

Abstract
The fibrosarcomatous transformation of dermatofibrosarcoma protuberans (DFSP) has been considered for some time to be associated with an adverse clinical outcome. However, the molecular and cellular mechanism underlying the tumor progression remains undetermined. As the chimeric gene, COL1A1-PDGFB, has been proposed to play an important role in the histogenesis of DFSP, we conducted a reverse transcription-polymerase chain reaction assay to ascertain whether the COL1A1-PDGFB fusion transcripts can be detected in both conventional DFSP and fibrosarcomatous components of DFSP with fibrosarcomatous areas (DFSP-FS), using a simple method of microdissection on sections of archival formalin-fixed, paraffin-embedded tumor specimens from six DFSP-FS cases. The COL1A1-PDGFB fusion transcripts could be detected in FS areas in five of the six cases, whereas conventional DFSP areas of all cases expressed the chimeric mRNA. A subsequent sequence analysis of the polymerase chain reaction products confirmed that the detected messages were derived from identical gene fusions in the two different components of each of the five cases. Our results verify that the COL1A1-PDGFB fusion transcripts are preserved in the FS areas of most DFSP-FSs. The expression of the fusion transcripts in both conventional DFSP and FS areas of DFSP-FS supports a common histogenesis of the two components.
Dermatofibrosarcoma protuberans (DFSP) is a fibrohistiocytic neoplasm of low-grade or intermediate malignancy that has a propensity for local recurrence but rarely metastasizes. 1 Histologically, several uncommon variants other than an ordinary form of DFSP have been recognized during the last two decades. These include Bednar tumor or pigmented DFSP, 2 myxoid DFSP, 3 granular cell DFSP, 4 DFSP with fibrosarcomatous areas (DFSP-FS), 5, 6 DFSP/DFSP-FS with foci of myoid/myofibroblastic differentiation, 7 DFSP with areas of giant cell fibroblastoma, 8 and an atrophic and plaque-like form of DFSP. 9 Among these variants, only the DFSP-FS variant has a prognostic significance, whereas most of the other variants simply represent morphological heterogeneity in DFSP with no distinct relationship to clinical behavior. The fibrosarcomatous transformation of DFSP has been considered a form of higher grade tumor progression and appears to be associated with an adverse clinical outcome. 6, 10, 11, 12
Cytogenetically, DFSP is characterized by a reciprocal translocation, t(17;22)(q22;q13), and a supernumerary ring chromosome derived from the translocation r(17;22). 13, 14, 15 Other rare forms, such as t(2;17) or t(9;22), have also been reported. 16, 17 Cloning of the r(17;22) or t(17;22) has revealed that the translocations result in a fusion of two genes, COL1A1 and PDGFB. 18 The COL1A1 gene is located in 17q21–22 and encodes the α1(I) chain of type I collagen. The PDGFB gene is located in 22q13 and encodes the B-chain of platelet-derived growth factor (PDGF) ligand. The fusion causes deregulation of the PDGFB gene by deleting its exon 1 and placing it under the direct control of the COL1A1 gene. This rearrangement leads to an unscheduled production of the growth factor, which seems to play an important role in the development of DFSP. In a more recent study, Greco et al provided direct evidence that the rearranged PDGFB gene could transform NIH3T3 cells in a model of an autocrine mechanism. 19
Detection of the COL1A1-PDGFB chimeric mRNA by a reverse transcription-polymerase chain reaction (RT-PCR) assay has also been proven to be a reliable and useful diagnostic marker for DFSP. 18, 20 Our recent approach showed that with some refinements, such as prolonged proteinase K treatment and selection of primers amplifying small target sequences, use of the molecular assay of RT-PCR was also feasible for detecting the fusion transcripts in archival formalin-fixed, paraffin-embedded tissues. 21
To our knowledge, the COL1A1-PDGFB fusion transcript has not been systematically analyzed in DFSP-FS, 21 and little is known about the status of the COL1A1-PDGFB fusion gene in the FS areas of DFSP-FS. Here, we examined six cases of DFSP-FS by RT-PCR to ascertain whether the chimeric gene is involved in the fibrosarcomatous transformation of DFSP. To separate the two different components of DFSP-FS, we conducted a method of simple microdissection using archival formalin-fixed, paraffin-embedded tissues.
Materials and Methods
Tumor Samples
Six cases of DFSP-FS were retrieved from the files of the Department of Pathology and Oncology, School of Medicine, University of Occupational and Environmental Health, and the Department of Pathology, University of Tokyo Hospital. One case had been included in a previous study. 21 The histological diagnosis of each DFSP-FS case was confirmed by two of the authors (T. I. and H. H.) by reviewing the original and/or reprepared sections stained with hematoxylin and eosin (H&E) according to the established diagnostic criteria. 6, 12 An immunohistochemical study was performed on deparaffinized sections using the standard immunoperoxidase avidin-biotin complex technique as described elsewhere, with a panel of antibodies against CD34 (QBEND 10, Immunotech, Marseilles, France), bcl-2 (DAKO, Kyoto, Japan), α-smooth muscle actin (1A4, Sigma, St. Louis, MO), muscle-specific actin (HHF35, Enzo, New York, NY), and desmin (D33, DAKO).
