

Abstract
Previously, we have demonstrated that the renaturation of heat denatured firefly luciferase is dependent upon the activity of Hsp90 in rabbit reticulocyte lysate. Here we demonstrate that this assay may identify inhibitors that obstruct the chaperone activity of Hsp90 either by direct binding to its N-terminal or C-terminal nucleotide binding sites or by interference with the ability of the chaperone to switch conformations. The assay was adapted and optimized for high-throughput screening. Greater than 20,000 compounds were screened to demonstrate the feasibility of using this assay on a large scale. The assay was reproducible (avg. Z-factor = 0.62) and identified 120 compounds that inhibited luciferase renaturation by greater than 70% at a concentration of 12.5 μg/mL. IC50 values for twenty compounds with varying structures were determined for inhibition of luciferase refolding and in cell-based assays for Hsp90 inhibition. Several compounds had IC50 values < 10 μM and represent a number of new lead structures with the potential for further development and optimization as potent Hsp90 inhibitors.
Keywords: Hsp90, luciferase refolding, high-throughput screening, inhibitors
1. Introduction
Many newly synthesized polypeptides rely upon molecular chaperones to facilitate their maturation into biologically active proteins.1 Mutant, and unstable or misfolded proteins also rely upon chaperones to achieve their native conformations and retain their activity. Hsp90 facilitates protein folding via interactions with a set of ancillary proteins (co-chaperones) and the binding/hydrolysis of ATP.2, 3
Hsp90’s reaction cycle involves its switching between at least three conformations that are enforced by the binding and hydrolysis of ATP (reviewed in 2-4,5-8). Hsp90 contains two nucleotide binding sites: an ATP-binding Bergerat fold within its N-terminus 9, 10, and a nucleotide binding site in its C-terminal domain.5, 8, 11, 12 The C-terminal domain of Hsp90 is also responsible for its stable dimerization 13, and contains sequences that regulate ATP hydrolysis by the N-termini.14-17 The N-terminal, C-terminal and central domains have all been implicated in the binding of protein clients.13, 18, 19
The known Hsp90 inhibitors, radicicol (RDC), geldanamycin (GA) and novobiocin, are produced by the mycoparasitic fungus Humicola fuscoatra, and the soil actinomycetes species Streptomyces hygroscopicus and Streptomyces spheroids, respectively (Figure 1). While GA, its derivatives and radicicol bind to the N-terminal ATP binding domain of Hsp90, novobiocin interacts with the C-terminus (reviewed in 20). GA and novobiocin have distinct effects on the conformation of Hsp90.5, 7 GA appears to hold Hsp90 in an open conformation in which all regions of the protein are hypersensitive to proteolysis and thus remain exposed. In contrast, novobiocin binding to the C-terminus induces an Hsp90 conformation that has all but one of the major proteolysis sites protected from cleavage.5
Figure 1.
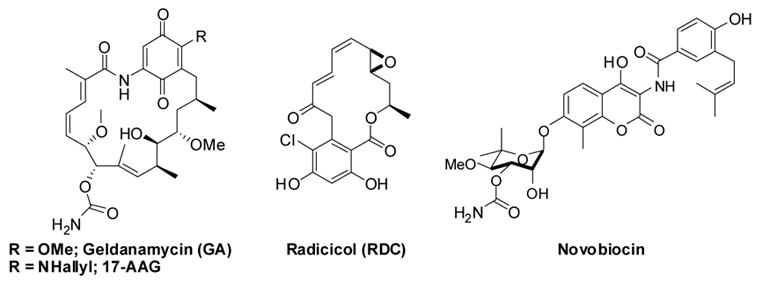
Natural Product Inhibitors of Hsp90.
In malignant cells, Hsp90 is overexpressed and is required to fold and maintain the activity of both native and mutated signal transduction proteins that are responsible for the uncontrolled proliferation of transformed cells.21 Although many molecular chaperones are required for cell viability, only Hsp90 has been shown to possess differential activity in cancerous vs. normal cells.22 More than 100 Hsp90-dependent client proteins have been identified and of these, 48 are directly related to oncogenesis 23 and are represented in all six hallmarks of cancer.22 Because so many oncogenic protein substrates are dependent upon the Hsp90 protein folding machinery for conformational maturation/activation, inhibitors of Hsp90 provide a combinatorial attack on multiple signaling pathways, such that its inhibition provides a mechanism for the simultaneous derailment of multiple signaling cascades. Consequently, Hsp90 has emerged as a promising target for the development of anti-tumor agents. Inhibitors of Hsp90 based on GA (e.g., 17-AAG), have entered more than 20 clinical trials for the treatment of cancer.24, 25
Additional observations suggest that Hsp90 inhibitors have the potential to prevent or reverse the progression of neurodegenerative diseases characterized by the accumulation of misfolded protein aggregates. Recent studies by Greengard and coworkers provided evidence that increased levels of Hsp90 and/or Hsp70 can refold phosphorylated tau aggregates and protect neuronal cells from Aβ-induced toxicity.26 Exposure of neuronal cells to GA increased expression of Hsps and promoted the rapid clearance of phosphorylated tau, providing protection from Aβ-induced toxicity in a dose-dependent manner.26 GA has also been shown to suppress huntingtin protein aggregation in cells by activating a specific heat shock response 27 and further evidence suggests that pharmacological inhibition of Hsp90 may represent a potential therapeutic strategy for the treatment of Parkinson’s disease.28
While initial results from clinical trials on the use of GA and its derivatives for the treatment of cancer appear promising, the low solubility and hepatotoxicity of these compounds limit their clinical potential.29 Because of the tremendous potential of Hsp90 inhibitors for the treatment of cancer, neurodegenerative disease and other disorders characterized by the accumulation of toxic protein aggregates, a means of identifying new, potent inhibitors of Hsp90 remains a high priority among investigators in this field. Herein we describe a robust, sensitive and simple high-throughput assay to identify Hsp90-inhibitors based on the Hsp90-dependent refolding of firefly luciferase in rabbit reticulocyte lysate.
