

Abstract
Idiopathic inflammatory myopathies (IIMs), comprising polymyositis, dermatomyositis, and inclusion-body myositis, are characterized by inflammatory cell infiltrates in skeletal muscle tissue, muscle weakness, and muscle fatigue. The cellular infiltrates often consist of T lymphocytes and macrophages but also, in some cases, B lymphocytes. Emerging data have led to improved phenotypic characterization of the inflammatory cells, including their effector molecules, in skeletal muscle, peripheral blood, and other organs that are frequently involved, such as skin and lungs. In this review we summarize the latest findings concerning the role of T lymphocytes, B lymphocytes, dendritic cells, and other antigen-presenting cells in the pathophysiology of IIMs.
Introduction
Idiopathic inflammatory myopathies (IIMs) are characterized by mononuclear inflammatory cell infiltrates in skeletal muscle tissue, by muscle weakness, and by muscle fatigue. They are often subclassified into three major groups on the basis of clinical and histopathological differences: polymyositis, dermatomyositis, and inclusion-body myositis. The cellular infiltrates in muscle tissue are mainly composed of T lymphocytes and macrophages but also, in some cases, B lymphocytes. This observation, together with frequently detected autoantibodies particularly in polymyositis and dermatomyositis, suggests that the inflammatory myopathies are immune-mediated; they are believed to be triggered by environmental factors in genetically susceptible individuals. The varying clinical features and the different predominating histopathological features such as localization and phenotypes of inflammatory infiltrates, or rimmed vacuoles as seen in inclusion-body myositis, suggest that there are different pathophysiological mechanisms leading to myositis. Despite these differences the inflammatory molecules produced in muscle tissue are highly similar in chronic inflammatory myopathies, suggesting that some molecular pathways are shared between the subsets of inflammatory myopathies.
In the inflammatory myopathies there are also signs of microvascular involvement. The involvement of microvessels was first reported in patients with dermatomyositis as capillary loss and recognized by the presence of the membrane attack complex (MAC) [1,2]. Later, activated capillaries with increased expression of adhesion molecules (intercellular cell-adhesion molecule-1 and/or vascular cell-adhesion molecule-1) and IL-1α were also seen in patients without skin rash, in polymyositis and inclusion-body myositis. Damage or activation of blood vessels could indicate that the microvessels are targets of the immune reaction in some subsets of patients with IIM.
It has long been recognized that the inflammatory cell infiltrates and muscle fiber damage are patchy and are sometimes not detected in muscle biopsies. This is a clinical problem in the diagnostic procedure. Moreover, the lack of correlation between the degree of inflammatory infiltrates and muscle weakness has led to a search for mechanisms other than immune-mediated muscle fiber damage that could cause muscle weakness. One such non-immune mechanism, endoplasmic reticulum stress, has been proposed, on the basis of observations both from human studies and from an animal model for myositis, the major histocompatibility complex (MHC) class I transgene [3]. These non-immune mechanisms have been addressed in a recent review paper [4].
New data are constantly emerging, leading to improved characterization of the phenotypes of the inflammatory cells and their effector molecules that are expressed in IIMs, not only in the major target organ, the skeletal muscle, but also in peripheral blood and in other organs that are frequently involved, such as skin and lungs. This increasing knowledge has a great potential to improve our understanding of the role of these inflammatory cells in disease mechanisms in IIMs. In this review we summarize the latest findings concerning the role of T lymphocytes, B lymphocytes, dendritic cells, and other antigen-presenting cells (APCs) in the pathophysiology of IIMs.
T lymphocytes
T lymphocyte function
T lymphocytes recognize antigens on APCs through the T-cell antigen receptor in a MHC-restricted fashion. Peptides from intracellular pathogens proliferating in the cytoplasm are carried to the cell surface by MHC class I molecules and presented to cytotoxic (CD8+) T lymphocytes, which once fully activated have the capacity to lyse infected target cells. In contrast, peptide antigens from pathogens in intracellular vesicles, and those derived from ingested extracellular bacteria and toxins, are carried to the cell surface by MHC class II molecules and presented to CD4+ T helper cells. These can then differentiate into effector cells, such as TH1, TH2, and TH17 cells [5]. Pathogens that accumulate inside macrophages and dendritic cells (DCs) tend to stimulate the differentiation of TH1 cells and the production of IgG antibodies by B lymphocytes. Conversely, extracellular antigens tend to stimulate the production of TH2, which can subsequently stimulate the production of IgA and IgE. TH17 is a recently described effector T lineage that has been suggested to contribute to chronic inflammatory settings. CD8+ T lymphocytes do not have as distinct sublineages and are cytotoxic cells working in a perforin/granzyme-dependent manner; interestingly, CD4+ lymphocytes can sometimes also display cytotoxic effector functions.
T lymphocytes in idiopathic inflammatory myopathies
Although prominent T lymphocyte infiltrates are not always found in muscle biopsies, two types of cellular infiltrate have been recognized in IIMs, one being endomysial inflammatory infiltrates consisting mainly of CD8+ T lymphocytes and macrophages invading non-necrotic muscle fibers expressing MHC class I antigens [6-8]. These are typically, but not exclusively, found in inclusion-body myositis and polymyositis. The other type of mononuclear cell infiltration is perivascular/perimysial and has become a characteristic of dermatomyositis; it consists predominantly of CD4+ T lymphocytes, occasionally together with B lymphocytes and macrophages [6,9]. The deposition of complement components is also mainly localized to the perivascular regions of muscular or cutaneous lesions. However, the 'classical' T lymphocyte picture in IIM is, as the authors say, much more complex and an oversimplification of reality [6,9]. Independent of T lymphocyte localization, their presence suggests an involvement of the adaptive immune system in these disorders (Figures 1 and 2).
Figure 1.
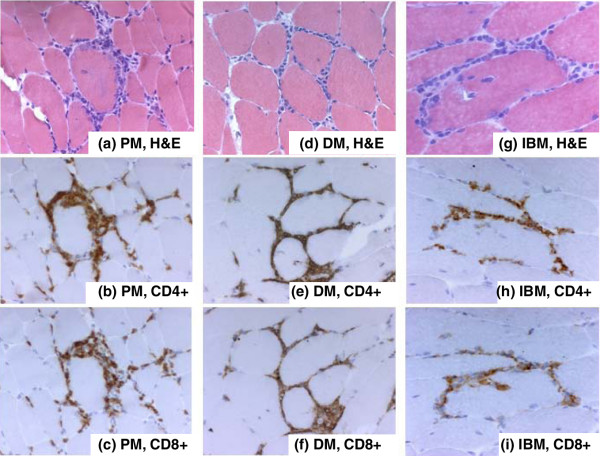
Endomysial localization of CD4+ and CD8+ T lymphocytes. Immunohistochemical staining of samples from patients with polymyositis (PM) (a-c), dermatomyositis (DM) (d-f), and inclusion-body myositis (IBM) (g-i). (a) Hematoxylin and eosin (H&E) staining to localize inflammatory cell infiltrates in a patient with polymyositis. (b) CD4+ T lymphocytes stained with a monoclonal SK3 mouse IgG1 antibody (Becton Dickinson, San Jose, CA, USA) in the same area as in (a) but further down in the biopsy. (c) CD8+ T lymphocytes stained with a monoclonal SK1 mouse IgG1 antibody (Becton Dickinson) in a consecutive section to that in (b). (d) H&E staining to localize inflammatory cell infiltrates in a patient with dermatomyositis. (e) CD4+ T lymphocytes stained with a monoclonal SK3 mouse IgG1 antibody in the same area as in (d) but further down in the biopsy. (f) CD8+ T lymphocytes stained with a monoclonal SK1 mouse IgG1 antibody in a consecutive section to that in (e). (g) H&E staining to localize inflammatory cell infiltrates in a patient with inclusion-body myositis. (h) CD4+ T lymphocytes stained with a monoclonal SK3 mouse IgG1 antibody in the same area as in (g) but further down in the biopsy. (i) CD8+ T lymphocytes stained with a monoclonal SK1 mouse IgG1 antibody in a consecutive section to that in (h). Original magnifications: ×250 (a-f) and ×312.5 (g-i).
