

Abstract
Estrogen deficiency is one of the most frequent causes of osteoporosis in women and a possible cause of bone loss in men. But the mechanism involved remains largely unknown. Estrogen deficiency leads to an increase in the immune function, which culminates in an increased production of tumor necrosis factor (TNF) by activated T cells. TNF increases osteoclast formation and bone resorption both directly and by augmenting the sensitivity of maturing osteoclasts to the essential osteoclastogenic factor RANKL (the RANK ligand). Increased T cell production of TNF is induced by estrogen deficiency via a complex mechanism mediated by antigen presenting cells and the cytokines IFNγ, IL-7 and transforming growth factor-β. The experimental evidence that suggests that estrogen prevents bone loss by regulating T cell function and the interactions between immune cells and bone is reviewed here.
Estrogen deficiency is the most frequent cause of bone loss in humans. Bone loss results both from decreased ovarian production of sex steroids and the increase in follicle stimulating hormone (FSH) production induced by estrogen deficiency. FSH is now known to directly stimulate the production of tumor necrosis factor (TNF), a potent osteoclastogenic cytokine from bone marrow granulocytes and macrophages [1,2]. While FSH is likely to play a relevant role in the mechanism by which natural and surgical menopause lead to bone loss, this article will focus on the direct (FSH independent) mechanisms by which estrogen deficiency causes bone loss.
Estrogen deficiency is experimentally induced by ovariectomy (ovx). The main effect of ovx is a marked stimulation of bone resorption, which is caused primarily by increased osteoclast (OC) formation, but estrogen deficiency also increases OC lifespan due to reduced apoptosis [3]. The net bone loss caused by increased OC number and life span is limited in part by a compensatory augmentation of bone formation within each remodeling unit. This event is a consequence of stimulated osteoblastogenesis fueled by an expansion of the pool of early mesenchymal progenitors, and by increased commitment of such pluripotent precursors toward the osteoblastic lineage [4]. In spite of stimulated osteoblastogenesis, the net increase in bone formation is inadequate to compensate for enhanced bone resorption because of an augmentation in osteoblast (OB) apoptosis, a phenomenon also induced by estrogen deficiency [5]. An additional event triggered by estrogen withdrawal that limits the magnitude of the compensatory elevation in bone formation is the increased production of inflammatory cytokines such as IL-7 and TNF, which limit the functional activity of mature OBs [6,7]. Increased bone resorption, trabecular thinning and perforation, and a loss of connection between the remaining trabeculae are the dominant features of the initial phase of rapid bone loss that follows the onset of estrogen deficiency [8]. This acute phase is followed by a long-lasting period of slower bone loss where the dominant microarchitectural change is trabecular thinning. This phase is due, in part, to impaired osteoblastic activity secondary to increased OB apoptosis [9].
OC formation is induced by the cytokines 'receptor activator of NF-κB ligand' (RANKL) and macrophage colony stimulating factor (M-CSF). These factors are produced primarily by bone marrow (BM) stromal cells and OBs [10], and activated T cells [11]. RANKL binds to RANK, a receptor expressed on OCs and OC precursors, and to osteoprotegerin, a soluble decoy receptor produced by numerous hematopoietic cells. Thus, osteoprotegerin, by sequestering RANKL and preventing its binding to RANK, functions as an anti-osteoclastogenic cytokine. M-CSF induces the proliferation of OC precursors, the differentiation of more mature OCs, and increases the survival of mature OCs. RANKL promotes the differentiation of OC precursors from an early stage of maturation into fully mature multinucleated OCs and activates mature OCs.