2. Results
2.1 Optimization of the luciferase refolding assay for high-throughput screen (HTS)
We have previously demonstrated that refolding of heat denatured luciferase is Hsp90-dependent.30 Since reticulocyte lysate is a rich source of Hsp90 and contains the full complement of co-chaperones, which rapidly refold luciferase into its native form, it represents a model quasi-physiological system in which to screen for Hsp90 inhibitors. Furthermore, luciferase has a very high quantum yield from its catalytic reaction making it a very sensitive assay for Hsp90-dependent protein refolding. GA, an N-terminal Hsp90 inhibitor prevented refolding of luciferase at submicromolar concentrations (IC50 ~ 0.2 μM, Figure 2). In addition, novobiocin which binds to the nucleotide binding site in the C-terminal domain of Hsp90 also inhibits the refolding of luciferase in a concentration-dependent fashion (IC50 = 400 μM). The IC50 values for chlorobiocin and coumermycin A1 were approximately 60 and 40 μM, respectively. This result correlates well with the observation that chlorobiocin and coumermycin A1 are 5 to 10 times more active than novobiocin for inducing the degradation of Hsp90-dependent clients HER-2 and Raf-1 in cell culture.31, 32 Thus, this assay has the capability of identifying inhibitors of Hsp90 that bind to either the N- or C-terminus of Hsp90.
Figure 2. Effect of Hsp90 inhibition on the renaturation of firefly luciferase in rabbit reticulocyte lysate.
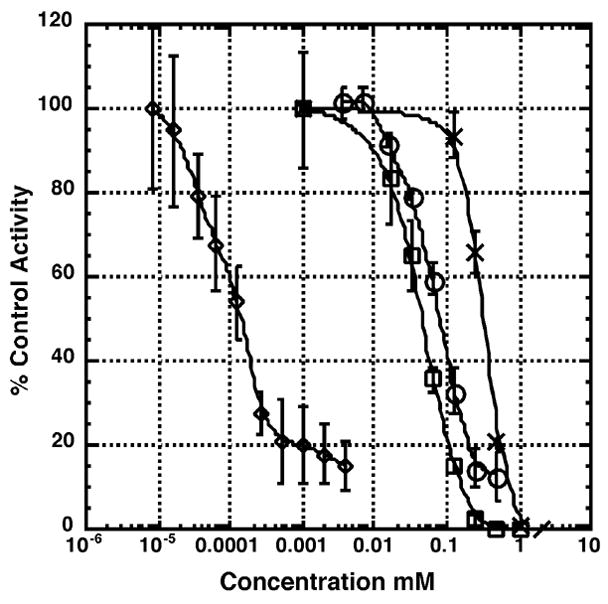
Thermally denatured luciferase was incubated in rabbit reticulocyte in the presence of DMSO (vehicle control) or the indicated concentrations of geldanamycin (◇), novobiocin (X), chlorobiocin (○), or coumermycin A1 (□). After 30 min, luciferase activity was measured as described under “Materials and Methods”. Reactions were carried out in triplicate and values expressed as percent of the DMSO control.
To adapt the luciferase refolding assay to an HTS format the optimization of a number of parameters was necessary. Since the hemoglobin present in reticulocyte lysate quenches light production, we determined the maximum dilution of reticulocyte lysate that gave optimal luciferase refolding activity and remained linear for a three hour incubation (Figure 3, lower panel). Next, we optimized the concentration of assay buffer additives (coenzyme A, Triton X-100, glycerol and DMSO) to minimize the decay of the luciferase glow reaction after its initial flash upon injection of the assay buffer. Conditions were selected under which the glow from the enzymatic reaction decayed less than 2% per min, such that the light production remained at 70–80% of the yield after the initial flash phase (Figure 3, upper panel). Under these conditions we observed less than a 2% decay in signal over a 1 min period, which is approximately the time required for the instrument to read 384 wells, with the first and last control wells being the second and 383rd wells read.