Microdissection
Microdissection was performed on sections cut from formalin-fixed, paraffin-embedded tissues essentially in the same way as previously described with some modifications. 22 Briefly, 12 sections were serially cut from a representative block at a thickness of 5 μm each with disposable microtome blades and collected on clean and sterile treated glass. To avoid cross-contamination of samples, a new microtome blade was used for each case. The first and the last sections of each case were stained with H&E for routine microscopic examination to verify adequacy of the tumor tissue in the specimen. The intermediate 10 consecutive sections were dewaxed and rehydrated through graded alcohol to DNase/RNase-free water. Sections were stained lightly with hematoxylin for easy identification. Different areas of DFSP and FS were cut out, respectively, from the sections using disposable sterile fine needles under a phase contrast microscope (STEMI SV8, Zeiss, Jena, Germany). Particular care was taken to avoid contamination of DFSP samples in the FS specimens and vice versa (Figure 1A and 1B) .
Figure 1.

Photomicrographs of dermatofibrosarcoma protuberans with fibrosarcomatous areas before (A) and after microdissection (B) in case 1. Asterisk represents a fibrosarcomatous area; triangle represents a conventional DFSP area (H&E; original magnification, ×2.5).
RT-PCR Assay
The microdissected tissue fragments were collected in an Eppendorf tube that contained 200 μl of lysis buffer and homogenized with a hand homogenizer. Then, 10 μl of proteinase K (100 mg/ml, Merck, Darmstadt, Germany) were added to the samples. After incubation at 55°C overnight, 1.0 ml of Trizol reagent (Gibco BRL, Gaithersburg, MD) was added to the samples, followed by 200 μl of chloroform. RNA extracts were treated with DNase I (Gibco BRL) for 15 minutes at 37°C. The DNase was then inactivated at 65°C for 10 minutes by adding 3 μl of 25 mmol/L ethylene diamine tetraacetate. Approximately 10 μl of the treated RNA extracts was reverse transcribed into cDNA using 1 μl of random primers (Gibco BRL). The integrity of RNA was evaluated by running a parallel PCR for a 127-bp fragment of the ubiquitously expressed porphobilinogen deaminase (PBGD) gene and a 247-bp fragment of the phosphoglycerate kinase (PGK) gene with the following primers: PBGD-S, 5′-TGTCTGGTAACGGCAATGCGGCTGCAAC-3′ and PBGD-A, 5′-TCAATGTTGCCACCACACTGTCCGTCT-3′, 23 and PGK-forward 5′-CAGTTTGGAGCTCCTGGAAG-3′ and PGK-reverse 5′-TGCAAATCCAGGGTGCAGTG-3′. 24 To amplify the COL1A1-PDGFB fusion transcripts, single-step PCR was carried out using a set of specific COL1A1 forward primers and a PDGFB reverse primer as previously described (Table 1) . 21 The primers corresponded to the α-helical domain of the COL1A1 gene (exon 6 through exon 49) and the exon 2 of PDGFB gene. The PCR profile consisted of 45 cycles of denaturation at 94°C for 1 minute, annealing at 66°C for 45 seconds, and elongation at 72°C for 50 seconds, followed by a final extension at 72°C for 10 minutes. In each PCR procedure, a no-reverse transcription control and a no-cDNA template control were included. The PCR products were visualized by ethidium bromide staining on a 2% agarose gel.
Table 1.