Figure 2.
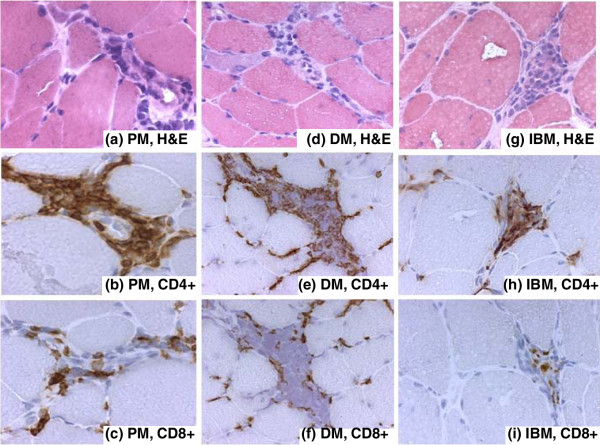
Perivascular localization of CD4+ and CD8+ T lymphocytes. Immunohistochemical staining of samples from patients with polymyositis (PM) (a-c), dermatomyositis (DM) (d-f), and inclusion-body myositis (IBM) (g-i). (a) Hematoxylin and eosin (H&E) staining to localize inflammatory cell infiltrates in a patient with polymyositis. (b) CD4+ T lymphocytes stained with a monoclonal SK3 mouse IgG1 antibody in the same area as in (a) but further down in the biopsy. (c) CD8+ T lymphocytes stained with a monoclonal SK1 mouse IgG1 antibody in a consecutive section to that in (b). (d) H&E staining to localize inflammatory cell infiltrates in a patient with dermatomyositis. (e) CD4+ T lymphocytes stained with a monoclonal SK3 mouse IgG1 antibody in the same area as in (d) but further down in the biopsy. (f) CD8+ T lymphocytes stained with a monoclonal SK1 mouse IgG1 antibody in a consecutive section to that in (e). (g) H&E staining to localize inflammatory cell infiltrates in a patient with inclusion-body myositis. (h) CD4+ T lymphocytes stained with a monoclonal SK3 mouse IgG1 antibody in the same area as in (g) but further down in the biopsy. (i) CD8+ T lymphocytes stained with a monoclonal SK1 mouse IgG1 antibody in a consecutive section to that in (h). Original magnifications: ×312.5 (a-c, g-i) and ×250 (d-f).
Cytotoxic CD8+ T lymphocytes can release three different cytotoxic proteins: perforin, granzyme, and granulysin. It is known from earlier studies that in polymyositis and inclusion-body myositis, CD8+ T lymphocytes and macrophages surrounding the non-necrotic muscle fibers expressing MHC class I antigen do express perforin. Perforin may cause a leak in the sarcolemmal surface through which granzymes could invade the sarcoplasm to initiate muscle fiber necrosis [10-12]. Recently, granulysin has also been demonstrated in both polymyositis and inclusion-body myositis [13]. The presence of granulysin-expressing CD8+ T lymphocytes tended to correlate with steroid resistance in polymyositis [13]. Interestingly, perforin/granzyme-expressing CD4+ T lymphocytes have also been demonstrated [10,14]. A schematic summary of the potential role of different immune cells in the context of chronic muscle inflammation is presented in Figure 3.
Figure 3.
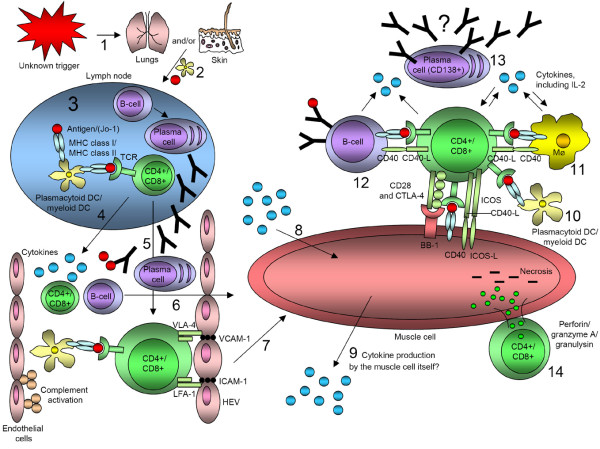
Hypothetical involvement of T lymphocytes, B lymphocytes, and dendritic cells (DCs) in idiopathic inflammatory myopathies. (1) An unknown trigger (for example viral infection or ultraviolet radiation) in the respiratory tract or through the skin leads to the cleavage of histidyl-tRNA synthetase by granzyme B through antiviral CD8+ T lymphocytes in the lungs. (2) Immature DCs carry receptors on its surface that recognize common features of many pathogens. When a DC takes up a pathogen in infected tissue it becomes activated and migrates to the lymph node. (3) Upon activation, the DC matures into a highly effective antigen-presenting cell (APC) and undergoes changes that enable it to activate pathogen-specific lymphocytes in the lymph node. T lymphocytes become activated and B lymphocytes, with active help from CD4+ T lymphocytes, proliferate and differentiate into plasma cells. (4) Activated DCs, T lymphocytes, and B lymphocytes could release cytokines into the bloodstream. (5) The activated T lymphocyte, on which the DC-MHC-antigen complex is bound, itself binds to specialized endothelial cells called high endothelial venules (HEV). For this purpose it uses the VLA-4 (very late activation antigen-4) and LFA-1 (lymphocyte function associated antigen-1) molecules on its surface to interact with adhesion molecules (vascular cell-adhesion molecule-1 (VCAM-1) and intercellular cell-adhesion molecule-1 (ICAM-1)) on HEVs, where they can penetrate into peripheral lymphoid tissues. (6,7) Naïve T lymphocytes and B lymphocytes that have not yet encountered their specific antigen circulate continuously from the blood into the peripheral lymphoid tissues. (8,9) Various cytokines from the bloodstream or produced locally could affect the muscle tissue or cell in many different ways. However, it is not clear whether the muscle cell itself could produce and release cytokines. (10–12) DCs, macrophages (Mϕ), and B lymphocytes can interact with T lymphocytes in various ways. T lymphocytes could possibly also bind to muscle cells through inducible co-stimulators (ICOS), CD40 ligand (CD40-L), CD28, and CTLA-4 (CD152) on T lymphocytes to ICOS ligand (ICOS-L), CD40, and BB-1 antigen on the muscle cell. In that fashion, the muscle cell would function as an APC. (13) Plasma cells (CD138+) can be found in the muscle tissue of certain subgroups of patients with idiopathic inflammatory myopathy, but whether these cells could produce autoantibodies locally is not yet known. (14) T lymphocytes have been shown to bind in close contact with muscle cells and to release perforin, granzyme A, and granulysin, which may cause necrosis of muscle tissue or cells.
