% purity. The fusion was successfully cleaved [3], but further purification of the cleaved CB2 by either ion-exchange- or size-exclusion chromatography did not produce sufficient purity and/or yield (Yeliseev and Krepkiy, unpublished). Therefore the introduction of another small affinity tag into the CB2-fusion to enable a second purification by affinity chromatography was highly desirable. In addition to the polyhistidine tag at the C-terminus of the fusion, we attached a small StrepTag directly to the N-terminus of CB2 [5]. The selection of a StrepTag for the second step of affinity chromatography was based on the following considerations. The Strep-tag is rather small (only eight amino acid residues), it is uncharged, and, therefore, not likely to perturb receptor structure and function. The Strep-tag binds to the chromatographic resin (StrepTactin-agarose) with high affinity, even in the presence of detergents; it dissociates from the resin upon addition of desthiobiotin under mild conditions. Finally, this affinity chromatography procedure is efficient and relatively inexpensive, and consequently suitable for the production of milligram quantities of functional protein [6].
The use of dual affinity tags for expression and purification of recombinant proteins has become an increasingly popular method that simplifies purification and yields homogenous preparations of the proteins of interest [7]. These tags are usually attached to the N- and C-terminal domains of the expressed proteins [8]. The use of two consecutive steps of affinity chromatography allows for the efficient removal of contaminating proteins, including products of proteolytic degradation of the fusion protein that lack either N- or C-terminal tags [9]. By combining these two separate purification steps, only the full-length, non-degraded polypeptide, containing both N- and C-terminal tags, is retained.
Materials and methods
Chemicals and reagents
[3H]CP55,940 was purchased from Perkin-Elmer. Restriction enzymes and DNA-modifying enzymes were obtained from New England Biolabs. The HisProbe-HRP kit for detection of polyhistidine fusion proteins was obtained from Pierce, the Ni-NTA resin from Qiagen, and the StrepTactin agarose resin from Novagen. The antibodies against CB2 were purchased from Cayman Chemicals, and antibodies against maltose-binding protein from New England Biolabs. Oligonucleotides were purchased from Operon Biosciences.
Cholesteryl hemisuccinate Tris salt (CHS), the detergents 3[(cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and n-dodecyl-β-D-maltoside (DM) were obtained from Anatrace. N-Octyl-β-D-glucopyranoside (OG) was purchased from Calbiochem.
The lipids 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DOPE-PEG) were obtained from Avanti Polar Lipids, Inc.
Expression vectors and strains
Escherichia coli strain BL21(DE3) was purchased from Stratagene. The plasmid for expression of hexahistidine-tagged Tev protease (pRK793) was a gift from Dr. D.S. Waugh (NCI-Frederick, NIH).
Construction of plasmids for CB2 expression
Plasmid pAY122 encodes the human CB2 fused to the E. coli maltose-binding protein at the N-terminus and to thioredoxin A from E. coli, followed by decahistidine tag at the C-terminus. The sequence of CB2 is flanked by two Tev protease recognition sites, such that CB2 can be released from the fusion protein by treatment with Tev protease. The StrepTag II sequence was placed upstream of CB2 to yield CB2 with the WSHPQFEK tag attached at the N-terminus after cleavage with Tev protease. The StrepTag II sequence was introduced by ligating the pAY107 [3] plasmid digested with BamHI, with the double-stranded oligonucleotide encoding the WSHPQFEK sequence. The oligonucleotides used to create the double-stranded fragment of DNA were: Stag2-For: 5′-phosphate-gatcgtggtcgcatccgcagtttgaaaaac-3′ and Stag2-Rev: 5′-phosphate-gatcgtttttcaaactgcggatgcgaccac-3′. To increase efficiency of selection for the insert-containing plasmid, the oligonucleotides were designed such that the BamHI site was removed from the resulting construct; the ligation products were treated with BamHI enzyme before transforming them into E. coli cells. The plasmid DNA from the resulting colonies was isolated and sequenced. The resulting plasmid (pAY113) encodes amino acid residues arginine and serine positioned in between the StrepTag II and the N-terminus of CB2. The presence of a charged residue (R) at the N-terminus of CB2 may result in undesirable side effects. Therefore the R residue was replaced with a G, generating a small linker sequence GS in between the StrepTag II and CB2 in plasmid pAY122. This was achieved by PCR-assisted site-directed mutagenesis of pAY113 using the site-directed mutagenesis kit from Stratagene and primers Gly1-For: 5′-ccgcagtttgaaaaaggatccgaggaatgctgg-3′ and Gly1-Rev: 5′-ccagcattcctcggatcctttttcaaactgcgg-3′. The resulting plasmid contains a BamHI site upstream of the CB2 sequence.
Plasmid pAY125 encodes StrepTag III and a repeat of the WSHPQFEK sequence positioned upstream of CB2. It was constructed by introducing the second WSHPQFEK sequence (with a small linker GGGS) into plasmid pAY122 digested with BamHI, by ligating a double stranded DNA generated by annealing two oligonucleotides Strep3-For: 5′-phosphate-gatcgggcggcgctagctggtcgcatccgcagtttgaaaaaggcggcg-3′ and Strep3-Rev: 5′-phosphate-gatccgccgcctttttcaaactgcggatgcgaccagctagcgccgccc-3′ (Fig. 1).