Additional cytokines, either produced by or regulating T cells, are responsible for the upregulation of OC formation observed in a variety of conditions, such as inflammation, and estrogen deficiency. One such factor is TNF, a cytokine that enhances OC formation by upregulating the stromal cell production of RANKL and M-CSF [12] and by augmenting the responsiveness of OC precursors to RANKL [13]. Furthermore, TNF stimulates OC activity and inhibits osteoblastogenesis [12], thus further driving an imbalance between bone formation and bone resorption. The relevance of TNF has been demonstrated in multiple animal models. For example, ovx fails to induce bone loss in mice lacking TNF or its type 1 receptor [14]. Likewise, transgenic mice insensitive to TNF due to the overexpression of a soluble TNF receptor [15], and mice treated with the TNF inhibitor TNF binding protein [16] are protected from ovx-induced bone loss.
The presence of increased levels of TNF in the BM of ovx animals and in the conditioned media of peripheral blood cells of postmenopausal women is well documented [17]. However, the cells responsible for this phenomenon had not been conclusively identified. Recent studies on highly purified BM cells have revealed that ovx increases the production of TNF by T cells, but not by monocytes [13], and that earlier identification of TNF production by monocytes was likely due to T cell contamination of monocytes purified by adherence. Thus, the ovx-induced increase in TNF levels is likely to be due to T cell TNF production. Attesting to the relevance of T cells in estrogen deficiency induced bone loss in vivo, measurements of trabecular bone by peripheral quantitative computed tomography and μ-computed tomography revealed that athymic T cell deficient nude mice are completely protected against the trabecular bone loss induced by ovx [13,14,18]. T cells are key inducers of bone-wasting because ovx increases T cell TNF production to a level sufficient to augment RANKL-induced osteoclastogenesis [13]. T cell produced TNF may further augment bone loss by stimulating T cell RANKL production. The specific relevance of T cell TNF production in vivo was demonstrated by the finding that while reconstitution of nude recipient mice with T cells from wild-type mice restores the capacity of ovx to induce bone loss, reconstitution with T cells from TNF deficient mice does not [14].
Ovx upregulates T cell TNF production by increasing the number of TNF producing T cells without altering the amount of TNF produced by each T cell [14]. Ovx causes an expansion of the T cell pool in the BM by increasing T cell activation, a phenomenon that results in increased T cell proliferation and life span. Ovx increases T cell activation by enhancing antigen presentation by BM macrophages [19] and dendritic cells. This phenomenon is a result of the ability of estrogen deficiency to upregulate the expression of major histocompatibility complex II and the costimulatory molecule CD80. Although the mechanism of T cell activation elicited by estrogen deficiency is similar to that triggered by infections, the intensity of the events that follow estrogen withdrawal is significantly less severe and this process should be envisioned as a partial increase in T cell autoreactivity to self-peptides resulting in a modest expansion in the pool of effector CD4+ cells.
The physiological inducer of major histocompatibility complex II expression is IFNγ, an inflammatory cytokine produced by helper T cells. Ovx increases T cell production of IFNγ through complex mechanisms that remain largely unknown. The relevance of IFNγ is shown by the failure of mice lacking the IFNγ receptor (IFNγR-/-) and IFNγ (IFNγ-/- mice) to sustain bone loss in response to ovx [19,20].
A mechanism by which estrogen deficiency upregulates the production of IFNγ is through repression of transforming growth factor (TGF)β production [18]. The production of TGFβ by bone and BM cells is directly stimulated by estrogen through binding of the activated estrogen receptor on a estrogen responsive element (ERE) element on the TGFβ promoter [21]. Thus, estrogen withdrawal leads to increased production of TGFβ in the BM. TGFβ receptors are expressed in T cells and TGFβ signaling in T cells leads to powerful repression of T cell activation and of their production of IFNγ. Thus, TGFβ blocks T cell activation both directly and by decreasing antigen presentation via diminished production of IFNγ.
Studies with a transgenic mouse that expresses a dominant negative form of the TGFβ receptor in T cells have allowed the significance of the repressive effects of this cytokine on T cell function in the bone loss associated with estrogen deficiency to be established [18]. This strain, known as CD4dnTGFβRII, is severely osteopenic due to increased bone resorption. More importantly, mice with T cell-specific blockade of TGFβ signaling are completely insensitive to the bone sparing effect of estrogen [18]. This phenotype results from a failure of estrogen to repress IFNγ production which, in turn, leads to increased T cell activation and T cell TNF production.