The T lymphocyte repertoire in blood seems to differ between polymyositis and dermatomyositis [15]. In peripheral blood of active dermatomyositis, a decreased percentage of CD3+ and CD8+ T lymphocytes and decreased IFN-γ expression by CD4+ and CD8+ T lymphocytes but an increase in B lymphocytes and IL-4-producing CD4+ T lymphocyte frequencies were found. These features were not seen in the inactive form of the disease. In patients with polymyositis with relapsing disease, markedly perturbed T cell repertoires were seen. The expanded T lymphocytes clonally displayed a memory phenotype, expressed intracellular perforin, and responded to stimulation by IL-2, which indicates that they have the potential for reactivation under appropriate conditions [16]. This suggests that a continuous autoantigen-driven process might be prominent in this disease and that in patients with polymyositis a relapse would more probably be associated with the reactivation of clones, which are present at disease onset, rather than with the emergence of new ones. The concept that clonally expanded muscle-infiltrating CD8+ T lymphocytes could recirculate into the blood is true not only for patients with polymyositis but also for those with inclusion-body myositis [17]. Another possibility for the recirculation of T lymphocytes seen in polymyositis and inclusion-body myositis might be that the same antigen, for example a virus, could independently induce the same expansion in the blood and in the muscle tissue if the antigen is present at both sites.
In patients with IIMs, T lymphocytes are found not only in inflammatory muscle tissues or blood but also in lungs in patients with interstitial lung disease (ILD), which is a frequent manifestation in patients with polymyositis and dermatomyositis; this was found in up to 60 to 70% of such patients when sensitive techniques such as high-resolution computed tomography and pulmonary function tests were employed [18]. ILD seems to be less common in patients with inclusion-body myositis, although no reports are available in which newly diagnosed patients have been investigated for lung involvement with these techniques. In the lungs CD8+ T lymphocytes were distributed both in and around lymphoid follicles and in the walls of normal-appearing alveoli, whereas CD4+ T lymphocytes and B lymphocytes were seen in association with lymphoid follicles. CD4+ T lymphocytes have also been demonstrated in reconstructed thick alveolar walls [19]. The CD4+/CD8+ ratio in bronchoalveolar lavage fluid was low [20,21]; most of the CD8+ T lymphocytes were of HLA-DR+ [21] and CD25+[22] types, suggesting that they were activated. In a small study [23], a low CD4+/CD8+ ratio was seen in bronchoalveolar lavage fluids in all patients with radiographic signs of ILD early in the disease course of patients with polymyositis or dermatomyositis. The function of these T lymphocytes is not known. It can only be speculated that they might interact with antigens (from viruses or other sources) expressed in various pulmonary cells (Figure 3). It is interesting in this context that there is a reported tissue variation of histidyl-tRNA synthetases with a higher expression in normal lungs than in other organs, and that patients with autoantibodies against histidyl-tRNA synthetase – anti-Jo-1 antibodies – have ILD in close to 100% of cases.
Furthermore, a restricted accumulation of T lymphocytes expressing selected T-cell antigen receptor V-gene segments was recorded in skeletal muscle and lung but not in peripheral blood, which suggests a common target antigen in these organs [23]. Moreover, a role for histidyl-tRNA synthetase in the disease mechanism is supported by the observation that this antigen can serve as a chemokine for DCs and T lymphocytes when cleaved by certain proteases [24].
Even though the histopathological picture in polymyositis and dermatomyositis is often different, a similar clonal expansion of T lymphocytes is seen in bronchoalveolar lavage fluid [25]. In addition, T lymphocytes have been extracted from muscle tissue of these patients. The established T cell lines showed a variable proportion of CD4+ and CD8+ T lymphocytes, which did not correlate with diagnosis [26]. Examples of CD4+ and CD8+ T lymphocyte localization in polymyositis, dermatomyositis, and inclusion-body myositis are shown in Figures 1 and 2. As demonstrated here, a similar pattern with a mixture of cell populations and both endomysial and perimysial localization may be found despite the diagnosis of myositis.
When evaluating the presence of CD8+ T lymphocytes in biopsies it is important to consider which reagents have been used. Although the CD8 molecule is the classic lineage marker for cytotoxic T lymphocytes, this is only true if the CD8 molecule consists of an α and a β subunit (CD8αβ) and not if it is a CD8αα homodimer. The homodimer can sometimes be found on activated CD4+ T lymphocytes [27], especially in the gastrointestinal tract. Thus if the reagent used to detect CD8+ T lymphocytes is an antibody against the α subunit it could detect both classical CD8+ T lymphocytes but also activated CD4+ T lymphocytes. This information was not always available when immunophenotyping of lymphocytes in muscle tissue was presented. However, if the analysis includes the detection of a cytotoxic agent this caveat is partly avoided. A summary of selected T lymphocyte studies in IIMs is presented in Table 1.
Table 1.
Selected articles on immunohistochemical localization of inflammatory cells in idiopathic inflammatory myopathies
| Reference | Polymyositis | Dermatomyositis | Inclusion-body myositis |
| T lymphocytes | |||
| [9] | 53 patients. T lymphocytes were least abundant at perivascular and most abundant at endomysial sites. The CD4/CD8 ratio was highest at perivascular sites and lowest at endomysial sites. The proportion of T lymphocytes estimated to be Ia+, or activated, was nearly twice as high at endomysial sites as at perivascular sites | 31 patients. The proportion of T lymphocytes was lowest at perivascular sites and highest at endomysial sites. The CD4/CD8 ratio was highest at perivascular sites and lowest at endomysial sites. Many inflammatory cells display the Ia marker, but only one-sixth of perivascular lymphocytes and one-fifth of endomysial T lymphocytes was Ia+, or activated | 48 patients. T lymphocytes were least abundant at perivascular sites and most abundant at endomysial sites. CD4/CD8 ratio was highest at perivascular sites and lowest at endomysial sites. The proportion of T lymphocytes estimated to be Ia+, or activated, was nearly twice as high at endomysial sites as at perivascular sites |
| [8] | 7 patients. Endomysial infiltrates of T lymphocytes were predominant. Partly invaded non-necrotic cells were also seen; some of these fibers also expressed MHC class I | 21 patients. Severe muscle fiber necrosis, predominant perivascular T lymphocyte infiltrates, fibrosis and perifascicular atrophy. MHC class I were localized to perifascicular fibers | - |
| [11] | 5 patients. Perforin was co-localized with granzyme A and occasionally invaded non-necrotic muscle fibers. The percentage of perforin+ cells among the endomysial CD8+ cell population was 9.9% | 5 patients. There were very few perforin+ and granzyme A-positive cells in DM | 2 patients. Perforin was co-localized with granzyme A and occasionally invaded non-necrotic muscle fibers. The percentage of perforin+ cells among the endomysial CD8+ cell population was 12.5% |
| [10] | 5 patients. Co-localization of perforin with CD8, CD4, CD3, and CD2 was analyzed. The general distribution of inflammatory cells was endomysial. The overall CD4/CD8 ratio was 1.1 and the total number of CD8+ inflammatory cells in 9 random microscopic fields was 236. More than 90% of perforin+ cells were CD2+CD3+, and perforin was expressed both in non-invasive interstitial T lymphocytes and in autoinvasive CD8+ T lymphocytes. Of all perforin+ cells, 60% were CD8+ and 40% CD4+. Conversely, 75% of the CD8+ cells and 50% of the CD4+ cells were perforin+. 43% of the CD8+ T lymphocytes that contacted a muscle fiber expressed perforin vectorially toward the target muscle fiber | 4 patients. Co-localization of perforin with CD8, CD4, CD3, and CD2 was analyzed. The general distribution of inflammatory cells was perivascular and perimysial. The overall CD4/CD8 ratio was 1.2 and the total number of CD8+ inflammatory cells in 9 random microscopic fields was 167. More than 90% of perforin+ cells were CD2+CD3+, and 50% of all perforin+ cells were CD4+ and 50% were CD8+. Conversely, 80% of the CD4+ cells and 90% of the CD8+ cells were perforin+. Perforin was distributed randomly in the cytoplasm of the inflammatory T lymphocytes | - |