Plasmid pAY127 encodes the CB2-fusion protein similar to the CB2-122, from which the TrxA sequence was removed. The resulting plasmid pAY127 encodes the MBP fused at the C-terminus to the Tev recognition site—to StrepTag II—to CB2—to second Tev recognition site—to decahistidine tag. This construct was generated by performing a polymerase chain reaction (PCR) using the QuickChange II XL site-directed mutagenesis kit (Stratagene) on a plasmid pAY122 with primers: 127-For: 5′-ttccagtctggtgg cggttctggtacccaccatcaccatcac-3′ and 127-Rev: 5′-gtgatggtg atggtgggtaccagaaccgccaccagactggaa-3′. The PCR conditions were as follows: 1 cycle at 95 °C for 1 min; 18 cycles of: 95 °C for 50 s, 60 °C for 50 s, 68 °C for 8 min; followed by incubation at 68 °C for 7 min. PfuUltra high fidelity DNA polymerase was used.
Plasmid pAY129 encodes the CB2-fusion protein similar to CB2-122, from which most of the sequence of the maltose-binding protein, and the first Tev recognition site were removed (see Fig. 1). Thus, in the resulting construct, the signal sequence of the MBP (first 26 amino acid residues) is fused via a small linker to the StrepTag II—to CB2—to Tev recognition site—to TrxA—to decahistidine tag. The construct was generated by performing PCR using the QuickChange II XL kit (Stratagene) on a plasmid pAY122 with primers: 129-For: 5′-ttttccgcctcggctctcgcccatatgtggtcgcatccgcagtttgaa-3′ and 129-Rev: 5′-ttcaaactgcggatgcgaccaca tatgggcgagagccgaggcggaaaa-3′. PCR conditions were the same as for constructing the plasmid pAY127.
Plasmid pAY131 is based on pAY129, from which the sequences of the TrxA and the TEV recognition site were removed. It encodes the fusion protein containing signal sequence of MBP fused to the StrepTag II—to CB2—to decahistidine tag. It was constructed by performing PCR on pAY129 using the QuickChange II XL mutagenesis kit and primers: 126-For: 5′-gatctagacctctctgattgcggtacccaccatcaccatcac-3′ and 126-Rev: 5′-gtgatggtgatggtgggtaccgcaatcagagaggtctagatc-3′. PCR conditions were the same as for plasmid pAY127.
Plasmid pAY130 is based on pAY125. It encodes a fusion protein consisting of the full-length MBP fused at the C-terminus to the Tev recognition site—to StrepTag III—to CB2—to decahistidine tag. It was generated by performing PCR using pAY125 as a template and the oligonucleotides: 126-For: 5′-gatctagacctctctgattgcggtacccaccatcac catcac-3′ and 126-Rev: 5′-gtgatggtgatggtgggtaccgcaatcagagag gtctagatc-3′. PCR conditions were the same as for plasmid pAY127.
Expression of CB2-fusion proteins and preparation of membranes
Expression of the recombinant CB2-fusion proteins was performed in E. coli BL21(DE3) cells grown in double-strength YT medium, supplemented with ampicillin (50 μg/ml) and 0.2% (w/v) glucose as described previously [3]. E. coli membranes from the cells expressing recombinant CB2 were prepared according to our previously published protocol [3] and stored at -80 °C until use.
Ligand-binding assays
The ligand-binding assays were performed using Millipore MultiScreen GF/B 96-well filter plates. Filters were treated with a 0.5% solution of polyethylenimine before use. For competition-binding assays, E. coli membranes (5 μg of protein per sample) were incubated with 1.43 nM [3H] CP55,940 and variable concentrations of unlabeled ligands for 1 h at 30 °C. The binding buffer consisted of 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 3 mM MgCl2, and 0.14% (w/v) BSA. For saturation binding, membranes were incubated with variable concentrations of [3H] CP55,940. Nonspecific binding was determined in the presence of 1 μM unlabeled CP55,940. Free and bound ligands were separated by rapid filtration through GF/B filters, and these filters were washed four times with 200 ml of cold binding buffer. Filters were transferred to 4 ml of ScintiSafe Plus scintillation counting fluid (Fisher) and radioactivity counted on a LS 6000IC counter (Beckman). Data from ligand binding were analyzed using Prism 4 software (GraphPad) as described previously [4].
For ligand-binding studies on purified CB2 that was reconstituted into a lipid matrix, we used essentially the same procedure as for non-purified CB2 in E. coli membranes, except that proteoliposomes contained much less total protein (5-20 ng of protein per sample) to compensate for the high fractional content of functional receptors.
Activation of G proteins in an in vitro coupled assay
Activation of G proteins by CB2-containing E. coli membranes was performed using the protocol previously reported in [3]. The G-protein components of the assay, Gαi and Gβ1γ2 were expressed in E. coli cells and in Sf9 cells, respectively, and purified according to previously published procedures [10,11].
Purification of recombinant CB2
Following solubilization of the fusion protein, CB2-125, from E. coli cells in buffer A (Tris-HCl buffer pH 7.5 containing 200 mM NaCl, 30% (v/v) glycerol and detergents 0.5% (w/v) CHAPS, 0.1% (w/v) DM, and 0.1% (w/v) CHS) according to the previously published procedure [3], CB2 was immobilized on a Ni-NTA column (Qiagen), washed with buffer A supplemented with 25 mM imidazole, and eluted with buffer B (buffer A supplemented with 250 mM imidazole). CB2-containing fractions were combined and concentrated in centrifugal spin concentrators (Orbital Biosciences) with a 70 kDa molecular mass cutoff. The protein was dialyzed against 50 mM Tris-HCl buffer pH 7.5 supplemented with 100 mM NaCl, 15% glycerol and detergents (buffer C), and cleaved with Tev protease overnight at 4 °C as described previously [3], with minor modifications that are described in the Results section.
The digested protein was loaded onto a StrepTactin Poros column (3 ml, EMD Biosciences) equilibrated with buffer C. The column was washed with four column volumes of buffer C, and the protein eluted with three column volumes of buffer C supplemented with 5 mM desthiobiotin. Elution fractions were combined, concentrated in centrifugal spin concentrators with a 30 kDa molecular mass cut off, and dialyzed against buffer A at 4 °C overnight. The final protein concentration was determined with a Bio-Rad DC kit. The concentrated protein was aliquoted, frozen in liquid nitrogen, and stored at -80 °C until use.