As a proof of principle, a somatic gene therapy approach was used to induce the overexpression of TGFβ1 in ovx mice. These experiments confirmed that elevation of the systemic levels of TGFβ prevents the bone loss and the increase in bone turnover induced by ovx [18].
Another mechanism by which estrogen regulates IFNγ production is through IL-7, a potent lymphopoietic cytokine and inducer of bone destruction in vivo [22]. IL-7 is produced primarily by bone marrow stromal cells and OBs, but the mechanism by which ovx increases IL-7 production and the exact source of this cytokine remain to be determined. The BM levels of IL-7 are significantly elevated following ovx [6,23,24], and in vivo IL-7 blockade, using neutralizing antibodies, is effective in preventing ovx induced bone destruction [6] by suppressing T cell expansion and T cell IFNγ production [23]. Indeed, the elevated BM levels of IL-7 contribute to the expansion of the T cell population in peripheral lymphoid organs through several mechanisms. Firstly, IL-7 directly stimulates T cell proliferation by lowering tolerance to weak self antigens. Secondly, IL-7 increases antigen presentation by upregulating the production of IFNγ. Thirdly, IL-7 and TGFβ inversely regulate the production of each other [25,26]. The factors that regulate T cell function and contribute to ovx induced bone loss are shown in Figure 1.
Figure 1.
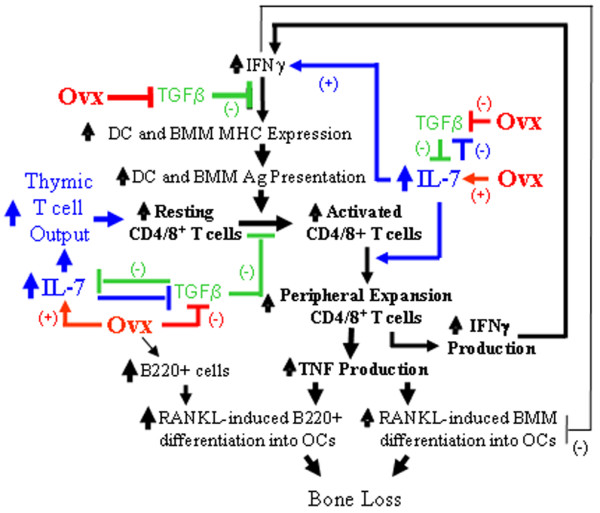
Factors that regulate T cell function and contribute to ovx induced bone loss. Ag, antigen; BMM, bone marrow macrophages; DC, dendritic cells; IFN, interferon; IL, interleukin; OC, osteoclast; ovx, ovariectomy; RANK, receptor activator of NF-κB; RANKL, RANKL ligand; TGF, transforming growth factor; TNF, tumor necrosis factor.
T cells differentiate in the thymus, an organ that undergoes progressive structural and functional declines with age, coinciding with increased circulating sex-steroid levels at puberty [27]. However, the thymus continues to generate new T cells even into old age. In fact, active lymphocytic thymic tissue has been documented in adults up to 107 years of age [28]. Under severe T cell depletion secondary to HIV infection, chemotherapy or BM transplant, an increase in thymic output (known as thymic rebound) becomes critical for long-term restoration of T cell homeostasis. For example, middle aged women treated with autologous BM transplants develop thymic hypertrophy and a resurgence of thymic T cell output that contributes to the restoration of a wide T cell repertoire [29], although the intensity of thymic rebound declines with age.