| [13] | 17 patients. The numbers of CD4, CD8, and CD56 cells per 1,000 muscle fibers at endomysial sites were 275 ± 140, 355 ± 150, and 25 ± 13 (means ± SD) in all cases. Granulysin was expressed in the cytoplasm of infiltrating cells, mostly CD4+ and CD8+ cells, only a few CD56+ cells at a higher level in endomysial sites than in perivascular sites. Notably, steroid-resistant patients with PM had a higher frequency of double-positive CD8 and granulysin at endomysial sites than steroid-resistant patients with IBM | - | 7 patients. The numbers of CD4, CD8, and CD56 cells per 1,000 muscle fibers at endomysial sites were 250 ± 120, 330 ± 200, and 31 ± 16 (means ± SD) in all cases. Granulysin was expressed in the cytoplasm of infiltrating cells, mostly CD4 and CD8+ cells, only a few CD56+ cells at a higher level at endomysial sites than at perivascular sites |
| [61] | 2 patients. CD8+ T lymphocytes were found to invade muscle fibers co-expressing ICOS-L and MHC class I. The endomysial ICOS-positive CD8+ and CD4+ T lymphocytes were seen at a similar frequency and ratio to those in patients with IBM | 2 patients. Most ICOS and ICOS-L positive cells were localized to the perimysial regions and connective tissue. ICOS-expressing CD4+ and CD8+ T lymphocytes were infrequent and present only in the perimysium and around blood vessels, but not among autoinvasive T lymphocytes | 20 patients. CD8+ T lymphocytes were found to invade ICOS-L and MHC class I co-expressing muscle fibers. ICOS-L expression was found on the surface of almost all fibers, including healthy myofibers away from inflammation. 1/20 or 2/20 autoinvasive CD8+ T lymphocytes were double- positive for ICOS and CD8. A similar percentage of CD4+ T lymphocytes, partly found in close contact with CD8+ T lymphocytes, also showed ICOS double immunoreactivity |
| B lymphocytes | |||
| [9] | 53 patients. B lymphocytes were most abundant at perivascular sites and least abundant at endomysial sites; however, only few B lymphocytes were expressed in comparison with patients with dermatomyositis | 31 patients. B lymphocytes were most abundant at perivascular sites and least abundant at endomysial sites | 48 patients. B lymphocytes were most abundant at perivascular sites and least abundant at endomysial sites; however, only few B lymphocytes were expressed compared with dermatomyositis |
| Polymyositis, dermatomyositis, and inclusion-body myositis, [34]; dermatomyositis, [49] | 3 patients. Occasional scattered B lymphocytes (CD19 and CD20) with a mean density of 2.8 cells/mm2 were seen. Plasma cells (CD138) showed a mean density of 11.0 cells/mm2. CD138+ cells were located predominantly in the endomysium. There were 3.9-fold more plasma cells than B lymphocytes | No data were given for 5 patients with dermatomyositis who were investigated for B lymphocytes (CD19 and CD20) and plasma cells (CD138). However, a comparison was done with another study in which it was shown that IBM muscle tissue had 0.84-fold B lymphocytes and 4.3-fold plasma cells | 16 patients. Occasional scattered B lymphocytes (CD19 and CD20) with a mean density of 4.2 cells/mm2 were seen. Plasma cells (CD138) showed a mean density of 17.2 cells/mm2. CD138+ cells were located predominantly in the endomysium. There were 4.1-fold more plasma cells than B lymphocytes |
| Dendritic cells | |||
| Polymyositis and inclusion- body myositis, [48]; dermatomyositis, [49] | 10 patients. Collections of myeloid DCs (BDCA-1+) were present in 90%, widely distributed across the section with additional high-density accumulations. High-density accumulations were typically endomysial and either surrounded myofibers and sometimes invaded apparently non-necrotic myofibers. They were also expressed in dense collections of cells (always some CD3+ cells in these dense collections) between myofibers. Hardly any plasmacytoid DCs (BDCA-2+) were seen | 14 patients. Plasmacytoid DCs (BDCA-2+) were seen in 10/14 patients and were mainly localized to endomysial and preivascular locations | 20 patients with IBM were investigated. Collections of myeloid DCs (BDCA-1+) were present in 17/20 patients widely distributed across the section with additional high-density accumulations. These high- density accumulations were typically endomysial and either surrounded myofibers and sometimes invaded apparently non-necrotic myofibers. They could also be expressed in dense collections of cells (always some CD3+ cells in these dense collections) between myofibers. Hardly any plasmacytoid DCs (BDCA-2+) were seen |
| Dermatomyositis, [49]; inclusion-body myositis, [48,49] | - | 14 patients. MxA was expressed in perifascicular areas. Eight patients showed MxA expression by myofibers in a preferentially perifascular distribution in both atrophic and normal-sized fibers but also in capillaries and other blood vessels in 13/14 patients. In regions of perifascicular atrophy all myofibers were positive | 20 patients. A quantitative study has been done between patients with IBM and DM who had not received immunosuppressive treatment. The numbers of myeloid and plasmacytoid DCs were similar in perivascular and perimysial areas in IBM. However, the endomysial locations of myeloid DC numbers were a mean of 3-fold higher than those of plasmacytoid DCs. In contrast, in DM there were 16-fold and 3-fold more plasmacytoid DCs than myeloid DCs in endomysial and perivascular sites, respectively |
| [47] | 6 patients. Immature DCs (CD1a) were mainly found in lymphocytic infiltrates in all patients. Mature DCs (DC-LAMP/CD83) were localized to perivascular infiltrates surrounding muscle fibers | 6 patients. Immature DCs (CD1a) were mainly found in lymphocytic infiltrates in all patients. Mature DCs (DC-LAMP/CD83) were localized to perivascular infiltrates surrounding muscle fibers | - |
The references were selected according to immunohistochemical data of localization of T lymphocytes, B lymphocytes, and dendritic cells (DCs) in skeletal muscle tissue of patients with polymyositis (PM), dermatomyositis (DM), and inclusion-body myositis (IBM). MHC, major histocompatibility complex; ICOS, inducible co-stimulator; ICOS-L, ICOS ligand.
A role of T lymphocytes in polymyositis and dermatomyositis is further supported by the clinical improvement after treatment with immunosuppressive drugs that are known to affect T lymphocytes. In contrast, patients with inclusion-body myositis rarely display improved muscle function after treatment with immunosuppressive drugs, which is why the role of T lymphocytes in disease mechanisms is more questionable in this form of myositis. Therapeutic agents that do not solely target T lymphocytes, but in all mentioned examples affect T lymphocyte populations, have been shown to be effective in polymyositis and dermatomyositis; these are methotrexate, cyclosporin A, tacrolimus, and anti-thymocyte globulin. Methotrexate is one of the most commonly used second-line immunosuppressive drugs given to patients with IIM. It is known to be well tolerated and effective in polymyositis and dermatomyositis, although no placebo-controlled trials have yet been performed [28]. There are also case series showing beneficial effects of tacrolimus and anti-thymocyte globulin [29,30]. In addition, topical cutaneous tacrolimus therapy has also effectively been applied to skin lesions in patients with dermatomyositis [31]. Although the clinical improvement with these drugs could indicate that T lymphocytes have a role in polymyositis and dermatomyositis, there are no data available to show that these therapies have effects on inflammatory cell infiltrates or molecular expression in muscle tissue that correlate with the clinical effects, which would strengthen such a hypothesis.