Figure 3. Properties of the optimized luciferase assay.
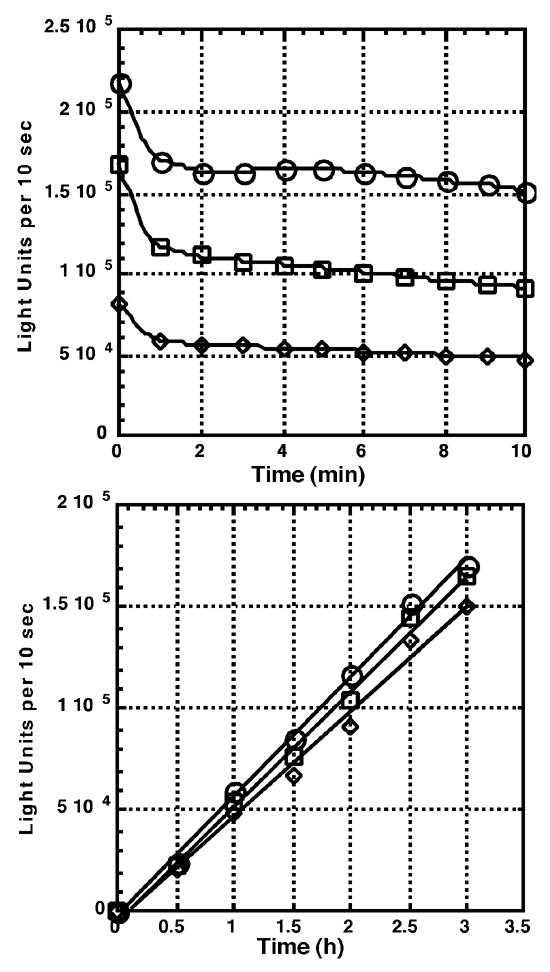
Luciferase renaturation was carried out under the conditions described under section 2.4 of the “Materials and Methods” but with a final volume of 100 μl. The amount of luciferase activity present in a 10 μl aliquot of the reaction was measured at the beginning of the assay (0 h) and at 0.5 h intervals for 3 h. Light units generated per 10 sec for each point of the time course were measured immediately after the injection of assay buffer (0 min), and every minute there after for 10 min. Lower Panel: light units generated per 10 sec for each 0.5 h sample integrated at 1 (○), 5 (□) and 10 (◇) min after injection of the assay buffer. Upper panel: light units generated per 10 sec integrated every 1 min after the injection of assay buffer for samples taken after 1 (◇), 2 (□), and 3 (○) h of incubation with the luciferase renaturation assay mix.
Sixty plates from The University of Kansas High Throughput Screening facility were assayed, representing over 20,000 compounds (Figure 4). The assay produced an average Z value of 0.62 (± 0.09 standard deviation; ± 0.009 standard error) for the sixty plates, indicating the results were statistically reliable. Compounds (186 hits) that inhibited luciferase activity by greater than 70% relative to the positive controls were re-screened to identify false positives (direct inhibitors of luciferase) and to obtain an initial estimate of the IC50 value for each compound. From the sixty plates assayed, 120 compounds were found to inhibit the Hsp90-dependent luciferase refolding activity of reticulocyte lysate at a concentration of 12.5 μg/ml or less. Of these, twenty compounds with varying chemical structures that displayed potent inhibition were selected for further characterization (Figure 5).
Figure 4.
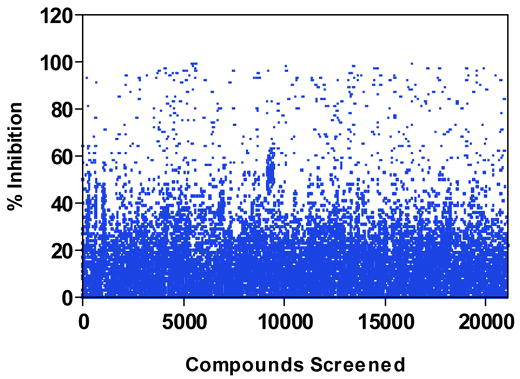
Scatter plot showing activities of the 21,240 screened compounds.
Figure 5.

Molecular Scaffolds Identified as Hsp90 Inhibitors via High-Throughput Screening.
2.2 Further Characterization of HTS hits
Lead compounds identified from the high-throughput screen were dissolved in DMSO and IC50 values for inhibition of firefly luciferase renaturation were determined (Table 1). A wide-range of IC50 values were obtained (0.12 to 120 μM) with most compounds inhibiting Hsp90-dependent refolding at the low micromolar level. The IC50s of compounds 11 and 18 were notably higher than expected from the initial screen. This discrepancy is likely due to a change in their chemical composition (possibly oxidation of the sulfur moiety), which occurred upon storage in 2.5% DMSO. We note that in additional screens done subsequent to the screen described here, two compounds were identified that were potent inhibitors of luciferase refolding, but were found to be inactive when the pure compounds were obtained from Chembridge (unpublished observations). The compounds were included in two additional independent screens, and the compounds were again identified as potent inhibitors of luciferase refolding, emphasizing the reproducibility of the assay. Thus, it is imperative that the hits be confirmed, and IC50 values be evaluated using pure fresh compounds.