Circular dichroism spectroscopy
Circular dichroism experiments were conducted on a JASCO J-810 spectropolarimeter. Spectra were recorded from 180 to 250 nm with a 1 nm resolution and 4 s of integration time. The CB2 samples had a concentration of 5-10 μM and were investigated in a quartz cuvette with a path length of 0.1 mm. The helical content of the protein sample was calculated according to [12].
Reconstitution of the purified receptor into a lipid matrix
The purified CB2-125 was reconstituted into a lipid matrix according to the following procedure: the lipids POPC (2 mg), SOPC (2 mg), and DOPE-PEG (400 μg) were co-dissolved in chloroform and the solvent removed in a stream of argon gas. The lipids were dispersed in 1 ml of buffer A containing 4.4 μg of cleaved purified CB2-125. The resulting mixed micelles were diluted in 20 ml of cold PBS buffer which resulted in formation of a proteoliposome fraction. The proteoliposomes (probably containing residual detergents) were concentrated by centrifugation at 170,000g for 2 h at 4 °C, and re-suspended in a small volume of PBS buffer supplemented with 10% sucrose. Small aliquots of this preparation were frozen in liquid nitrogen and kept at -80°C.
Results
Effects of fusion partners/affinity tags on expression levels of CB2
We fused the eight amino acid-long StrepTag II (or a double repeat of that tag, StrepTag III) to the N-terminus of CB2. The sequence WSHPQFEK does not introduce an additional charge to CB2, is reasonably hydrophilic, and thus not likely to significantly affect structural properties and biological activity of the receptor. Digesting the fusion protein CB2-122 (Fig. 1) with Tev protease exposes the Strep-tag for binding to the affinity resin StrepTactin (EMD Biosciences).
Although the StrepTag II was successfully used for purification of several soluble proteins [9], the chromatographic purification and cleavage of the hydrophobic CB2 is different because it must be performed on detergent micelles. The conditions for binding of recombinant, Strep-tag-containing proteins on a StrepTactin resin, as recommended by the manufacturer, do not include the use of high concentrations of detergents like CHAPS, DM, and CHS; therefore it was necessary to test whether a single StrepTag sequence would bind with sufficient affinity to the resin in the presence of these detergents. To increase the binding affinity of the tagged protein to the resin, we introduced a repeat of the WSHPQFEK sequences (separated by a small GGGS linker) in two of the recombinant constructs, CB2-125 and CB2-130 (Fig. 1).
In two constructs (CB2-130 and CB2-131) the second Tev recognition site (downstream of the CB2) was deleted, and the decahistidine tag fused directly to the C-terminus of CB2. Since the TrxA is also removed from these constructs, it was possible to study the effects of TrxA on expression levels. In construct CB2-127 the TrxA sequence is removed but the second Tev site is still present, thus allowing examination of the effects of the Tev recognition sequence on expression levels of the fusion protein.
In constructs 129 and 131 the signal sequence of MBP (S, Fig. 1) is fused to the StrepTag II followed by the CB2 sequence.
The expression levels of the various CB2-fusions were assessed by Western blot analysis (Fig. 2). The presence of the His-tag, CB2, and MBP in the fusion proteins was visualized by probing protein extracts from bacterial plasma membranes with HisProbe reagent, anti-CB2 or anti-MBP antibody, respectively. Introduction of the StrepTag II (construct 122) or StrepTag III (construct 125) into the fusion did not change expression levels of the CB2-fusion significantly (compare with expression of the parental construct, CB2-107). On the other hand, removal of the TrxA sequence (constructs 127 and 130) resulted in a 1.5- to 2- fold decrease in yield. Also, the proportion of non-specifically cleaved fusion (band at MW ∼65 kDa), that stains with both anti-CB2 and anti-MBP antibodies, increased significantly for constructs 127 and 130, suggesting that the absence of the TrxA from the fusion results in destabilization of CB2 which makes it more vulnerable to proteolytic degradation. The most dramatic reduction of expression levels, however, was observed when the full-length MBP was substituted for the signal sequence of MBP (construct CB2-129) at the N-terminus of the CB2-fusion. Expression yields decreased at least 10- to 20-fold. Removal of both MBP and TrxA (construct 131) resulted in a further reduction of expression levels such that detection of CB2 by Western blot analysis was barely possible.
Fig. 2.
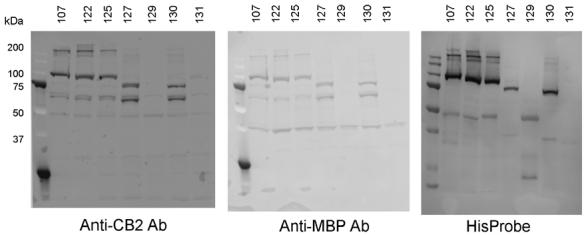
Western blot analysis of the CB2-fusion proteins (107, 122, 125, 127, 129, 130, and 131). The recombinant receptor was expressed in E. coli BL21(DE3) cells upon induction with 0.5 mM IPTG at 20 °C for 40 h. Cells were collected by centrifugation and cell membranes obtained as described in Materials and methods. After separation on 10% SDS-PAGE, proteins were electroblotted onto nitrocellulose membranes and probed with either anti-His tag HisProbe reagent or with antibodies against CB2 or MBP.