B lymphocytes
B lymphocyte function
B lymphocytes have a major role in the immunological pathogenesis of autoimmune diseases. Not only can their differentiated progeny, plasma cells [32,33], synthesize and secret large quantities of immunoglobulins, they could also regulate other cell types, secrete cytokines, and present antigens. B lymphocyte activation requires both binding of an antigen by surface immunoglobulin – the B-cell receptor – and an interaction with antigen-specific T lymphocytes (CD4+). CD4+ T lymphocytes then can induce vigorous B lymphocyte proliferation and direct the differentiation of the clonally expanded progeny of naïve B lymphocytes into either plasma cells or memory B lymphocytes. Both cytokines released by CD4+ T lymphocytes and somatic hypermutation of V-region genes can influence antibody isotype switching or the antigen-binding properties of the antibody, respectively, resulting in the production of antibodies of various isotypes that can be distributed to various body compartments.
B lymphocytes in idiopathic inflammatory myopathies
In 1984 and 1990, Arahata and Engel showed that B lymphocytes were more frequent in perivascular sites than in endomysial sites [6,9]. Furthermore, B lymphocytes were more common in muscle tissue from patients with dermatomyositis than from those with polymyositis or inclusion-body myositis. This observation was further supported by another study in which perivascular B lymphocytes were only found in patients with dermatomyositis but not in patients with polymyositis or inclusion-body myositis [8]. In contrast, a recent study by Greenberg and colleagues [34] demonstrated the presence of differentiated B lymphocytes in the form of CD138+ plasma cells predominantly in the endomysium of muscle tissue of patients with polymyositis and inclusion-body myositis (no patients with dermatomyositis were included in this study). A local antigen-driven response has been shown to be present in inflammatory myopathies. Clonal expansion, class-switched significant somatic mutation, and variation of local B lymphocyte and plasma cell maturation could occur within the skeletal muscle [35]. These characteristics are hallmarks of an antigen-driven response, which would mean that T lymphocytes are not the only cell that could drive an intramuscular antigen-specific autoimmunity in inflammatory myopathies.
In peripheral blood, activated B lymphocytes (RP105-negative B cells) were distinctly increased in patients with dermatomyositis in comparison with those with polymyositis [36]. In another study, peripheral blood mononuclear cells from patients with dermatomyositis, but not from those with with polymyositis, produced significant amounts of immunoglobulins in vitro [37].
Thus, earlier studies based on cellular expression in muscle tissue and, to some extent, the investigations of peripheral blood suggest that B lymphocytes are important in dermatomyositis but may have a less important role in polymyositis. However, more recent data using staining markers to detect plasma cells indicated a role of B lymphocytes in polymyositis and inclusion-body myositis as well [34]. Autoantibodies are present in 60 to 70% of patients with polymyositis or dermatomyositis, supporting a role for B lymphocytes in these disease entities, but less often in patients with inclusion-body myositis. Some autoantibodies found in patients with myositis, such as anti-Ro/SSA, anti-La/SSB, anti-Scl-70, and anti-U1 ribonucleo-protein, are also found in other autoimmune diseases, whereas others, especially anti-synthetase antibodies (for example anti-histidyl-tRNA synthetase or anti-Jo-1), anti-Mi2, and anti-signal-recognition particle are more specific for myositis. The myositis-specific autoantibodies are often associated with distinct clinical manifestations, such as the anti-synthetase syndrome characterized by myositis, ILD, arthritis, Raynaud's phenomenon, and skin changes called 'mechanic's hands'. The most frequent myositis-specific autoantibody is anti-Jo-1, which is more common in patients with polymyositis but may also be present in patients with dermatomyositis [38,39]. The newly discovered autoantibody anti-p155 seems to be associated more often with dermatomyositis and para-neoplastic dermatomyositis, and its frequency is similarly high in children (29%) and adults (21%) (with para-neoplasy the frequency is 75%) [40].
There are also reports on autoantibodies in patients with inclusion-body myositis. In one of these, an increased frequency of serum monoclonal antibodies reactive to a muscle constituent was demonstrated [41].
The functional role of plasma cells in muscle in polymyositis and inclusion-body myositis is not yet fully elucidated. B lymphocytes and plasma cells could, beside antibody secretion or their role as APCs, function as stimulatory cells for other immune cells. In dermatomyositis, CD4+ T lymphocytes could release IL-17 and IFN-γ after both polyclonal and oligoclonal activation to acquire a plasma cell-like morphology that is associated with a high secretory activity, similar to that of plasma cells secreting immunoglobulins [42]. The different role of immune cells in the context of muscle inflammation is presented in Figure 3.
More recent support for a role of B lymphocytes is the good clinical effect seen with B cell depletion. This was first reported in a pilot study that included patients with corticosteroid-refractory dermatomyositis who were treated with rituximab (Rituxan®), an anti-CD20 monoclonal antibody that is approved for treatment of some rheumatic diseases [43]. Later short-term beneficial effects with rituximab were also demonstrated in a case report of two patients with refractory polymyositis and one with dermatomyositis [44].
So far, only few studies have investigated and further addressed the roles of B lymphocytes and plasma cells in myositis. It is, however, certain that further research focusing on the functional role of B lymphocytes and specific autoantibodies in IIMs is warranted; this could provide pivotal insights in the disease mechanisms for a possible future specific targeted treatment strategy. A comparison of expression in B lymphocytes in IIMs is presented in Table 1.
Dendritic cells and other antigen-presenting cells
Dendritic cell function
Tissue DCs that have internalized particulate and soluble antigens at the site of inflammation are induced to mature, and an innate immune response is triggered. These cells are activated through receptors that signal the presence of pathogen components or cytokines. DCs are also responsible for T lymphocyte activation by migrating to the lymph node and providing both antigen presentation and co-stimulation. After maturation, DCs lose their ability to capture new antigens and thus there is a continuous flow of DCs from tissue to draining secondary lymphoid organs after challenge with antigen. In their mature activated form, DCs are the most potent APCs for naïve T lymphocytes. Both macrophages and B lymphocytes also have the capacity to function as APCs, but the ability of DCs to take up, process, and present a variety of pathogens and antigens makes them the most important activators of naïve T lymphocytes.
Dendritic cells and other antigen-presenting cells in idiopathic inflammatory myopathies
The presence of T lymphocytes in all subsets of IIMs indicates a permanent immune response that requires the presence of APCs. DCs are central to the development of innate and adaptive immune responses. Two main classes of DC have been classified, myeloid and plasmacytoid DCs. Myeloid DCs are potent APCs and have a function in the adaptive immune system. They are capable of capturing, processing, and presenting antigens and thereby stimulating lymphocytes to a specific immune response. In contrast, plasmacytoid DCs are important in the innate immune system and can produce large amounts of IFN-α and IFN-β, both of which have several functions including the stimulation of cells to produce specialized protein as a defense against pathogens. IFN-α can transform healthy monocytes into cells with properties of DCs; this was shown in sera from patients with active systemic lupus erythematosus [45]. IFN-α can also contribute to plasma cell differentiation and could therefore be important in the generation and sustenance of antibody responses [46].
Until recently, only few data were available on DCs in IIMs, but the results of some very recent studies have partly revealed important insights on this issue. Physiologically, DCs do not appear in normal muscle tissue, whereas the use of new markers for immature and mature DCs (CD1a and DC-LAMP/CD83, respectively) enabled the immature DCs to be detected in lymphocytic infiltrates in both polymyositis and dermatomyositis muscle tissue samples [47]. Local DCs have recently been demonstrated in all subsets of IIMs [48]: in muscle specimens from patients with inclusion-body myositis and patients with polymyositis, myeloid DCs were present in substantial numbers, frequently surrounding and sometimes invading intact myofibers. They were part of a dense collection of cells that also included T lymphocytes. In dermatomyositis muscles, an increased number of plasmacytoid DCs was found in comparison with the amount of myeloid DCs [48]. The importance of the plasmacytoid DCs in activating the type I interferon system in dermatomyositis was suggested by its association with the transcriptional response of the IFN-α and IFN-β inducible genes and by the fact that the interferon-induced MxA protein was expressed in muscle tissue of these patients [49]. MxA expression was demonstrated in capillaries and in perifascicular myofibers, which are characteristic sites of dermatomyositis pathology [49].