Sequences of the COL1A1 and PDGFB primers
| Primer | Nucleotide sequence |
|---|---|
| COL1A1(forward) | |
| Exon 5 | 5′-GCCGAGATGGCATCCCTGG-3′ |
| Exon 8 | 5′-TCAGGGTGCTCGAGGATTGC-3′ |
| Exon 11 | 5′-AAGGCTTCCAAGGTCCCCCTGG-3′ |
| Exon 15 | 5′-GGTGCTCGTGGAAATGATGG-3′ |
| Exon 17 | 5′-AAGGTCCCCAGGGTGTGCG-3′ |
| Exon 20 | 5′-AACCTGGTGCTCCTGGCAGC-3′ |
| Exon 23 | 5′-AAGCTGGTCGTCCCGGTGAAGC-3′ |
| Exon 26 | 5′-AAGGCTGGAGAGCGAGGTGTTC-3′ |
| Exon 29 | 5′-TGCTGGCAAAGATGGAGAGG-3′ |
| Exon 32 | 5′-TGAACGTGGTGCAGCTGGTCTTC-3′ |
| Exon 35 | 5′-TCCCACTGGAGCTCGTGG-3′ |
| Exon 38 | 5′-TGCTCCTGGAGCCAAAGGTGC-3′ |
| Exon 40 | 5′-TGCTGGCGAGAAAGGATCCCCTG-3′ |
| Exon 43 | 5′-TGGCAAGAGTGGTGATCGTGG-3′ |
| Exon 46 | 5′-TGGCTTCTCTGGCCTCCAGGG-3′ |
| Exon 49 | 5′-ACCTCAAGAGAAGGCTCACGATGG-3′ |
| PDGFB(reverse) | 5′-ATCAAAGGAGCGGATCGAGTGGTC-3′ |
Sequence Analysis
To analyze the type of breakpoints, 2 μl of the PCR products were cloned into a pCR2.1 vector by a TA cloning kit (Invitrogen, Carlsbad, CA). The plasmid DNA was then transfected into DH-5α (Toyobo, Osaka, Japan) and incubated overnight. White clones were picked and screened by PCR to confirm the presence of inserts. Sequence analyses were performed using an automated sequencing system, ALFexpress DNA sequencer (Pharmacia Biotech, Uppsala, Sweden). The results were analyzed by a computer using GeneWorks Release 2.5.1 software (Oxford Molecular Group Inc., Campbell, CA) and the data of the GenBank database.
Results
Histologically, the conventional DFSP areas of DFSP-FS were composed of a monotonous proliferation of small to medium-sized spindle cells arranged in a distinct storiform or cartwheel pattern, frequently showing honeycomb-like infiltration into the adjacent adipose tissue (Figure 2A) . The fibrosarcomatous areas consisted of a cellular proliferation of plump atypical spindle cells with hyperchromatic nuclei arranged in variably interlacing fascicles, often displaying a herringbone appearance (Figure 2B) . FS areas showed a significantly higher mitotic rate with an average of 14.7 mitoses per 10 high-power fields (HPFs), whereas in conventional DFSP areas, the mitotic rate was 0.7 mitoses per 10 HPFs. The transition between the two areas was generally distinct in the present cases. FS areas in three cases also contained foci of myoid/myofibroblastic differentiation, as previously described. 7, 25 Immunohistochemically, tumor cells in conventional DFSP areas were diffusely positive for CD34 in all of the cases, whereas FS areas had CD34-positive tumor cells in two of the six cases, one of which showed only focal and weak staining. Both components were negative for bcl-2, α-smooth muscle actin, muscle-specific actin, and desmin in all of the cases, although scattered myoid nodules in three cases showed an actin-positive, desmin-negative immunoreactivity. 25
Figure 2.
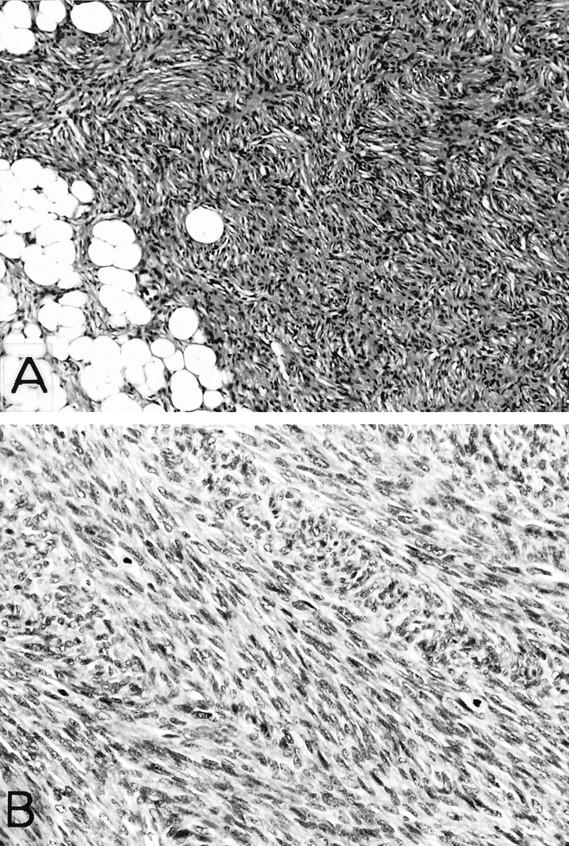
Histological appearance of DFSP-FS. A: Distinct storiform pattern in a conventional DFSP area with characteristic honeycomb infiltration of the adjacent fat tissue (case 4; H&E; original magnification, ×25). B: Herringbone structure in a fibrosarcomatous area of DFSP-FS (case 4; H&E; original magnification, ×50).