Table 1.
| Entrya | Anti-proliferationb | Her2 ELISAc | Luc. Refoldingd | |
|---|---|---|---|---|
| MCF-7 | SkBr3 | SkBr3 | ||
| 1 | >100e | 42 ± 4 | 67 ± 8 | 13 ± 4 |
| 2 | 41 ± 12 | 55 ± 8 | 64 ± 15 | NI |
| 3 | >100 | >100 | >100 | NI |
| 4 | >100 | >100 | >100 | 15 ± 0.6 |
| 5 | >100 | >100 | >100 | 4.4 ± 1.5 |
| 6 | 4 ± 1 | 23 ± 0.5 | 33 ± 6 | 36 ± 2 |
| 7 | 44 ± 8 | 55 ± 1 | 61 ± 1 | 15 ± 0.6 |
| 8 | >100 | >100 | >100 | 55 ± 9 |
| 9 | 3.0 ± 0.8 | 2.4 ± 1.1 | 4.4 ± 2.1 | 16 ± 1 |
| 10 | >100 | >100 | >100 | 2.1 ± 0.2 |
| 11 | 25 ± 11 | 9.0 ± 0.3 | 8.9 ± 3.4 | 7.0 ± 0.4 |
| 12 | 19 ± 4 | 7.8 ± 0.3 | 4.7 ± 1.7 | 120 ± 8 |
| 13 | >100 | >100 | >100 | 22 ± 0.5 |
| 14 | 93 ± 7 | 76 ± 12 | 44 ± 4.5 | 23 ± 8 |
| 15 | 2.1 ± 0.8 | 15 ± 1 | 12.5 ± 3.6 | 1.9 ± 0.3 |
| 16 | 9.1 ± 4 | 15 ± 6.2 | 10.9 ± 0.4 | 6.7 ± 2 |
| 17 | >100 | 25 ± 3.6 | 21.3 ± 14 | 15 ± 12 |
| 18 | >100 | >100 | >100 | 190 ± 4 |
| 19 | 34 ± 13 | 39 ± 2.1 | 51.6 ± 7 | 4.5 ± 0.3 |
| 20 | >100 | >100 | >100 | 0.12 ± 0.6 |
Compounds correspond to scaffolds shown in Figure 5.
Anti-proliferation assays were performed as described in Section 5.8.
Her2 ELISA assays were performed as described in Section 5.9.
Luciferase refolding assays were performed as described in Section 5.7.
Values represent Mean IC50 ± SE of at least two separate experiments performed in triplicate (μM).
Subsequently, we measured the ability of the compounds to inhibit the growth rate of MCF-7 and SkBr3 cells, two distinct human breast cancer cell lines (Table 1). Seven of the compounds identified in the screen (3, 4, 5, 8, 10, 13, 18 and 20), did not demonstrate anti-proliferative activity against either cell line at concentrations up to 100 μM. Interestingly, 1 and 17 were inactive in MCF-7 cells, however, they displayed growth inhibition against SkBr3 cells (IC50 = 42 and 25 μM, respectively) suggesting a potential for the development of cell type selective Hsp90 inhibitors. Of the remaining compounds, the IC50 values for inhibition of luciferase refolding generally correlated well with the concentration of the compounds required to inhibit cell proliferation. Several compounds (6, 9, 12, 15 and 16) had IC50 values < 10 μM and represent potential lead scaffolds for optimization.
To further characterize the properties exhibited by the compounds identified by HTS, we determined their ability to induce degradation of the Hsp90-dependent protein Her-2 via an ELISA assay. The degradation of Her2 correlated strongly with the observed anti-proliferative effects of the compounds linking anti-proliferation activity to Her2 degradation and Hsp90 inhibition. Two compounds, 9 and 15, representing novel molecular scaffolds and shown to be Hsp90 inhibitors were further analyzed for their ability to induce degradation of Hsp90 clients, AKT, Raf-1, and Her2 (Figure 6). Treatment of MCF-7 cells with compound 9 and 15 induced the degradation of AKT, Raf-1 and Her2, an observation consistent with their ability to inhibit Hsp90 function.
Figure 6.