Ligand binding to recombinant CB2
The functionality of the recombinant receptor and the density of ligand-binding sites in the membrane preparations were determined with a ligand-binding assay using the specific agonist [3H] CP55,940 (Table 1, Fig. 3). The data confirmed results of the Western blot analysis. In particular, removal of the TrxA from the fusion decreased the density of ligand-binding sites of bacterial membranes expressing CB2. Removal of the MBP sequence (constructs 129 and 131) resulted in a decrease of ligand binding to almost undetectable levels, confirming results of the Western blot analysis. Constructs containing MBP had Kd-values similar to values of the native receptor for several well characterized cannabinoid ligands, like the endogenous ligand anadamide, the synthetic ligands CP55,940 and HU210, and tetrahydrocannabinol (Table 1). This confirms the structural integrity of the recombinant CB2-fusion in bacterial membranes.
Table 1.
Ligand binding on the membranes of E. coli cells expression various CB2-fusion constructs
| Construct | Bmax, pmol/mg protein |
Ki (nM) |
|||||
|---|---|---|---|---|---|---|---|
| CP55,940 | HU210 | WIN 55,212-2 | SR144,528 | Δ9-THC | Anandamide | ||
| 107 | 11.9 | 1.5-5.6 | 0.9 | 2.9 | 1.0 | 42 | 860 |
| 122 | 10.8 | 5.5 | 1.5 | 5.4 | 5.3 | 61 | 1196 |
| 125 | 12.1 | 1.5-4.0 | 0.8-1.5 | 2.5 | 4.4 | 61 | 1843 |
| 130 | 7.1 | 2.5-6.9 | 2.2 | 2.6-13.3 | 5.7 | 85 | 1198 |
| 127 | 8.7 | 3.9-4.0 | 0.9-1.2 | 3.7 | 4.3 | 108-287 | 1470 |
| 129 | ≤1 | 6.2 | nd | nd | nd | nd | nd |
| 131 | Nd | nd | nd | nd | nd | nd | nd |
Five micrograms of total membrane proteins were used per assay. nd, not determined.
Fig. 3.
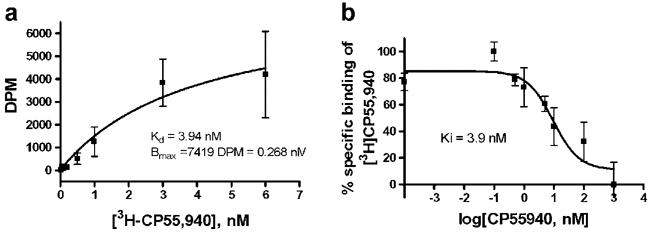
(a) Saturation binding of [3H]CP55,940 and (b) competition binding of cannabinoid ligands on membranes of E. coli BL21 expressing CB2-125.
Activation of G proteins in an in vitro coupled assay
An even more stringent test for functionality of the recombinant CB2-fuison is the performance of the protein in an in vitro coupled assay with binding of the subunits of cognate G proteins (Gαi1, Gβ1γ2) upon receptor activation. The assay was performed on E. coli membranes expressing CB2-125 (Fig. 4). We observed a dose-dependent activation of G proteins in response to binding of specific cannabinoid agonist CP55,940. The results clearly show that the CB2-fusion also activates cognate G proteins in a dose-dependent manner.
Fig. 4.
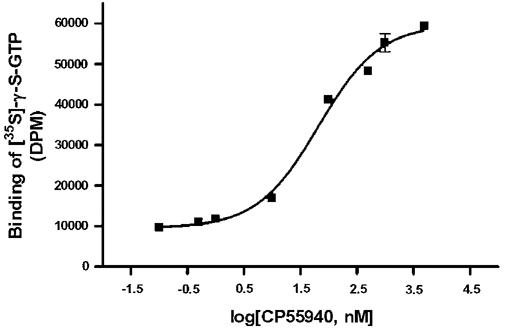
Activation of G proteins by the recombinant CB2-fusions in E. coli membranes.
TEV protease cleavage of the fusion protein
The CB2-125 fusion was purified from E. coli cells by Ni-NTA chromatography according to a previously published procedure [3]. In order to remove the expression tags from the CB2-fusion, the purified protein was treated with Tev protease. Since the buffers used for solubilization and purification of CB2 contain detergents (CHAPS, CHS,DM), salts and glycerol, the effects of these compounds on the activity of TEV protease was investigated. It was reported previously that the tobacco etch virus protease is partially inhibited by detergents in a dose-dependent fashion [13].
The influence of the following conditions on cleavage efficiency of the Tev protease was studied: concentration of NaCl, imidazole, glycerol, detergents (CHAPS, DM, CHS, dodecylphosphocholine), temperature, and duration of incubation. The efficiency of the proteolytic digestion of the CB2-fusion was quantified by measuring relative intensities of CB2 bands in Western blots and comparing them to intensity of the full-length CB2-fusion. The fusion protein (CB2-125) and the recombinant Tev protease were mixed at a molar ration of 5:1 in a Tris-HCl buffer supplemented with salts and detergents, and incubated at either 4, 20 or 30 °C. Aliquots of the reaction mixture were withdrawn at several time points, and the protein composition of the mixture analyzed by SDS-PAGE and Western blot, probed either with antibodies against CB2 or with Streptactin conjugated to HRP. Relative intensities of the bands in SDS-PAGE and of immunoreactive bands in Western blot were quantified by the computer program ImageQuant (Molecular Dynamics). The results of these experiments are summarized in Table 2.
Table 2.