The COL1A1-PDGFB fusion transcripts were detected in both of the DFSP and FS areas in five of the six DFSP-FS cases (cases 1, 2, 4, 5, and 6; 83.3%), and only in the DFSP areas in one case (case 3). The amplification assay yielded distinctive fragments of 105, 106, and 258 bp in length (Figure 3) . Nucleotide sequence analysis of the PCR products showed that the exon 2 of the PDGFB gene was fused in-frame to variable regions of the COL1A1 gene. In this study, the breakpoints of the COL1A1 gene were located in exon 25 (cases 5 and 6), exon 32 (cases 1, 3, and 4), and exon 46 (case 2) (Figure 4) . In each of the five cases, the breakpoint of the fusion gene in the FS areas was the same as that in the conventional DFSP areas. In case 3, the repeated RT-PCR assay failed to detect the fusion transcript in the sample of the pure FS areas, although the transcript was detectable in the specimen of ordinary DFSP. 21 The PBGD gene transcripts could be amplified in 11 samples (91.7%), whereas the PGK gene transcripts were detected in only 4 samples (33.3%). The details of the results are summarized in Table 2 .
Figure 3.

Expression of COL1A1-PDGFB fusion transcripts (A) and PBGD gene mRNA (housekeeping gene; B) in each sample of DFSP-FS. The number of the lane is identical to the case number in Table 2 . The double bands in lane 5D were likely to be formed by DNA conformation changes, because the DNA sequences of the PCR products in both upper and lower bands in lane 5D were confirmed to be identical by the sequence analysis. M, 100-bp DNA ladder; D, conventional DFSP areas; F, fibrosarcomatous areas; N, negative control.
Figure 4.
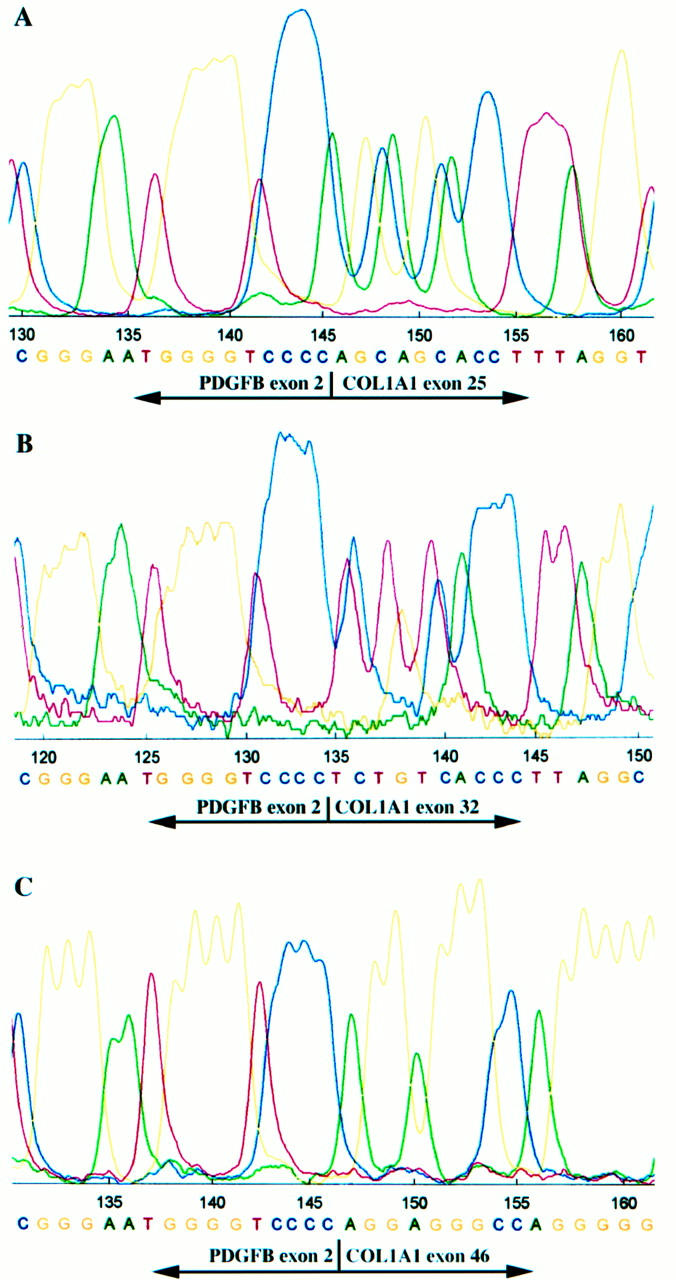
Nucleotide sequence analysis of PCR products showing three types of COL1A1-PDGFB fusion transcripts composed of junctions between PDGFB exon 2 and COL1A1 exon 25 (A), exon 32 (B), or exon 46 (C).
Table 2.