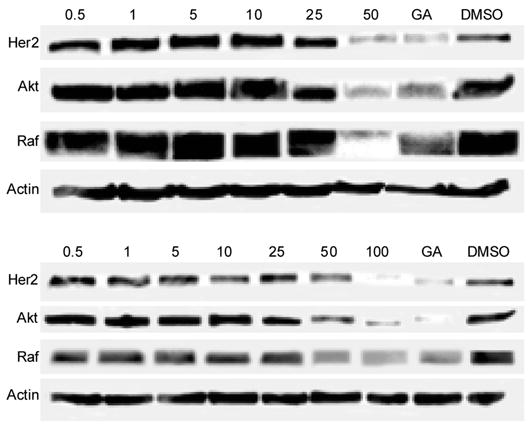
Induced degradation of Hsp90 client proteins via compounds 9 (upper panel) and 15 (lower panel). The compounds, at varying concentrations, (μM) were evaluated for their ability to downregulate Her2, Akt and Raf-1 as described in the methods section. Geldanamycin (GA, 500 nM) and DMSO were used as positive and negative controls, respectively.
3. Discussion
The data presented in this report demonstrate that the luciferase renaturation assay for Hsp90-dependent protein refolding is well-suited for high-throughput screening to identify novel molecular scaffolds as Hsp90 inhibitors. The assay is robust, sensitive and gives statistically significant Z-values. In addition, this assay has the capacity to detect compounds that inhibit Hsp90 by binding to either of the two nucleotide-binding sites present at the N- and C-termini of the protein, which represents the first Hsp90 assay developed for these properties. Twenty compounds with chemically diverse scaffolds were identified as valid HTS hits and chosen for further characterization in cellular based assays. Subsequent characterization of the compounds in these assays identified numerous scaffolds that maintained their inhibitory activity in cell culture. Compounds that did not induce Her2 degradation (via Her2 ELISA) or affect proliferation are potentially unable to diffuse through the lipid bilayer, may bind other cellular proteins/enzymes or may be rapidly metabolized in the cell, thus lowering the amount of available compound to bind Hsp90. In addition, the ability of these compounds to inhibit SkBr3 cell proliferation correlated directly with their ability to stimulate the turnover of Her2.
The compounds identified via this HTS provide a number of new lead structures with the potential for further development and optimization as potent Hsp90 inhibitors. Of the compounds identified, several (2, 5, 7, 15 and 19) consist of three aromatic rings separated by a variety of different linkers, structures very similar to the biaryl pyrazole series of N-terminal inhibitors identified by several other groups.33-35 In particular, the ~ 4-fold increase in potency displayed by 15 compared with the similar thiazole 7 provides important information for the further optimization of this heterocyclic core. Compounds 9 and 15 appear to represent unique chemical scaffolds not previously shown to inhibit Hsp90.
4. Conclusions
The first high-throughput assay has been developed to identify small molecule inhibitors of either the N- or C-terminus of Hsp90. A screen of ~20,000 diverse compounds identified 120 small molecules as potent Hsp90 inhibitors. Of these, twenty representative compounds were further characterized in cell culture and several novel scaffolds were identified as potent inhibitors of Hsp90 in vitro. However, it should be noted that utility of this assay extends beyond its ability to identify Hsp90 inhibitors, as the renaturation of luciferase in reticulocyte lysate has been shown to be dependent not only upon the action of Hsp90, but also on the actions of Hsp70 and the co-chaperones Hsp40, Hop and p23.30, 36-40 Thus, this assay has the potential to identify inhibitors for any of these proteins. Additional studies are currently underway to confirm the target of several of these compounds and to determine if they have a distinctive mode of binding compared to GA and novobiocin. The results from such studies will be reported in due course.
5. Experimental
5.1 Materials
Rabbit reticulocyte lysate (1:2, lysis of 1 volume of packed cells in 2 volumes of deionized water) was purchased from Green Hectares (Oregon, WI). Firefly luciferase (L-9506), luciferin, molecular biology grade acetylated bovine serum albumin, ATP, Coenzyme A and novobiocin were purchased from Sigma. Chlorobiocin and coumermycin A1 were provided by the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute, NIH. Stability buffer: 25 mM Tricine HCl (pH 7.8), 8 mM MgSO4, 0.1 mM EDTA, 10% glycerol, 1% Triton X-100, and 10 mg/mL acetylated BSA. Cold mix: 100 mM TrisHCl pH 7.7, 10 mM Mg(OAc)2, 375 mM KCl, 15 mM ATP, and 25 mM creatine phosphate. Creatine phosphokinase (CPK) was prepared as a 10 mg/mL stock in 50% glycerol. Experimental compounds representing hits from the high-throughput assay were purchased from Chembridge and ChemDiv.
5.2 Preparation of denatured luciferase
Luciferase was dissolved in stability buffer lacking glycerol and Triton X-100 at a concentration of 0.5 mg/mL. After the luciferase was completely dissolved, 10% glycerol, and 1% Triton X-100 were added, and the luciferase was denatured by heating at 41 °C for 10 min. The activity of luciferase was continuously monitored at one min intervals during this procedure to avoid its irreversible aggregation that occurs upon over-heating or prolonged incubation at elevated temperatures.41
5.3 Preparation of denatured luciferase reagent and reticulocyte lysate for 384 well assay
Luciferase reagent (10 mL) was prepared by mixing 8 mL of cold mix, 0.8 mL of 10 mg/mL CPK, 1.075 mL of deionized water; and 0.125 mL of denatured luciferase. Three volumes (1:2) of rabbit reticulocyte lysate were diluted with one volume of 20 mM Tris-HCl (pH 7.4) containing 75 mM KCl. These reagents were snap frozen in liquid nitrogen after preparation and stored at –80 °C. For long term storage the diluted reticulocyte lysate was stored in liquid nitrogen. After thawing, the reticulocyte lysate was centrifuged at 25,000g for 20 min to remove any particulates.