Cleavage of CB2-125 fusion protein with TEV protease
| Parameter of incubation | Proportion of un-cleaved CB2-fusion (%) |
|||
|---|---|---|---|---|
| Full-length | Cleaved at one Tev site | Full-length | Cleaved at one Tev site | |
| 4 h incubation | 20 h incubation | |||
| Temperature | ||||
| 4 °C | 3 | 10 | 1 | 5 |
| 20 °C | 2 | 5 | 1 | 5 |
| pH | ||||
| 7.5 | 2 | 10 | 2 | 5 |
| 8.0 | 2 | 10 | 1 | 5 |
| Concentration of triple detergent | ||||
| 1× | 5 | 10 | nd | 5 |
| 2× | 5 | 15 | 3 | 10 |
| 6× | 5 | 20 | 2 | 15 |
| Concentration of DPC | ||||
| 3 mM | 3 | 15 | nd | 5 |
| 30 mM | 3 | 20 | 2 | 10 |
| 60 mM | 10 | 30 | 5 | 15 |
| Concentration of glycerol | ||||
| 0% | nd | 15 | nd | 5 |
| 5% | 2 | 15 | nd | 5 |
| 15% | 3 | 15 | nd | 5 |
| 30% | 5 | 20 | nd | 10 |
| Concentration of NaCl | ||||
| 0% | 2 | 10 | 2 | 5 |
| 50 mM | 2 | 10 | 2 | 5 |
| 100 mM | 5 | 20 | nd | 10 |
| 200 mM | 10 | 25 | 5 | 15 |
| Concentration of imidazole | ||||
| 0 | nd | 5 | nd | 5 |
| 50 mM | 2 | 10 | nd | 5 |
| 100 mM | 3 | 10 | 2 | 5 |
| 200 mM | 10 | 20 | 5 | 15 |
Ni-NTA-purified CB2 in triple detergent was diluted 20-fold in the respective buffer and mixed with 0.2 (mol/mol) TEV protease. nd, not determined.
Preparations of purified CB2 after Ni-NTA column chromatography contain high concentrations (200-250 mM) of imidazole. Therefore the effects of this compound on the cleavage reaction were studied as well. The presence of imidazole at a concentration of 200 mM inhibited the reaction significantly. The content of the uncleaved and partly cleaved CB2-fusion was higher than 20%, even after 20 h of incubation. A reduction of imidazole concentration to less than 50 mM allowed the cleavage reaction to proceed to completion, with only minor fractions of partly cleaved protein remaining (Fig. 5).
Fig. 5.
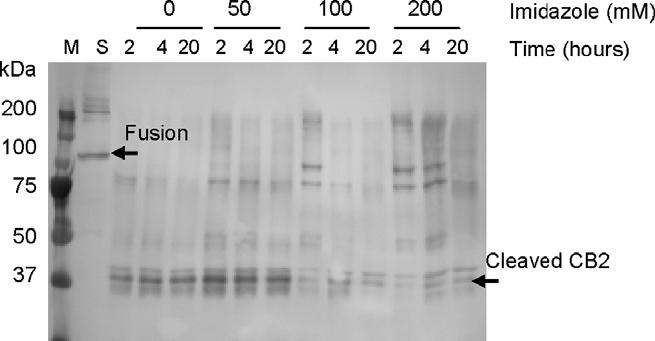
Cleavage of CB2-125 fusion with TEV protease. The reaction was performed by mixing pre-purified (Ni-NTA chromatography) micellar solutions of CB2-125 fusion with Tev protease at a molar ratio of 5:1 in buffer C supplemented with indicated amounts of imidazole. The CB2-fusion and cleaved CB2 are indicated by arrows.
Detergents are essential for protein solubilization and chromatographic purification. After purification on a Ni-NTA coloumn, the CB2-fusion could be routinely concentrated with a Centricon microconcentrator. The latter procedure also concentrates detergents to various extent (Kimura et al., unpublished). Therefore we investigated the influence of the concentration of the “triple detergent” mix (CHAPS, DM, CHS) on the cleavage reaction. An increase of detergent concentrations to 1% CHAPS, 0.2% DM, 0.2% CHS (2× detergent mix) did not result in a noticeable inhibition of the reaction. A further increase (3% CHAPS, 0.6% DM, 0.6% CHS) reduced cleavage of the CB2-fusion to 50-60% after 4 h of incubation. Likewise, we observed that Tev protease tolerates presence of DPC up to concentrations of 3-9 mM, while a concentration of 30 mM and higher resulted in significant inhibition.
Glycerol up to a concentration of 15% v/v did not alter efficiency of cleavage of the CB2-fusion, but 30% glycerol inhibited the reaction significantly. After 4 h of incubation, approximately 20-30% of the fusion remained unprocessed; after 20 h of incubation at 4 °C, as much as 10% of the fusion polypeptide was still present in the reaction mixture. NaCl at concentrations up to 100 mM did not affect the efficiency of cleavage of the CB2-fusion. However, the presence of 200 mM NaCl led to a noticeable reduction of cleavage reaction rates: 15-20% of the fusion was still unprocessed after 4 h of cleavage. After 20 h of incubation about 10% of the full-length non-cleaved CB2-fusion remained.
The influence of temperature on cleavage efficiency was also studied. An increase in temperature from 4 to 20 °C resulted in nearly 100% cleavage of the fusion protein at the 2nd TEV recognition site (downstream of CB2 sequence) after only 2 h of incubation. Extending the duration of the reaction at this temperature beyond 2 h did not raise efficiency of cleavage at the 1st TEV site (upstream of the CB2 sequence).
An increase in the pH of the reaction buffer from 7.5 to 8.0 did not result in any noticeable decrease of cleavage efficiency.
In summary, almost all components in the solubilization/purification buffers inhibited the TEV cleavage reaction in a concentration-dependent fashion. Therefore all subsequent cleavage reactions were performed in 50 mM Tris-HCl buffer, pH 7.5, containing 100 mM NaCl, 15% glycerol and a 1× concentration of the triple detergent mix. The reaction was performed at 4 °C for 12-20 h. Imidazole was removed, and concentrations of salts and glycerol adjusted by dialyzing the protein solution against two changes of Tris-HCl buffer containing additives at the concentrations reported above.