Clinicopathological and Molecular Characteristics of DFSP-FS
| Case no. | Age (yr)/sex | Site | Histology | CD34 | PBGD | PGK | COL1A1 - PDGFB |
|---|---|---|---|---|---|---|---|
| 1 | 14 /M | Thigh | DFSP | + | − | − | Exon 32 - Exon 2 |
| FS | − | + | − | Exon 32 - Exon 2 | |||
| 2 | 39 /M | Chest wall | DFSP | + | + | + | Exon 46 - Exon 2 |
| FS | − | + | + | Exon 46 - Exon 2 | |||
| 3 | 47 /F | Chest wall | DFSP | + | + | − | Exon 32 - Exon 2 |
| FS | − | + | − | / | |||
| 4 | 65 /F | Abdominal wall | DFSP | + | + | + | Exon 32 - Exon 2 |
| FS | − | + | + | Exon 32 - Exon 2 | |||
| 5 | 45 /M | Subclavicular | DFSP | + | + | − | Exon 25 - Exon 2 |
| FS | + | + | − | Exon 25 - Exon 2 | |||
| 6 | 40 /F | Chest wall | DFSP | + | + | − | Exon 25 - Exon 2 |
| FS | + | + | − | Exon 25 - Exon 2 |
PBGD, prophobilinogen deaminase; PGK, phosphoglycerate kinase; DFSP, conventional dermatofibrosarcoma protuberans areas; FS, fibrosarcomatous areas.
Discussion
Fibrosarcomatous change in DFSP is a rare event. In 1951, Penner first documented a case of metastasizing DFSP that contained areas histologically indistinguishable from fibrosarcoma. 26 Due to the rarity of the lesion, the initial description was followed by a limited number of studies, most of which were case reports. 27, 28, 29 In 1988, Wrotnowski et al described a series of six cases of DFSP-FS and proposed that fibrosarcomatous change within DFSP represented a secondarily developed neoplasm that was distinct from the surrounding DFSP, 5 but they did not find any significant difference in the subject of a prognosis between ordinary DFSP and DFSP-FS. In contrast, Ding et al stressed the unfavorable outcome of DFSP-FS in their report of nine cases of DFSP-FS. 6 They noted that patients with DFSP-FS had a higher rate of local recurrence, a shorter prerecurrence interval, and an increased incidence of distant metastasis than patients with ordinary DFSP. Connelly and Evans found a similar recurrence rate in their study of six cases and confirmed the adverse outcome of DFSP-FS. 10 More recently, two studies of a large series of DFSP-FS have supported that fibrosarcomatous change within DFSP represents a high-grade transformation and is associated with a worse prognosis than ordinary DFSP. 11, 12 The adverse clinical outcome of DFSP-FS has been supposed to be correlated with the higher cell proliferative activity in the high-grade FS areas. Several studies have demonstrated that mitotic rates in the FS areas of DFSP-FS were significantly higher than in the DFSP areas. 6, 11, 12, 30 Pizarro et al briefly reported that Ki-67 values correlated with their light microscopic classification of DFSP into DFSP-FS, hypercellular DFSP, and ordinary DFSP. 11 Similarly, Hisaoka et al also found an elevated value for the Ki-67 labeling index and the flow cytometric proliferative index in the FS areas of DFSP-FS. 30
Immunohistochemically, tumor cells in conventional DFSP areas of DFSP-FS are usually diffusely positive for CD34, whereas tumor cells in FS areas tend to lose their expression or to show only focal and weak staining. This different pattern of CD34 immunoreactivity has led some investigators to consider the antigen as a useful marker in recognizing FS areas. 31 However, it is noteworthy that there are some cases of DFSP-FS with FS areas showing diffuse staining of CD34, and ordinary DFSP or conventional DFSP areas in DFSP-FS that rarely lose or only focally show expression of the antigen. In the current series, FS areas expressed the antigen in two of the six cases (33.3%), one of which showed a diffuse staining. In the report by Diaz-Cascajo et al, three of the four cases were positive for CD34 in FS areas. 32
Recently, cytogenetic and molecular studies have provided valuable information with respect to the tumorigenesis of several soft tissue sarcomas. 33 Approaches in DFSP have revealed that the specific ring chromosomes derived from translocation (17;22) and reciprocal translocation t(17;22) fused in-frame the COL1A1 gene on chromosome 17 and the PDGFB gene on chromosome 22. 18 Greco et al demonstrated that the deregulated PDGFB could transform NIH3T3 cells, and an autocrine mechanism involving the PDGFB signaling pathway has been suggested to be related to the development and progression of DFSP. 19
Although the COL1A1-PDGFB fusion transcript has been studied in DFSP and its juvenile form, giant cell fibroblastomas, it has not been systematically investigated in the DFSP-FS variant. In the present study, we have demonstrated that the COL1A1-PDGFB fusion transcripts were detectable not only in conventional DFSP areas but also in FS areas in most cases of DFSP-FS. The sequence analysis of the PCR products confirmed that the amplified messages were derived from identical gene fusions in the two components of each case. This finding indicates that the COL1A1-PDGFB chimeric gene derived from t(17;22) or r(17;22) is still involved in the fibrosarcomatous transformation of DFSP and that the deregulated PDGFB continues to act as a growth factor in the process of tumor progression. Significance of the loss of expression of the COL1A1-PDGFB fusion transcripts in the fibrosarcomatous areas of one of the six DFSP-FS in this study, which was included in our previous paper, 21 is difficult to interpret. The result may be simply a false negative.