5.4 Luciferation Renaturation Assay
Black 384 well plates containing 20 μL of 2.5% DMSO in water in the first two 16 lane columns within the plate (negative and positive controls), and screening compounds (25 μg/mL) dissolved in 2.5% DMSO in the remaining 22 columns were utilized. Tris-buffered saline (10 μl; 20 mM Tris-HCl pH 7.5, 150 mM NaCl) containing 1% hemoglobin and 4% bovine serum albumin (TBS/HbBSA) was add to the first column as the negative control. Diluted reticulocyte lysate (10 μL) was dispensed to the remainder of the plate. The refolding reaction was initiated by the injection of 10 μL of the denatured luciferase mix to each well to give the following final concentrations: 20 mM Tris pH 7.7, 3 mM ATP, 5 mM creatine phosphate, 2.0 mM Mg(OAc)2, 75 mM KCl, 0.2 mg/mL CPK and 1.5 μg/mL denatured luciferase. Plates were centrifuged for one min to assure complete mixing of the components. After incubation at room temperature for three hours, luciferase activity was measured by injection of 40 μL of assay buffer (75 mM Tricine-HCl (pH 7.8), 24 mM MgSO4, 0.3 mM EDTA, 2 mM DTT, 313 μM D-luciferin, 640 μM coenzyme A, 0.66 mM ATP, 150 mM KCl, 10% (v/v) Triton X-100, 20% (v/v) glycerol and 3.5 % DMSO). Light emission from each well was read with a 0.1 sec integration time using a Perkin-Elmer EnVision 2101 plate reader 3 to 12 minutes after addition of the assay buffer depending on the number of plates being assayed.
5.5 Control assay to identify direct inhibitors of native luciferase
After the initial screen, duplicate plates were prepared containing compounds that were found to inhibit luciferase renaturation by ~70%. Serial dilutions (1:3) of each compound starting at a concentration of 25 μg/mL in 2.5% DMSO (20 μl) were dispensed into six adjacent wells. The effect of each compound on the activity of luciferase was determined by the addition of 10 μL of native lucifease mix (prepared as described above, but not denatured) in modified stability buffer (25 mM Tricine HCl pH 7.8, 8 mM MgSO4, 0.1 mM EDTA, 30% glycerol, 3% Triton X-100, and 10 mg/mL BSA with 0.8 ng of native lucifease). Luciferase activity was measured after addition of 30 μL of assay buffer containing 4 mg/mL BSA. Plates were read immediately after dispensing the assay buffer as described above.
5.6 Initial estimate of the IC50 of compounds ability to inhibit Hsp90-dependent luciferase renaturation
The second set of plates that were prepared with serial dilutions of each drug was assayed for inhibition of luciferase renaturation as described above in 5.4. The concentration-dependent inhibition of luciferase refolding was used to estimate the IC50 of each compound. The positive control (2.5% DMSO) and negative control (TBS/HbBSA) were used as the limits for 0% and 100% inhibition, respectively.
5.7 IC50 determination for inhibitory activity of compounds
Hsp90-dependent refolding of firefly luciferase in rabbit reticulocyte lysate was carried out as previously described with minor modifications.42 Denatured luciferase (0.5 mg/mL) was prepared as described above. Reactions were carried out in triplicate at room temperature in 96 well microtitre plates, and experiments were repeated at least twice. Each well contained 1 μL of DMSO or experimental compound dissolved in DMSO to give the desired final concentration: 4 μL of cold mix, 0.4 μl of 10 mg/mL creatine phosphokinase, 5.5 μL of deionized water, and 0.1 μL of denatured luciferase. The refolding reaction was initiated by the injection of 10 μL of reticulocyte lysate (1:2) previously diluted with an equal volume of 20 mM Tris-HCl (pH 7.4) containing 75 mM KCl. After a 30 min incubation at 25 °C, luciferase activity was measured upon injection of 100 μL of assay buffer (25 mM Tricine-HCl (pH 7.8), 8 mM MgSO4, 0.1 mM EDTA, 33 μM DTT, 470 μM D-luciferin, 240 μM coenzyme A, and 0.5 mM ATP) followed by a 10 sec integration time to measure relative light unit (RLU) production using a LMaxII (Molecular Devices) microplate reader.. The IC50 value was calculated as the concentration required to inhibit the recovery of luciferase activity by 50% relative to the DMSO control. The effect of experimental compounds on the activity of native luciferase was also assayed as a control.