We have also found MxA expression in muscle tissue in dermatomyositis, but also in polymyositis and inclusion-body myositis, supporting a role of the type 1 interferon in all subsets of myositis. We therefore could not confirm a specific MxA staining pattern able to be used as a diagnostic tool that then could distinguish dermatomyositis from the other subsets of myositides, as suggested by Greenberg and colleagues [34]; this therefore needs to be pursued further. A comparison between expression patterns of DCs in IIMs is presented in Table 1.
However, the detailed function of both myeloid and plasmacytoid DCs in IIMs has still not been fully clarified.
Greenberg and colleagues [34] proposed two hypotheses: first, the presence of myeloid DCs invading the non-necrotic-seeming myofiber regions suggests the occurrence of active phagocytosis, endocytosis, pinocytosis, or receptor-mediated uptake of antigen, activities for which these cells are specialized; and second, that some of these cells are actively presenting antigen and activating T lymphocytes locally within muscle tissue rather than in the lymph node [48].
In addition, there is some evidence that chemokine receptors on DCs can promote autoimmune reactions. This is supported by the observation mentioned above that some autoantigens, such as aminoacyl tRNA synthetases, may exhibit chemotactic properties for activated monocytes, T lymphocytes, and immature DCs (not mature DCs). They could therefore have a capacity to attract inflammatory cells, including immature DCs, to infiltrate affected muscle cells. Taken together, these results suggest that antigens delivered to receptors on immature DCs are potent immunogens capable of breaking self-tolerance and able to induce autoimmune diseases [24,50].
As mentioned above, macrophages and B lymphocytes can also function as APCs. In addition to those, other cell types can acquire this function, although usually without reaching the same efficacy as a 'professional' APC [51]. Emerging evidence from experiments in vitro and in vivo suggests that muscle fibers could function as antigen-specific cells [52-54]. A schematic summary of the different roles of immune cells in the context of muscle inflammation is presented in Figure 3.
Whether muscle cells actually do function as APCs still remains uncertain. Muscle fibers are incapable of expressing the 'classical' co-stimulatory molecules B7-1 and B7-2, but some studies indicate that skeletal muscle cells and human myoblasts still have the possibility of acting as APCs with a cell-to-cell contact between BB-1 antigen on the one hand, and CD28 and CTLA-4 on CD4+ or CD8+ T lymphocytes on the other [53-55]. However, whether muscle cells that express BB-1 simultaneously could express MHC class I and/or class II antigens is still not clarified. Furthermore, cDNA for the BB-1 antigen has not been isolated. BB-1 is selectively induced by treatment of myoblasts with pro-inflammatory cytokines such as IFN-γ or tumor necrosis factor [55]. As both these cytokines have been detected in muscle tissue from patients with IIM [56-58], the muscle environment in the inflamed muscle could possibly provide the necessary signals on muscle fibers for an antigen presentation to T lymphocytes.
Once a naïve T lymphocyte has been activated, it expresses several proteins to sustain or modify the co-stimulatory signal for the clonal expansion and differentiation of T lymphocytes. Several co-stimulatory signal systems have been identified in muscle tissue from patients with IIM, such as CD40-CD40 ligand (CD40-L), inducible co-stimulator (ICOS)-ICOS ligand (ICOS-L), B7RP-1, B7h, and B7-H2. When a co-stimulatory receptor is bound by its ligand, an activating signal is transmitted to the T lymphocyte. This also activates the APC to express B7 molecules, which further stimulate T lymphocyte proliferation. Interestingly, muscle cells and myofibers have been shown to express some of these co-stimulatory signals, namely CD40-L and ICOS-L (ICOS-L both expressed and functional [59]) during pathological conditions [59,60]. Schmidt and colleagues [61] demonstrated that muscle fibers expressing MHC class I, taken from patients with inclusion-body myositis, caused an upregulation of ICOS-ICOS-L molecules in association with enhanced perforin expression by autoinvasive CD8+ T lymphocytes, which could exert a cytotoxic effect. This further supports the hypothesis that muscle fibers could work as APCs.
Conclusion
The molecular basis of IIMs in humans, as in many other autoimmune rheumatic diseases, is heterogeneous, involving several complex cellular components that probably contribute to differences in disease susceptibility, clinical and histopathological phenotype, and severity. Although this heterogeneity makes the study of the pathogenesis of IIMs extraordinarily complex, it might also provide distinct avenues for novel therapeutic interventions. Controlling the immune response is as complex as its launching. An essential feature of physiological immune response is its self-limitation, by which it is attenuated by several mechanisms. We have only just started to understand the orchestrated life of T lymphocytes, B lymphocytes, and DCs in IIMs, but there are still many unanswered questions about how this usually effective system can go awry and result in false immune-mediated reactions.
On the basis of detailed immunohistochemical studies on muscle biopsies, two major types of inflammatory infiltrate were observed: endomysial and perivascular/perimysial. In endomysial infiltrates there was a striking dominance of CD8+ T lymphocytes, which could even be the predominating infiltrating cell type, followed by macrophages and CD4+ T lymphocytes. These infiltrates often surrounded non-necrotic fibers and sometimes seemed to invade the fibers (Figure 1). This observation suggests an immune reaction that targets muscle fibers. The perivascular infiltrates, in contrast, were dominated by CD4+ T lymphocytes and macrophages, and sometimes the presence of B lymphocytes suggested an immune reaction that targets micro-vessels (Figure 2). A role for B lymphocytes as well as one for CD4+ T lymphocytes in the pathogenesis of IIMs is supported by frequently detected autoantibodies in polymyositis and dermatomyositis but less often in inclusion-body myositis. These autoantibodies are both non-specific (frequently also being found in other autoimmune disease) and myositis-specific [38,39]. A role for CD4+ T lymphocytes in the disease mechanism is further supported by the genetic association with HLA-DRB1*0301, DQA1*0501, and DQB1*0201, which was particularly seen for subgroups of patients with autoantibodies.
The endomysial infiltrates were reported to be characteristic features of polymyositis and inclusion-body myositis, whereas the perivascular infiltrates were associated with patients with dermatomyositis. However, there are cases with a less distinct localization of infiltrates or with combined endomysial and perivascular cellular infiltrates [3,6]. Moreover, in some cases the inflammatory cell infiltrates are diffusely spread in the tissue, whereas in other cases the infiltrates are very small or are not found at all. In addition, the perivascular changes may be seen in patients without a skin rash, whereas endomysial infiltrates are occasionally seen in cases with a skin rash.
Taken together, these results indicate that there may be two major pathways: one leading to cellular infiltrates with predominating endomysial localization, and another with a predominantly perivascular localization often with microvessel involvement and capillary loss. The latter is more often seen in patients with a skin rash and dermatomyositis, but there seems to be an overlap between clinical phenotypes, histopathology, and immunotypes. These observations suggest that there might be more than just one factor that determines the histopathological and clinical phenotypes, for example genes and environment.
During the past few years, the results of several advanced histopathological, molecular, functional, and medical studies have provided new data that could be of importance in understanding the immune mechanisms in IIMs. Taken together, they demonstrate the complexity of involvement of the immune system in these diseases and suggest that both the innate and adaptive immune systems are involved in dermatomyositis, polymyositis, and inclusion-body myositis. This complexity of T lymphocyte populations in muscle tissue in clinical subsets of myositis was demonstrated in the original observations of different cellular subsets in muscle tissue by Arahata and Engel [6,7,9] and is exemplified in Figures 1 and 2. These observations make it necessary to revise the 'old historical' hypothesis on the pathogenesis of IIMs. Recently a dispute over the most appropriate and accurate diagnostic criteria has erupted, including the importance of the histopathological picture and the localization of immune cells [62]. Other phenotypes such as autoantibody may also be important for the classification of disease subsets that will help to enhance our understanding of disease mechanisms and thereby improve the treatment and prognosis for these patients. A revision of classification criteria has recently been started in an international interdisciplinary collaboration that we believe will facilitate these efforts.