Cytogenetic studies on DFSP-FS are extremely limited. Recently, Hamada et al supposed, by the method of fluorescence in situ hybridization (FISH), that a gain of chromosome 17 developed in high-grade groups, recurrent or large-sized DFSP, and DFSP with the possibility of a progression to DFSP-FS. 34 The authors speculated that extra copies of chromosome 17 may be derived from t(17;22) or supernumerary ring chromosomes. However, the cellular and molecular mechanism underlying the fibrosarcomatous transformation of DFSP is still not fully clarified. Some investigators have suggested that the high-grade tumor progression of DFSP was associated with alterations in the p53 pathway, such as overexpression of p53 protein by a mutated gene and mdm2 overexpression. 30, 35 We should further investigate whether there are other additional cytogenetic or molecular aberrations that might be involved in the process of fibrosarcomatous progression of DFSP.
Fibrosarcoma is considered to be a rare malignant tumor of fibroblasts. Fibrosarcomas are divided by patient age into adult fibrosarcoma (AFS) and congenital or infantile fibrosarcoma (CFS). The latter has a better prognosis than the former, with a very low metastatic rate. Cytogenetically, CFS is characterized by relatively consistent numerical chromosome changes, such as trisomy 8, 11, 17, and 20. 36 Furthermore, recent studies have shown that CFS contained a novel recurrent t(12;15)(p13;q25), resulting in a gene fusion of ETV6-NTRK3. 37 Similar rearrangements were also demonstrated in congenital mesoblastic nephroma (CMN), and a close relationship between these two entities has been suggested. 38 In contrast, the reports on cytogenetic analysis in AFS showed complex karyotypes with no consistent rearrangements, although a few examples had nonrandom chromosomal changes, such as t(2;19). 39 Now, we need to ascertain whether there are cases with the COL1A1-PDGFB fusion gene among cases of classical AFS, because many AFS cases have clinical features, including anatomical location preponderantly in the trunk, similar to those of DFSP/DFSP-FS.
In conclusion, we have shown that fibrosarcomatous areas in DFSP-FS also expressed the COL1A1-PDGFB fusion transcripts, indicating that the chimeric gene COL1A1-PDGFB may continue to act as a growth factor in the tumor progression of DFSP. The expression of the fusion transcripts in both conventional DFSP and FS areas in DFSP-FS supports a common histogenesis of the two components. Moreover, the gene fusion may provide a potential diagnostic marker for DFSP-FS.
Acknowledgments
We thank Professor Yasuyuki Sasaguri and Dr. Shohei Shimajiri, University of Occupational and Environmental Health, for helping us design the primers used in this study and for critical and encouraging comments, and Professor Shinji Itoyama, Saitama Medical School, for providing paraffin-embedded tissues of a DFSP-FS case (case 6). We also thank Miss Atsuko Tanaka for her sophisticated technical assistance.
Address reprint requests to Dr. Hiroshi Hashimoto, Department of Pathology and Oncology, School of Medicine, University of Occupational and Environmental Health, 1–1 Iseigaoka, Yahatanishi-ku, Kitakyushu 807-8555, Japan. E-mail: hiroshi@med.uoeh-u.ac.jp.
Footnotes
Supported in part by 1998 Grants-in-Aid from the Ministry of Education, Science, Sports and Culture (08070229) and the Vehicle Racing Commemorative Foundation.
J. W. and W. S. are research fellows in the Shanghai University of Occupational and Environmental Health Joint Project on the Cytogenetic Study of Soft Tissue and Bone Tumors.