Affinity purification of CB2 after cleaving the CB2-fusion
After purification of the CB2-fusion on a Ni-NTA column and removal of the expression tags by cleaving with Tev protease, it was necessary to isolate the cleaved CB2 from the reaction mixture. This was done according to the following protocol: before applying the reaction products to the StrepTactin affinity resin, products of the TEV digest were passed through a small Ni-NTA column to capture the hexahistidine-tagged Tev protease and the decahistidine-tagged C-terminal part of the fusion (TrxA-His-10). The full-length as well as partly digested CB2-fusion, harboring a decahistidine tag, were also retained on the Ni-NTA resin and thus removed from the reaction mix. This purification was performed in the presence of 20 mM imidazole to minimize the non-specific capturing of CB2 on the resin. The flowthrough from the Ni-NTA resin, containing the CB2, was applied onto a small (3 ml) StrepTactin column that was pre-equilibrated with buffer C. The resin was washed three times with three column volumes of buffer (100 mM Tris-HCl pH 7.5, 15% glycerol, 100 mM NaCl, 0.5% CHAPS, 0.1% CHS, 0.1% DM) and the protein eluted with three column volumes of the same buffer, supplemented with 5 μM desthiobiotin. The eluted fractions were combined and concentrated on a centrifugal concentrator. Purity of the resulting CB2 was confirmed by SDS-PAGE (Fig. 6). The yield of purified CB2 from 10 L of bacterial culture expressing the CB2-125 fusion protein was typically 4-5 mg (Table 3). The yield of the functional receptor was determined after reconstitution of the purified CB2 into the lipid matrix (see section on ligand-binding studies on reconstituted receptor below). The specific activity of the receptor preparations at various stages of purification could not be reliably determined due to the difficulties in separating the detergent-solubilized receptor from the hydrophobic radioligand that associates with detergent micelles.
Fig. 6.
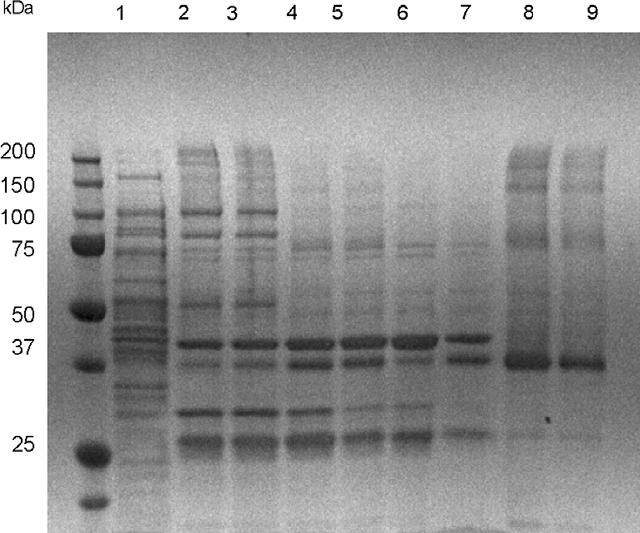
Purification of CB2-125. Lane 1, crude extract before 1st Ni-NTA chromatography; lane 2, concentrated fractions after 1st Ni-NTA chromatography; lane 3, fractions after dialysis against buffer B; lane 4, proteins after TEV protease digest; lane 5, combined fractions (flowthrough and wash) from the 2nd Ni-NTA column; lane 6, flowthrough from the StrepTactin column; lane 7, wash fraction from the StrepTactin column; lanes 8 and 9, eluate fractions from StrepTactin column, after concentration (5 μg and 2.5 μg of protein per lane, respectively).
Table 3.
Purification of CB2-125 from 10 L of E. coli BL21(DE3) cells
| Purification step | Volume (ml) | Total protein (mg) | Recovery a (%) | Specific activity, pmol of binding sites per mg of protein |
|---|---|---|---|---|
| Supernatant (crude extract after ultracentrifugation) | 600 | 8950 | 100 | 12.1 pmol/ mg proteinb |
| Combined fractions from Ni-NTA affinity column | 5.3 | 13.5 | 80 | |
| Combined fractions after dialysis/ TEV digest | 8.2 | 13.2 | 78 | |
| Flowthrough/ wash fraction from the second Ni-NTA column | 12 | 7.24 | 64 | |
| StrepTactin column | 5.8 | 4.2 | 44 | 11,400 pmol/mg proteinc |
Recovery was determined by measuring relative intensities of CB2 bands in Western blots.
Activity measured in cytoplasmic membranes prepared from the E. coli BL21(DE3) cells expressing CB2-125.
Specific activity as determined by saturation binding of [3H] CP55,940 to the proteoliposomes containing purified CB2-125.
Similar to the purification of CB2 from the CB2-125-fusion (StrepTag III), we cleaved and purified CB2 from CB2-122 (StrepTag II). Although expression levels of constructs CB2-122 and CB2-125 were almost identical (see Fig. 2 and Table 1), the yields of CB2 isolated by StrepTactin chromatography were consistently lower than yields from CB2-125 by about 30-40%. It was observed that significant losses of CB2 occurred because of poor retention of CB2 on the StrepTactin column. Substantial amounts of CB2 leaked from the column while it was washed with buffer C (results not shown). Therefore, in all subsequent experiments the construct containing StrepTag III was used rather than StrepTag II.
Characterization of the purified CB2 by CD-spectroscopy
The purified CB2 was dialysed against 50 mM Tris/HCl buffer, pH 7.4, containing 200 mM NaCl, 30% (v/v) glycerol, and the triple detergent mix. Circular dichroism spectra were recorded in a cell with a path length of 0.1 mm. The short path length was critical for the suppression of light scattering effects which would have prevented measurements in the spectral region of less than 210 nm. The α-helical content of the protein, calculated as described in Materials and methods, was ∼54%, confirming that the purified receptor retained a high helical content in micellar solution (Fig. 7).