Abbreviations
APC = antigen-presenting cell; DC = dendritic cell; ICOS = inducible co-stimulator; ICOS-L = ICOS ligand; IFN = interferon; IIM = idiopathic inflammatory myopathy; IL = interleukin; ILD = interstitial lung disease; MAC = membrane attack complex; MHC = major histocompatibility complex.
Competing interests
The authors declare that they have no competing interests.
References
- Whitaker JN, Engel WK. Vascular deposits of immunoglobulin and complement in idiopathic inflammatory myopathy. N Engl J Med. 1972;286:333–338. doi: 10.1056/NEJM197202172860701. [DOI] [PubMed] [Google Scholar]
- Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol. 1990;27:343–356. doi: 10.1002/ana.410270402. [DOI] [PubMed] [Google Scholar]
- Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, Chang J, Dwivedi S, Mitsak M, Chen YW, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- Grundtman C, Lundberg IE. Pathogenesis of idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2006;8:188–195. doi: 10.1007/s11926-996-0024-4. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193–208. doi: 10.1002/ana.410160206. [DOI] [PubMed] [Google Scholar]
- Emslie-Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol. 1989;20:224–231. doi: 10.1016/0046-8177(89)90128-7. [DOI] [PubMed] [Google Scholar]
- Pedrol E, Grau JM, Casademont J, Cid MC, Masanes F, Fernandez-Sola J, Urbano-Marquez A. Idiopathic inflammatory myopathies. Immunohistochemical analysis of the major histocompatibility complex antigen expression, inflammatory infiltrate phenotype and activation cell markers. Clin Neuropathol. 1995;14:179–184. [PubMed] [Google Scholar]
- Engel AG, Arahata K, Emslie-Smith A. Immune effector mechanisms in inflammatory myopathies. Res Publ Assoc Res Nerv Ment Dis. 1990;68:141–157. [PubMed] [Google Scholar]
- Goebels N, Michaelis D, Engelhardt M, Huber S, Bender A, Pongratz D, Johnson MA, Wekerle H, Tschopp J, Jenne D. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996;97:2905–2910. doi: 10.1172/JCI118749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo S, Koga R, Goto K, Nakamura K, Arai M, Tamaki M, Sugita H, Nonaka I, Arahata K. Immunohistochemical analysis of perforin and granzyme A in inflammatory myopathies. Neuromuscul Disord. 1994;4:219–226. doi: 10.1016/0960-8966(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–982. doi: 10.1016/S0140-6736(03)14368-1. [DOI] [PubMed] [Google Scholar]
- Ikezoe K, Ohshima S, Osoegawa M, Tanaka M, Ogawa K, Nagata K, Kira JI. Expression of granulysin in polymyositis and inclusion-body myositis. J Neurol Neurosurg Psychiatry. 2006;77:1187–1190. doi: 10.1136/jnnp.2005.081810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach A, Kohler H, Rappl G, Abken H. Human CD4+ T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J Immunol. 2006;177:5668–5675. doi: 10.4049/jimmunol.177.8.5668. [DOI] [PubMed] [Google Scholar]
- Benveniste O, Cherin P, Maisonobe T, Merat R, Chosidow O, Mouthon L, Guillevin L, Flahault A, Burland MC, Klatzmann D, et al. Severe perturbations of the blood T cell repertoire in polymyositis, but not dermatomyositis patients. J Immunol. 2001;167:3521–3529. doi: 10.4049/jimmunol.167.6.3521. [DOI] [PubMed] [Google Scholar]
- Benveniste O, Herson S, Salomon B, Dimitri D, Trebeden-Negre H, Jean L, Bon-Durand V, Antonelli D, Klatzmann D, Boyer O. Long-term persistence of clonally expanded T cells in patients with polymyositis. Ann Neurol. 2004;56:867–872. doi: 10.1002/ana.20293. [DOI] [PubMed] [Google Scholar]
- Dimitri D, Benveniste O, Dubourg O, Maisonobe T, Eymard B, Amoura Z, Jean L, Tiev K, Piette JC, Klatzmann D, et al. Shared blood and muscle CD8+ T-cell expansions in inclusion body myositis. Brain. 2006;129:986–995. doi: 10.1093/brain/awl020. [DOI] [PubMed] [Google Scholar]
- Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63:297–301. doi: 10.1136/ard.2003.006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamadori I, Fujita J, Kajitani H, Bandoh S, Tokuda M, Ohtsuki Y, Yoshinouchi T, Okahara M, Yamaji Y, Tanimoto Y, et al. Lymphocyte subsets in lung tissues of interstitial pneumonia associated with untreated polymyositis/dermatomyositis. Rheumatol Int. 2001;21:89–93. doi: 10.1007/s00296-001-0146-y. [DOI] [PubMed] [Google Scholar]
- Sauty A, Rochat T, Schoch OD, Hamacher J, Kurt AM, Dayer JM, Nicod LP. Pulmonary fibrosis with predominant CD8 lymphocytic alveolitis and anti-Jo-1 antibodies. Eur Respir J. 1997;10:2907–2912. doi: 10.1183/09031936.97.10122907. [DOI] [PubMed] [Google Scholar]
- Kourakata H, Takada T, Suzuki E, Enomoto K, Saito I, Taguchi Y, Tsukada H, Nakano M, Arakawa M. Flowcytometric analysis of bronchoalveolar lavage fluid cells in polymyositis/dermatomyositis with interstitial pneumonia. Respirology. 1999;4:223–228. doi: 10.1046/j.1440-1843.1999.00179.x. [DOI] [PubMed] [Google Scholar]
- Kurasawa K, Nawata Y, Takabayashi K, Kumano K, Kita Y, Takiguchi Y, Kuriyama T, Sueishi M, Saito Y, Iwamoto I. Activation of pulmonary T cells in corticosteroid-resistant and -sensitive interstitial pneumonitis in dermatomyositis/polymyositis. Clin Exp Immunol. 2002;129:541–548. doi: 10.1046/j.1365-2249.2002.01933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund PWJ, Fathi M, Rasmussen E, Grunewald J, Tornling G, Lundberg IE. Restricted TCR BV gene usage in lungs and muscle tissue of patients with idiopathic inflammatory myopathies. Arthrithis Rheum. 2007;56:372–383. doi: 10.1002/art.22293. [DOI] [PubMed] [Google Scholar]
- Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, Casciola-Rosen L, Hartlein M, Kron M, Yang D, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196:781–791. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chino Y, Murata H, Goto D, Matsumoto I, Tsutsumi A, Sakamoto T, Ohtsuka M, Sekisawa K, Ito S, Sumida T. T cell receptor BV gene repertoire of lymphocytes in bronchoalveolar lavage fluid of polymyositis/dermatomyositis patients with interstitial pneumonitis. Int J Mol Med. 2006;17:101–109. [PubMed] [Google Scholar]
- Hohlfeld R, Engel AG. Coculture with autologous myotubes of cytotoxic T cells isolated from muscle in inflammatory myopathies. Ann Neurol. 1991;29:498–507. doi: 10.1002/ana.410290509. [DOI] [PubMed] [Google Scholar]
- O'Donovan MR, Jones DR, Robins RA, Li KF, Shim HK, Zheng Z, Arlett CF, Capulas E, Cole J. Co-cultivation of CD4+ and CD8+ human T-cells leads to the appearance of CD4 cells expressing CD8 through de novo synthesis of the CD8 α-subunit. Hum Immunol. 1999;60:1018–1027. doi: 10.1016/S0198-8859(99)00098-1. [DOI] [PubMed] [Google Scholar]
- Vencovsky J, Jarosova K, Machacek S, Studynkova J, Kafkova J, Bartunkova J, Nemcova D, Charvat F. Cyclosporine A versus methotrexate in the treatment of polymyositis and dermatomyositis. Scand J Rheumatol. 2000;29:95–102. doi: 10.1080/030097400750001897. [DOI] [PubMed] [Google Scholar]