References
- 1.Enzinger FM, Weiss SW: Fibrohistiocytic tumors. Enzinger FM Weiss SW eds. Soft Tissue Tumors. 1995, :pp 325-349 Mosby, St. Louis [Google Scholar]
- 2.Dupree WB, Langloss JM, Weiss SW: Pigmented dermatofibrosarcoma protuberans (Bednar tumor): a pathologic, ultrastructural, and immunohistochemical study. Am J Surg Pathol 1985, 9:630-639 [DOI] [PubMed] [Google Scholar]
- 3.Frierson HF, Cooper PH: Myxoid variant of dermatofibrosarcoma protuberans. Am J Surg Pathol 1983, 7:445-450 [DOI] [PubMed] [Google Scholar]
- 4.Banerjee SS, Harris M, Eyden BP, Hamid BNA: Granular cell variant of dermatofibrosarcoma protuberans. Histopathology 1990, 17:375-378 [DOI] [PubMed] [Google Scholar]
- 5.Wrotnowski U, Cooper PH, Shmookler BJ: Fibrosarcomatous change in dermatofibrosarcoma protuberans. Am J Surg Pathol 1988, 12:287-293 [DOI] [PubMed] [Google Scholar]
- 6.Ding J, Hashimoto H, Enjoji M: Dermatofibrosarcoma protuberans with fibrosarcomatous areas: a clinicopathologic study of nine cases and a comparison with allied tumors. Cancer 1989, 64:721-729 [DOI] [PubMed] [Google Scholar]
- 7.Calonje E, Fletcher CDM: Myoid differentiation in dermatofibrosarcoma protuberans and its fibrosarcomatous variant: clinicopathologic analysis of 5 cases. J Cutan Pathol 1996, 23:30-36 [DOI] [PubMed] [Google Scholar]
- 8.Beham A, Fletcher CDM: Dermatofibrosarcoma protuberans with areas resembling giant cell fibroblastoma: report of two cases. Histopathology 1990, 17:165-167 [DOI] [PubMed] [Google Scholar]
- 9.Davis DA, Sanchez RL: Atrophic and plaquelike dermatofibrosarcoma protuberans. Am J Dermatopathol 1998, 20:498-501 [DOI] [PubMed] [Google Scholar]
- 10.Connelly JH, Evans HL: Dermatofibrosarcoma protuberans: a clinicopathologic review with emphasis on fibrosarcomatous areas. Am J Surg Pathol 1992, 16:921-925 [PubMed] [Google Scholar]
- 11.Pizarro GB, Fanburg JC, Miettinen M: Dermatofibrosarcoma protuberans (DFSP) with fibrosarcomatous transformation: re-explored (abstract). Mod Pathol 1997, 10:13A [Google Scholar]
- 12.Mentzel T, Beham A, Katenkamp D, Dei Tos AP, Fletcher CDM: Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol 1998, 22:576-587 [DOI] [PubMed] [Google Scholar]
- 13.Pedeutour F, Simon MP, Minoletti F, Barcelo G, Terrier-Lacombe MJ, Combemale P, Sozzi G, Ayraud N, Turc-Carel C: Translocation t(17;22)(q22;q13) in dermatofibrosarcoma protuberans: a new tumor-associated chromosome rearrangement. Cytogenet Cell Genet 1996, 72:171-174 [DOI] [PubMed] [Google Scholar]
- 14.Pedeutour F, Simon MP, Minoletti F, Sozzi G, Pierotti MA, Hecht F, Turc-Carel C: Ring 22 chromosomes in dermatofibrosarcoma protuberans are low-level amplifiers of chromosome 17, and 22 sequences. Cancer Res 1995, 55:2400-2403 [PubMed] [Google Scholar]
- 15.Naeem R, Lux ML, Huang SF, Naber SP, Corson JM, Fletcher JA: Ring chromosomes in dermatofibrosarcoma protuberans are composed of interspersed sequences from chromosome 17 and 22. Am J Pathol 1995, 147:1553-1558 [PMC free article] [PubMed] [Google Scholar]
- 16.Sinovic J, Bridge JA: Translocation (2;17) in recurrent dermatofibrosarcoma protuberans (letter). Cancer Genet Cytogenet 1994, 75:156-157 [DOI] [PubMed] [Google Scholar]
- 17.Sonobe H, Furihata M, Iwata J, Ohtsuki Y, Chikazawa M, Taguchi T, Shimizu K: Dermatofibrosarcoma protuberans harboring t(9;22)(q32;q12.2). Cancer Genet Cytogenet 1999, 110:14-18 [PubMed] [Google Scholar]
- 18.Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre JM, Terrier-Lacombe MJ, Mandahl N, Craver RD, Blin N, Sozzi G, Turc-Carel C, O’Brien KP, Kedra D, Fransson I, Guibaud C, Dumanski JP: Deregulation of the platelet-derived growth factor b-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet 1997, 15:95-98 [DOI] [PubMed] [Google Scholar]
- 19.Greco A, Fusetti L, Villa R, Sozzi G, Minoletti F, Mauri P, Pierotti M: Transforming activity of the chimeric sequence formed by the fusion of collagen gene COL1A1 and the platelet derived growth factor b-chain gene in dermatofibrosarcoma protuberans. Oncogene 1998, 17:1313-1319 [DOI] [PubMed] [Google Scholar]