Fig. 7.
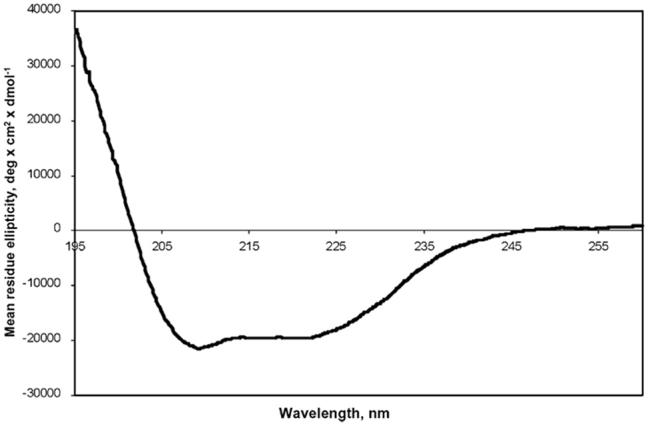
Circular dichroism spectrum of purified CB2-125 in 50 mM Tris-HCl pH 7.4, 200 mM NaCl containing 10% (v/v) glycerol and triple detergent mix (0.5% CHAPS, 0.1% DM, 0.1% CHS).
Ligand-binding studies on reconstituted CB2
The reconstitution of CB2 into a lipid matrix was performed as described in Materials and methods. The proteoliposomes were pelleted by centrifugation and resuspended in 50 mM Tris buffer pH 7.5 containing 10% sucrose. Ligand binding to CB2 was studied, using the same procedure as for the ligand binding to the CB2-fusion proteins in the plasma membrane of E. coli, before purification. For ligand competition studies, proteoliposomes were incubated with [3H] CP55,940, exposed to a series of concentrations of unlabeled, competing ligand, and the reaction mixture passed through a Whatman GF/B filter and the radioactivity retained on the filter was counted. For saturation binding experiments, the reconstituted receptor was incubated with a series of concentrations of radioactively labeled CP55,940, both in the absence and presence of a saturating concentration of unlabeled CP55,940. The samples were filtered through a Whatman glass-fiber filter, and the total amount of retained radioactivity, as well as the non-specific retention of the radioactively labeled ligand was determined. Both ligand competition and saturation binding confirmed that at least 25-35% of the reconstituted receptor retained functionality (Fig. 8). The dissociation constant for the specific cannabinoid agonist CP55,940 was in the low nanomolar range, similar to values for the native receptor in mammalian tissues.
Fig. 8.
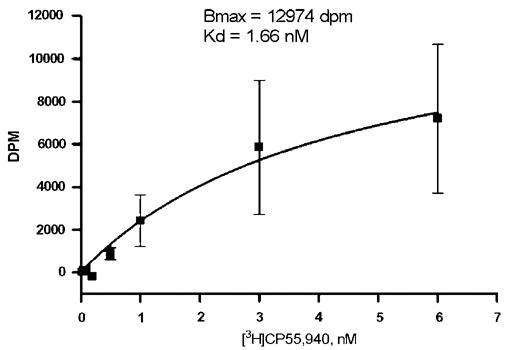
Saturation binding of [3H]CP55,940 on CB2-125, reconstituted into a lipid matrix.
Discussion
We have studied the effects of using MBP and TrxA as a fusion partners for expression of the peripheral cannabinoid receptor, CB2, in E. coli. The presence of the full-length MBP at the N-terminal part of the fusion appears to be essential for high-level expression (>1 mg of protein per liter of culture) of CB2. The removal of most of the sequence of “mature” MBP, which was achieved by fusing the CB2 directly to the signal sequence of MBP (first 26 amino acid residues), resulted in a dramatic decrease, 10- to 20-fold, in the levels of the fusion protein, although a small fraction of the CB2-129 fusion protein in E. coli membranes still retained its ability to bind cannabinoid ligands. The presence of TrxA as a C-terminal part of the fusion appears to raise expression levels of CB2 1.5- to 2-fold. On the other hand, the presence of the small affinity Strep-tag or the Tev recognition sequence did not have any adverse effects on expression levels. Thus, by comparing expression levels and ligand binding characteristics of several CB2-fusion constructs harboring different combinations of tags, we confirm that the presence of both MBP and TrxA leads to the increased accumulation of functional receptor in bacterial plasma membranes.
We have demonstrated the feasibility of expressing CB2-fusions containing two affinity tags within its sequence in E. coli. The fusion was successfully cleaved and CB2 was purified in milligram quantities using a second step of affinity chromatography. A hexahistidine tag at the C-terminus of the CB2-fusion protein enabled purification of the full-length fusion to better than 80%, while the Strep-tag was instrumental for purification of CB2 to a final purity of 80-90% after cleaving the fusion. The purification of the recombinant protein with the StrepTag III gave higher yields than for CB2 with the StrepTag II. The constructs carrying two WSHPQFEK sequences, rather than one, had higher affinity to the StrepTactin resin, either because binding via two tags is stronger or because one of the tags was easier accessible. The binding capacity of the StrepTactin resin was at least 1.5-2 mg of the recombinant protein per ml of resin, despite the presence of detergents and of high concentrations of glycerol.
We improved efficiency of cleaving the CB2-fusion at the two Tev proteolytic sites in the presence of detergents by removing imidazole, lowering the concentrations of NaCl to 100 mM, and that of glycerol to 15% (v/v) by dialysis. The Tev protease tolerated concentrations of up to 1.0% CHAPS, 0.2% CHS, and 0.2% DM.