- Ochi S, Nanki T, Takada K, Suzuki F, Komano Y, Kubota T, Miyasaka N. Favorable outcomes with tacrolimus in two patients with refractory interstitial lung disease associated with polymyositis/dermatomyositis. Clin Exp Rheumatol. 2005;23:707–710. [PubMed] [Google Scholar]
- Mitsui T, Kuroda Y, Kunishige M, Matsumoto T. Successful treatment with tacrolimus in a case of refractory dermatomyositis. Intern Med. 2005;44:1197–1199. doi: 10.2169/internalmedicine.44.1197. [DOI] [PubMed] [Google Scholar]
- Lampropoulos CE, DP DC. Topical tacrolimus treatment in a patient with dermatomyositis. Ann Rheum Dis. 2005;64:1376–1377. doi: 10.1136/ard.2004.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stashenko P, Nadler LM, Hardy R, Schlossman SF. Expression of cell surface markers after human B lymphocyte activation. Proc Natl Acad Sci USA. 1981;78:3848–3852. doi: 10.1073/pnas.78.6.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63:1424–1433. [PubMed] [Google Scholar]
- Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, Bregoli L, O'Connor KC, Amato AA. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65:1782–1787. doi: 10.1212/01.wnl.0000187124.92826.20. [DOI] [PubMed] [Google Scholar]
- Bradshaw EM, Orihuela A, McArdel SL, Salajegheh M, Amato AA, Hafler DA, Greenberg SA, O'Connor KC. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol. 2007;178:547–556. doi: 10.4049/jimmunol.178.1.547. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Koarada S, Tada Y, Ushiyama O, Morito F, Suzuki N, Ohta A, Horiuchi T, Miyake K, Nagasawa K. Difference in B cell activation between dermatomyositis and polymyositis: analysis of the expression of RP105 on peripheral blood B cells. Ann Rheum Dis. 2001;60:1137–1140. doi: 10.1136/ard.60.12.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge G, Faith A, Saunders C, Dubowitz V. A comparative study of in vitro proliferative responses to mitogens and immunoglobulin production in patients with inflammatory muscle disease. Clin Exp Rheumatol. 1989;7:27–33. [PubMed] [Google Scholar]
- Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, Miller FW. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70:360–374. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, Grondal G, Hietarinta M, Isenberg D, Kalden JR, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001;60:116–123. doi: 10.1136/ard.60.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O'Hanlon TP, Miller FW, Rider LG, Childhood Myositis Heterogeneity Study Group; International Myositis Collaborative Study Group A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682–3689. doi: 10.1002/art.22164. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Illa I, Gallardo E, Juarez C. Inclusion body myositis and paraproteinemia: incidence and immunopathologic correlations. Ann Neurol. 1997;41:100–104. doi: 10.1002/ana.410410116. [DOI] [PubMed] [Google Scholar]
- Page G, Sattler A, Kersten S, Thiel A, Radbruch A, Miossec P. Plasma cell-like morphology of Th1-cytokine-producing cells associated with the loss of CD3 expression. Am J Pathol. 2004;164:409–417. doi: 10.1016/S0002-9440(10)63131-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601–607. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- Noss EH, Hausner-Sypek DL, Weinblatt ME. Rituximab as therapy for refractory polymyositis and dermatomyositis. J Rheumatol. 2006;33:1021–1026. [PubMed] [Google Scholar]
- Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/S1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- Page G, Chevrel G, Miossec P. Anatomic localization of immature and mature dendritic cell subsets in dermatomyositis and polymyositis: interaction with chemokines and Th1 cytokine-producing cells. Arthritis Rheum. 2004;50:199–208. doi: 10.1002/art.11428. [DOI] [PubMed] [Google Scholar]
- Greenberg SA, Pinkus GS, Amato AA, Pinkus JL. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve. 2007;35:17–23. doi: 10.1002/mus.20649. [DOI] [PubMed] [Google Scholar]
- Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M, et al. Interferon-α/β-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–678. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- Oppenheim JJ, Yang D, Biragyn A, Howard OM, Plotz P. Chemokine receptors on dendritic cells promote autoimmune reactions. Arthritis Res. 2002;4(Suppl 3):S183–188. doi: 10.1186/ar574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Curnow SJ, Willcox N, Vincent A. Induction of primary immune responses by allogeneic human myoblasts: dissection of the cell types required for proliferation, IFNγ secretion and cytotoxicity. J Neuroimmunol. 1998;86:53–62. doi: 10.1016/S0165-5728(98)00013-7. [DOI] [PubMed] [Google Scholar]
- Murata K, Dalakas MC. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol. 1999;155:453–460. doi: 10.1016/s0002-9440(10)65141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata KY, Sugie K, Takamure M, Ueno S. Expression of the costimulatory molecule BB-1 and its receptors in patients with scleroderma-polymyositis overlap syndrome. J Neurol Sci. 2002;205:65–70. doi: 10.1016/S0022-510X(02)00309-X. [DOI] [PubMed] [Google Scholar]
- Behrens L, Kerschensteiner M, Misgeld T, Goebels N, Wekerle H, Hohlfeld R. Human muscle cells express a functional costimulatory molecule distinct from B7.1 (CD80) and B7.2 (CD86) in vitro and in inflammatory lesions. J Immunol. 1998;161:5943–5951. [PubMed] [Google Scholar]
- Lundberg I, Brengman JM, Engel AG. Analysis of cytokine expression in muscle in inflammatory myopathies, Duchenne dystrophy, and non-weak controls. J Neuroimmunol. 1995;63:9–16. doi: 10.1016/0165-5728(95)00122-0. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Molecular immunology and genetics of inflammatory muscle diseases. Arch Neurol. 1998;55:1509–1512. doi: 10.1001/archneur.55.12.1509. [DOI] [PubMed] [Google Scholar]
- Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40:865–874. doi: 10.1002/art.1780400514. [DOI] [PubMed] [Google Scholar]
- Wiendl H, Mitsdoerffer M, Schneider D, Melms A, Lochmuller H, Hohlfeld R, Weller M. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible co-stimulatory molecule, ICOSL: implications for the pathogenesis of inflammatory myopathies. Brain. 2003;126:1026–1035. doi: 10.1093/brain/awg017. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kawaguchi Y, Harigai M, Takagi K, Ohta S, Fukasawa C, Hara M, Kamatani N, et al. Increased CD40 expression on muscle cells of polymyositis and dermatomyositis: role of CD40-CD40 ligand interaction in IL-6, IL-8, IL-15, and monocyte chemoattractant protein-1 production. J Immunol. 2000;164:6593–6600. doi: 10.4049/jimmunol.164.12.6593. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Rakocevic G, Raju R, Dalakas MC. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain. 2004;127:1182–1190. doi: 10.1093/brain/awh148. [DOI] [PubMed] [Google Scholar]
- Hengstman GJ, van Engelen BG. Polymyositis, invasion of non-necrotic muscle fibres, and the art of repetition. BMJ. 2004;329:1464–1467. doi: 10.1136/bmj.329.7480.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]