5.8 Anti-proliferative effect of Hsp90 Inhibitors
MCF-7 cells were maintained in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium:Ham’s F-12 (Gibco) supplemented with non-essential amino acids, L-glutamine (2 mM), streptomycin (500 μg/mL), penicillin (100 units/mL) and 10% fetal bovine serum. SkBr3 cells were maintained in McCoy’s 5A media (Iwakata & Grace modification, Cellgro) with L-glutamine supplemented with streptomycin (500 μg/mL), penicillin (100 units/mL), and 10% fetal bovine serum. Cells were grown to confluence in a humidified atmosphere (37 °C, 5% CO2). Cells (2000/well, 100 μL) were seeded in 96-well plates and allowed to attach overnight (37 °C, 5% CO2). Compounds or GA at varying concentrations in DMSO (vehicle) were added (1% DMSO final concentration) and cells returned to the incubator (37 °C, 5% CO2) for 72 h. At 72 h, the number of viable cells was determined using an MTS/PMS cell proliferation kit (Promega) per the manufacturer’s instructions. Cells incubated in 1% DMSO were used as 100% proliferation and values were adjusted accordingly.
5.9 Her2 ELISA of Hsp90 Inhibitors
An ELISA to determine the ability of the HTS hits to degrade Hsp90 client proteins was performed as previously described with minor modifications.43 Briefly, SkBr3 cells were grown as described above and seeded (3000 cells/well) in 96-well plates and allowed to attach overnight (37°C, 5% CO2). Compounds, at varying concentrations, were added and the plates returned to the incubator for 24 h. After 24 h, the number of cells in each well were counted under the microscope to insure that there was no significant reduction in cell number compared to the DMSO control. Media was removed and the cells washed three times with ice-cold buffer (PBS with 1% Tween). Methanol (−20°C) was added and the plates placed at 4°C for 10 min to permeabilize and fix the cells. The plates were washed again with ice-cold buffer and incubated in blocking buffer (5% BSA in PBST) for 1 h at rt. The plates were incubated with a Her2 specific antibody (rabbit IgG; 1:500 dilution in blocking buffer) at 4°C overnight. The plates were washed again and incubated at room temperature for 2 h in the presence of an HRP-conjugated anti-rabbit IgG (1:1000 in blocking buffer). Plates were rinsed, chemiluminescent reagent was added and the plates immediately read on a luminometer (Molecular Devices).
5.10 Western Blot Analyses of HTS Hits
MCF-7 cells were seeded (1×106/plate) in culture dishes and allowed to attach overnight. Compound 9 and 15 were added at varying concentration and incubated (37°C, 5% CO2) for 36 h. Cells were harvested and analyzed for Hsp90 client protein degradation as described previously.44
Acknowledgments
The authors gratefully acknowledge support of this project by the NIH (R01 CA114393), and the Oklahoma Agricultural Experiment Station (Project No. 1975).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young JC, Barral JM, Ulrich Hartl F. Trends Biochem Sci. 2003;28:541. doi: 10.1016/j.tibs.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 2.Pratt WB, Toft DO. Exp Biol Med (Maywood) 2003;228:111. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 3.Pearl LH, Prodromou C. Adv Prot Chem. 2002;59:157. doi: 10.1016/s0065-3233(01)59005-1. [DOI] [PubMed] [Google Scholar]
- 4.Picard D. Cell Mol Life Sci. 2002;59:1640. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Biochemistry. 2004;43:8217. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]
- 6.Shao J, Irwin A, Hartson SD, Matts RL. Biochemistry. 2003;42:12577. doi: 10.1021/bi035138j. [DOI] [PubMed] [Google Scholar]
- 7.Hartson SD, Thulasiraman V, Huang W, Whitesell L, Matts RL. Biochemistry. 1999;38:3837. doi: 10.1021/bi983027s. [DOI] [PubMed] [Google Scholar]
- 8.Soti C, Racz A, Csermely P. J Biol Chem. 2001;277:7066. doi: 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- 9.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Cell. 1997;89:239. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 10.Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90:65. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 11.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J Biol Chem. 2000;275:37181. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 12.Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, Briand C, Makarov AA, Catelli MG, Peyrot V. J Biol Chem. 2002;277:12208. doi: 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- 13.Yamada S, Ono T, Mizuno A, Nemoto TK. Eur J Biochem. 2003;270:146. doi: 10.1046/j.1432-1033.2003.03375.x. [DOI] [PubMed] [Google Scholar]