We had reported earlier that CB2-fusions are successfully expressed and purified from E. coli [3]. However, the presence of the large expression tags more than doubled the protein’s molecular weight which is detrimental for structural studies. Therefore, removal of expression tags and purification of CB2 to a polypeptide with the (almost) intact sequence of its counterpart in human cells are essential for subsequent NMR experiments.
The expression and purification procedures yielded CB2 of high purity for reconstitution into a lipid matrix. Functionality of the receptor was preserved, as demonstrated by ligand-binding studies on the reconstituted CB2. We estimate that the amount of functional receptor after reconstitution was in the range from 25 to 50%. At this point we are uncertain if this is due to imperfections of the reconstitution process or to a loss of protein function during purification. We are, however, aware of deficiencies in the reconstitution protocol that may result in protein loss. Investigations to improve reconstitution are ongoing. In addition to that, there are certain technical difficulties in performing ligand-binding assays with the receptor reconstituted into liposomes that may potentially result in underestimate of the number of active sites. Cannabinoid ligands are highly hydrophobic compounds, and in order to deliver them to the binding sites on the receptor a “carrier” protein (BSA) is used. The presence of multilammelar structures in the proteoliposome preparation may result in significant difficulties in delivering the ligand to all receptor binding sites. Another possible problem is that the pore size of the glass-fiber filters used in filter-binding assay may not be sufficiently small to retain all proteoliposomes, thus resulting in losses of receptor-ligand complexes during rapid filtration procedure. Pre-treatment with polyethyleneimine may somewhat increase the retention of the proteoliposomes on a filter, but can not solve this problem entirely.
The results of ligand-binding studies (on both E. coli membranes and on reconstituted CB2) and on activation of G proteins in an in vitro coupled assay suggest that the presence of a small affinity tag (StrepTag) at the N-terminus of CB2 does not interfere with the function of CB2, and thus is not likely to perturb the structure of the recombinant receptor. The size of the cleaved and purified CB2 (40 kDa) is within range of NMR structural studies.
Acknowledgments
The authors thank Dr. R. Grisshammer (NIDDK, NIH) who kindly provided plasmid constructs; Dr. D. Waugh (NCI, NIH-Frederick) for providing the strain for expression of Tev protease; Dr. J. Northup (NIDCD, NIH) for help with performing G-protein coupled assays; Dr. W. Teague (NIAAA, NIH) for critical reading of the manuscript. This work was supported by the Intramural Research Program of the NIAAA, NIH.
Footnotes
- GPCR
- G protein-coupled receptors
- MBP
- maltose-binding protein
- TrxA
- thioredoxin A
- CHS
- cholesteryl hemisuccinate Tris salt.
References
- [1].Cavasotto CN, Orry AJW, Abagyan RA. Structure-based identification of binding sites, native ligands and potential inhibitors for G-protein coupled receptors. Proteins: Struct. Funct. Genet. 2003;51:423–433. doi: 10.1002/prot.10362. [DOI] [PubMed] [Google Scholar]
- [2].Bertin B, Freissmuth M, Breyer RM, Schutz W, Strosberg AD, Marullo S. Functional expression of the human serotonin 5-HT1A Receptor in Escherichia coli - ligand-binding properties and interaction with recombinant G-protein alpha-subunits. J. Biol. Chem. 1992;267:8200–8206. [PubMed] [Google Scholar]
- [3].Yeliseev AA, Wong KK, Soubias O, Gawrisch K. Expression of human peripheral cannabinoid receptor for structural studies. Protein Sci. 2005;14:2638–2653. doi: 10.1110/ps.051550305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Krepkiy D, Wong KK, Gawrisch K, Yeliseev A. Bacterial expression of functional, biotinylated peripheral cannabinoid receptor CB2. Protein Expr. Purif. 2005;49:60–70. doi: 10.1016/j.pep.2006.03.002. [DOI] [PubMed] [Google Scholar]
- [5].Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Comparison of affinity tags for protein purification. Protein Expr. Purif. 2005;41:98–105. doi: 10.1016/j.pep.2005.01.019. [DOI] [PubMed] [Google Scholar]
- [6].Junttila MR, Saarinen S, Schmidt T, Kast J, Westermarck J. Single-step Strep-tag (R) purification for the isolation and identification of protein complexes from mammalian cells. Proteomics. 2005;5:1199–1203. doi: 10.1002/pmic.200400991. [DOI] [PubMed] [Google Scholar]
- [7].Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- [8].Arnau J, Lauritzen C, Petersen GE, Pedersen J. Current strategies for the use of affinity tag and tag removal for the purification of recombinant proteins. Protein Expr. Purif. 2006;48:1–13. doi: 10.1016/j.pep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- [9].Cass B, Pham PL, Kamen A, Durocher Y. Purification of recombinant proteins from mammalian cell culture using a generic double-affinity chromatography scheme. Protein Expr. Purif. 2005;40:77–85. doi: 10.1016/j.pep.2004.10.023. [DOI] [PubMed] [Google Scholar]
- [10].Mumby SM, Linder ME. Methods in Enzymology. Academic Press; 1994. Myristoylation of G-protein α subunits; pp. 254–268. [DOI] [PubMed] [Google Scholar]
- [11].Glass M, Northup JK. Agonist selective regulation of g proteins by cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- [12].Chen YH, Yang JT, Chau KH. Determination of helix and betaform of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- [13].Mohanty AK, Simmons CR, Wiener MC. Inhibition of tobacco etch virus protease activity by detergents. Protein Expr. Purif. 2003;27:109–114. doi: 10.1016/s1046-5928(02)00589-2. [DOI] [PubMed] [Google Scholar]