- 20.O’Brien KP, Seroussi E, Dal Cin P, Sciot R, Mandahl N, Fletcher JA, Turc-Carel C, Dumanski JP: Various regions within the alpha-helical domain of the COL1A1 gene are fused to the second exon of the PDGFB gene in dermatofibrosarcomas and giant-cell fibroblastomas. Genes Chromosomes Cancer 1998, 23:187-193 [PubMed] [Google Scholar]
- 21.Wang J, Hisaoka M, Shimajiri S, Morimitsu Y, Hashimoto H: Detection of COL1A1-PDGFB fusion transcripts in dermatofibrosarcoma protuberans by reverse transcription-polymerase chain reaction using archival formalin-fixed, paraffin-embedded tissues. Diagn Mol Pathol 1999, 8:113-119 [DOI] [PubMed] [Google Scholar]
- 22.Gupta SK, Douglas-Jones AG, Morgan JM: Microdissection of stained archival tissue. J Clin Pathol 1997, 50:218-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finke J, Fritzen R, Ternes P, Lange W, Dölken G: An improved strategy and a useful housekeeping gene for RNA analysis from formalin-fixed, paraffin-embedded tissues by PCR. Biotechniques 1993, 14:448-453 [PubMed] [Google Scholar]
- 24.Argani P, Zakowski MF, Klimstra DS, Rosai J, Ladanyi M: Detection of the SYT-SSX chimeric RNA of synovial sarcoma in paraffin-embedded tissue and its application in problematic cases. Mod Pathol 1998, 11:65-71 [PubMed] [Google Scholar]
- 25.Morimitsu Y, Hisaoka M, Okamoto S, Hashimoto H, Ushijima M: Dermatofibrosarcoma protuberans and its fibrosarcomatous variant with areas of myoid differentiation: a report of three cases. Histopathology 1998, 32:547-551 [PubMed] [Google Scholar]
- 26.Penner DW: Metastasizing dermatofibrosarcoma protuberans: a case report. Cancer 1951, 4:1083-1086 [DOI] [PubMed] [Google Scholar]
- 27.Hagedorn M, Thomas C, von Kannen W: Dermatofibrosarcoma protuberans mit Übergang in ein sogenanntes Fibrosarkom. Dermatologica 1974, 149:84-89 [PubMed] [Google Scholar]
- 28.Ishii T, Koide O: An autopsy case of metastasizing dermatofibrosarcoma protuberans. Acta Pathol Jpn 1975, 25:503-515 [DOI] [PubMed] [Google Scholar]
- 29.Grouls V, Hienz HA: Dermatofibrosarcoma protuberans: transition to fibrosarcoma. Z Hautkr 1985, 60:1690-1701 [PubMed] [Google Scholar]
- 30.Hisaoka M, Okamoto S, Morimitsu Y, Tsuji S, Hashimoto H: Dermatofibrosarcoma protuberans with fibrosarcomatous areas: molecular abnormalities of the p53 pathway in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Virchows Arch 1998, 433:323-329 [DOI] [PubMed] [Google Scholar]
- 31.Goldblum JR: CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med 1995, 119:238-241 [PubMed] [Google Scholar]
- 32.Diaz-Cascajo C, Weyers W, Borrego L, Inarrea JB, Borghi S: Dermatofibrosarcoma protuberans with fibrosarcomatous areas: a clinico-pathologic and immunohistochemic study in four cases. Am J Surg Pathol 1997, 19:562-567 [DOI] [PubMed] [Google Scholar]
- 33.Dei Tos AP, Dal Cin P: The role of cytogenetics in the classification of soft tissue tumours. Virchows Arch 1997, 431:83-94 [DOI] [PubMed] [Google Scholar]
- 34.Hamada M, Hirakawa N, Fukuda T, Furue M, Hori Y, Tsuneyoshi M: A progression to dermatofibrosarcoma protuberans with a fibrosarcomatous component: a special reference to the chromosomal aberrations. Pathol Res Pract 1999, 195:451-460 [DOI] [PubMed] [Google Scholar]
- 35.Goldblum JR, Frank TS, Poy EL, Weiss SW: p53 mutations and tumor progression in well-differentiated liposarcoma and dermatofibrosarcoma protuberans. Int J Surg Pathol 1995, 3:35-42 [Google Scholar]
- 36.Schofield DE, Fletcher JA, Grier HE, Yunis EJ: Fibrosarcoma in infants and children: application of new techniques. Am J Surg Pathol 1994, 18:14-24 [DOI] [PubMed] [Google Scholar]
- 37.Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PHB: A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998, 18:184-187 [DOI] [PubMed] [Google Scholar]
- 38.Rubin BP, Chen CJ, Morgan TW, Xiao S, Grier HE, Kozakewich HP, Perez-Atayde AR, Fletcher JA: Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion. Cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol 1998, 153:1451-1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Limon J, Szadowska A, Iliszko M, Babinska M, Mrozek K, Jaskiewicz J, Kopacz A, Roszkiewicz A, Debiec-Rychter M: Recurrent chromosome changes in two adult fibrosarcomas. Genes Chromosomes Cancer 1998, 21:119-123 [DOI] [PubMed] [Google Scholar]