- 14.Chadli A, Bouhouche I, Sullivan W, Stensgard B, McMahon N, Catelli MG, Toft DO. Proc Natl Acad Sci U S A. 2000;97:12524. doi: 10.1073/pnas.220430297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richter K, Muschler P, Hainzl O, Buchner J. J Biol Chem. 2001;276:33689. doi: 10.1074/jbc.M103832200. [DOI] [PubMed] [Google Scholar]
- 16.Owen BA, Sullivan WP, Felts SJ, Toft DO. J Biol Chem. 2002;277:7086. doi: 10.1074/jbc.M111450200. [DOI] [PubMed] [Google Scholar]
- 17.Wegele H, Muschler P, Bunck M, Reinstein J, Buchner J. J Biol Chem. 2003;278:39303. doi: 10.1074/jbc.M305751200. [DOI] [PubMed] [Google Scholar]
- 18.Scheibel T, Weikl T, Buchner J. Proc Natl Acad Sci U S A. 1998;95:1495. doi: 10.1073/pnas.95.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Mol Cell. 2003;11:647. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 20.Blagg BS, Kerr TD. Med Res Rev. 2005 doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 21.Pearl LH. Curr Opin Genet Dev. 2005;15:55. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. Nature. 2003;425:407. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Burrows F. J Mol Med. 2004;82:488. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 24.Banerji U, Judson I, Workman P. Curr Cancer Drug Targets. 2003;3:385. doi: 10.2174/1568009033481813. [DOI] [PubMed] [Google Scholar]
- 25.Sausville EA, Tomaszewski JE, Ivy P. Curr Cancer Drug Targets. 2003;3:377. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 26.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Proc Natl Acad Sci U S A. 2003;100:721. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Hum Mol Genet. 2001;10:1307. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 28.Shen HY, He JC, Wang Y, Huang QY, Chen JF. J Biol Chem. 2005;280:39962. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 29.Sharp S, Workman P. Advances in cancer research. 2006;95:323. doi: 10.1016/S0065-230X(06)95009-X. [DOI] [PubMed] [Google Scholar]
- 30.Thulasiraman V, Matts RL. Biochemistry. 1996;35:13443. doi: 10.1021/bi9615396. [DOI] [PubMed] [Google Scholar]
- 31.Marcu MG, Schulte TW, Neckers L. J Natl Cancer Inst. 2000;92:242. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 32.Burlison JA, Blagg BS. Organic letters. 2006;8:4855. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 33.McDonald E, Jones K, Brough PA, Drysdale MJ, Workman P. Current topics in medicinal chemistry. 2006;6:1193. doi: 10.2174/156802606777812086. [DOI] [PubMed] [Google Scholar]
- 34.Barril X, Beswick MC, Collier A, Drysdale MJ, Dymock BW, Fink A, Grant K, Howes R, Jordan AM, Massey A, Surgenor A, Wayne J, Workman P, Wright L. Bioorg Med Chem Lett. 2006;16:2543. doi: 10.1016/j.bmcl.2006.01.099. [DOI] [PubMed] [Google Scholar]
- 35.Brough PA, Barril X, Beswick M, Dymock BW, Drysdale MJ, Wright L, Grant K, Massey A, Surgenor A, Workman P. Bioorg Med Chem Lett. 2005;15:5197. doi: 10.1016/j.bmcl.2005.08.091. [DOI] [PubMed] [Google Scholar]
- 36.Grenert JP, Johnson BD, Toft DO. J Biol Chem. 1999;274:17525. doi: 10.1074/jbc.274.25.17525. [DOI] [PubMed] [Google Scholar]
- 37.Johnson BD, Chadli A, Felts SJ, Bouhouche I, Catelli MG, Toft DO. J Biol Chem. 2000;275:32499. doi: 10.1074/jbc.M005195200. [DOI] [PubMed] [Google Scholar]
- 38.Johnson BD, Schumacher RJ, Ross ED, Toft DO. J Biol Chem. 1998;273:3679. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- 39.Schumacher RJ, Hansen WJ, Freeman BC, Alnemri E, Litwack G, Toft DO. Biochemistry. 1996;35:14889. doi: 10.1021/bi961825h. [DOI] [PubMed] [Google Scholar]
- 40.Schumacher RJ, Hurst R, Sullivan WP, McMahon NJ, Toft DO, Matts RL. J Biol Chem. 1994;269:9493. [PubMed] [Google Scholar]
- 41.Thulasiraman V, Matts RL. In: Methods in Molecular Biology: Bioluminescent Protocols. LaRossa R, editor. Humana Press, Inc; Totowa, NJ: 1997. Chapt. 11. [Google Scholar]
- 42.Thulasiraman V, Xu Z, Uma S, Gu Y, Chen JJ, Matts RL. Eur J Biochem. 1998;255:552. doi: 10.1046/j.1432-1327.1998.2550552.x. [DOI] [PubMed] [Google Scholar]
- 43.Huezo H, Vilenchik M, Rosen N, Chiosis G. Chem Biol. 2003;10:629. doi: 10.1016/s1074-5521(03)00144-3. [DOI] [PubMed] [Google Scholar]
- 44.Avila C, Hadden MK, Ma Z, Kornilayev BA, Ye QZ, Blagg BS. Bioorg Med Chem Lett. 2006;16:3005. doi: 10.1016/j.bmcl.2006.02.063. [DOI] [PubMed] [Google Scholar]