Restoration of thymic function after castration occurs in young [30] as well as in very old rodents [31]. Similarly, ovx increases the thymic export of naïve T cells [23]. Indeed, stimulated thymic T cell output accounts for approximately 50% of the increase in the number of T cells in the periphery, while the remaining 50% is due to enhanced peripheral expansion. Similarly, thymectomy decreases by approximately 50% the bone loss induced by ovx, thus demonstrating that the thymus plays a previously unrecognized causal effect in ovx-induced bone loss in mice. The remaining bone loss is a consequence of the peripheral expansion of naïve and memory T cells [23]. This finding, which awaits confirmation in humans, suggests that estrogen deficiency-induced thymic rebound may be responsible for the exaggerated bone loss in young women undergoing surgical menopause or for the rapid bone loss characteristic of women in their first five to seven years after natural menopause. Indeed, an age-related decrease in estrogen deficiency-induced thymic rebound could mitigate the stimulatory effects of sex steroid deprivation and explain why the rate of bone loss in postmenopausal women diminishes as aging progresses.
Conclusion
Remarkable progress has been made in elucidating the crosstalk between the immune system and bone and in uncovering the mechanism by which sex-steroids, infection and inflammation lead to bone loss by disregulating T lymphocyte function in animal models. If the findings in experimental animals are confirmed in humans, it will, perhaps, be appropriate to classify osteoporosis as an inflammatory, or even an autoimmune condition and certainly new therapeutic 'immune' targets will emerge.
Abbreviations
BM = bone marrow; ERE = estrogen responsive element; FSH = follicle stimulating hormone; IFN = interferon; IL = interleukin; M-CSF = macrophage colony stimulating factor; OB = osteoblast; OC = osteoclast; ovx = ovariectomy; RANK = receptor activator of NF-κB; RANKL = RANKL ligand; TGF = transforming growth factor; TNF, tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
References
- Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, Zaidi S, Zhu LL, Yaroslavskiy BB, Zhou H, et al. FSH directly regulates bone mass. Cell. 2006;125:247–260. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Sun L, Kumar TR, Blair HC, Zaidi M. Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc Natl Acad Sci USA. 2006;103:14925–14930. doi: 10.1073/pnas.0606805103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J Clin Invest. 1998;101:1942–1950. doi: 10.1172/JCI1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110:1643–1650. doi: 10.1172/JCI200215687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, Nanes MS. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141:3956–3964. doi: 10.1210/en.141.11.3956. [DOI] [PubMed] [Google Scholar]
- Eriksen EF, Hodgson SF, Eastell R, Cedel SL, O'Fallon WM, Riggs BL. Cancellous bone remodeling in type I (post-menopausal) osteoporosis: quantitative assessment of rates of formation, resorption, bone loss at tissue and cellular levels. J Bone Miner Res. 1990;5:311–319. doi: 10.1002/jbmr.5650050402. [DOI] [PubMed] [Google Scholar]
- Riggs BL, Parfitt AM. Drugs used to treat osteoporosis: the critical need for a uniform nomenclature based on their action on bone remodeling. J Bone Miner Res. 2005;20:177–184. doi: 10.1359/JBMR.041114. [DOI] [PubMed] [Google Scholar]
- Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050–5055. doi: 10.1210/en.142.12.5050. [DOI] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveirados-Santos AJ, Van G, Itie A, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Nanes MS. Tumor necrosis factor-alpha: molecular and cellular mechanisms in skeletal pathology. Gene. 2003;321:1–15. doi: 10.1016/S0378-1119(03)00841-2. [DOI] [PubMed] [Google Scholar]
- Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, Pacifici R. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest. 2000;106:1229–1237. doi: 10.1172/JCI11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, Pacifici R. Up-regulation of TNF-producing T cells in the bone marrow: A key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci USA. 2001;98:13960–13965. doi: 10.1073/pnas.251534698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammann P, Rizzoli R, Bonjour JP, Bourrin S, Meyer JM, Vassalli P, Garcia I. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J Clin Invest. 1997;99:1699–1703. doi: 10.1172/JCI119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimble R, Bain S, Pacifici R. The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. J Bone Miner Res. 1997;12:935–941. doi: 10.1359/jbmr.1997.12.6.935. [DOI] [PubMed] [Google Scholar]
- Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. 2006;116:1186–1194. doi: 10.1172/JCI28550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Qian WP, Dark K, Toraldo G, Lin AS, Guldberg RE, Flavell RA, Weitzmann MN, Pacifici R. Estrogen prevents bone loss through transforming growth factor beta signaling in T cells. Proc Natl Acad Sci USA. 2004;101:16618–16623. doi: 10.1073/pnas.0404888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, Sierra O, Pacifici R. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci USA. 2003;100:10405–10410. doi: 10.1073/pnas.1533207100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, Weitzmann MN, Pacifici R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang NN, Venugopalan M, Hardikar S, Glasebrook A. Identification of an estrogen response element activated by metabo-lites of 17b-estradiol and raloxifene. Science. 1996;273:1222–1225. doi: 10.1126/science.273.5279.1222. [DOI] [PubMed] [Google Scholar]
- Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, Suda T. Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci USA. 1997;94:9360–9365. doi: 10.1073/pnas.94.17.9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MR, Shepherd R, Leavey JK, Gao Y, Grassi F, Schnell FJ, Qian WP, Kersh GJ, Weitzmann MN, Pacifici R. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc Natl Acad Sci USA. 2005;102:16735–16740. doi: 10.1073/pnas.0505168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg MK, Svensson J, Venken K, Chavoshi T, Andersson N, Moverare Skrtic S, Isaksson O, Vanderschueren D, Carlsten H, Ohlsson C. Liver-derived IGF-I is permissive for ovariectomy-induced trabecular bone loss. Bone. 2006;38:85–92. doi: 10.1016/j.bone.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Huang M, Sharma S, Zhu LX, Keane MP, Luo J, Zhang L, Burdick MD, Lin YQ, Dohadwala M, Gardner B, et al. IL-7 inhibits fibrob-last TGF-beta production and signaling in pulmonary fibrosis. J Clin Invest. 2002;109:931–937. doi: 10.1172/JCI200214685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinett SM, Huang M, Dhanani S, Economou JS, Wang J, Lee P, Sharma S, Dougherty GJ, McBride WH. Down-regulation of murine fibrosarcoma transforming growth factor-beta 1 expression by interleukin 7. J Natl Cancer Inst. 1995;87:593–597. doi: 10.1093/jnci/87.8.593. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Sempowski GD, Wells AF, Hale LP. The human thymus during aging. Immunol Res. 2000;22:253–261. doi: 10.1385/IR:22:2-3:253. [DOI] [PubMed] [Google Scholar]
- Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. 1985;22:563–575. doi: 10.1111/j.1365-3083.1985.tb01916.x. [DOI] [PubMed] [Google Scholar]
- Hakim FT, Memon SA, Cepeda R, Jones EC, Chow CK, Kasten-Sportes C, Odom J, Vance BA, Christensen BL, Mackall CL, et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005;115:930–939. doi: 10.1172/JCI200522492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden AC, Moser MT, Tri SD, Mercader M, Kuntz SM, Dong H, Hurwitz AA, McKean DJ, Celis E, Leibovich BC, et al. Augmentation of T cell levels and responses induced by androgen deprivation. J Immunol. 2004;173:6098–6108. doi: 10.4049/jimmunol.173.10.6098. [DOI] [PubMed] [Google Scholar]
- Sutherland JS, Goldberg GL, Hammett MV, Uldrich AP, Berzins SP, Heng TS, Blazar BR, Millar JL, Malin MA, Chidgey AP, et al. Activation of thymic regeneration in mice and humans following androgen blockade. J Immunol. 2005;175:2741–2753. doi: 10.4049/jimmunol.175.4.2741. [DOI] [PubMed] [Google Scholar]