

Abstract
To understand the genetic basis of breast cancer in a comprehensive way, purported precursor lesions need to be analyzed at a large number of genetic marker loci and compared with each other and with the invasive components. However, the microscopic size of most of these lesions and the very small amount of material that can be obtained through microdissection limit the number of loci that can be included in the analysis. To address this issue, a multiplex genotyping approach has been developed. With this approach, polymorphic sequences at 28 marker loci were amplified simultaneously from the microdissected components in 5-μm paraffin-embedded breast tissue sections. The genotypes of the lesions were determined after resolving the amplified allelic products by denaturing gradient gel electrophoresis. Because the material isolated from each lesion in a single 5-μm section was sufficient for several 28-locus assays and several successive tissue sections with the same set of lesions may be prepared, it is possible to determine the genotype of each lesion at hundreds of genetic marker loci that may well cover the human genome. Analyzing a sufficient number of cases may yield information that could be used to understand the genetic basis of breast cancer development in a comprehensive way.
Epithelial proliferation in the ductal system of the human breast has been classified according to its severity. The terminology used is based on the organizational patterns of cell groups (architecture) and on characteristics of individual cells comprising these groups (cytology). The terminology for ductal epithelial proliferation from the mildest to the most severe types follows this sequence: ductal hyperplasia (DH), atypical ductal hyperplasia (ADH), ductal carcinoma in situ (DCIS). Epidemiological studies of large numbers of patients with these lesions followed for up to 20 years suggest that DH, ADH, and DCIS confer a progressively increasing relative risk for the development of invasive breast cancer (IC).1, 2, 3, 4, 5, 6, 7 Many patients with IC have proliferative lesions in their breast tissue adjacent to the IC, providing further support for a precursor-product relationship and the stepwise progression of breast cancer. Genetic analysis of individual lesions of different morphology may provide direct insight into these footprints of breast cancer development and the underlying mechanisms.
Lesions at stages of different degrees of cancer risk and those at the same stages, but with different morphological patterns, are often observed within the breast tissue. To learn the causes of different lesions, it is necessary to isolate them from each other and to analyze them separately. The first genetic analysis of individual lesions isolated by microdissection from breast tissue was reported in 1993.8 Since then, microdissection has become a powerful tool and has been used in many studies. Without microdissection, only the predominant invasive components in the primary tumors may be studied. However, because of extensive admixture of cancer cells and normal cells, results from these studies are often associated with a certain degree of ambiguity. With microdissection, contaminating cells may be minimized or eliminated from the invasive components. More importantly, in this way, microscopic lesions may be isolated and analyzed separately. Such an advance has made it possible to examine, at the molecular level, the relationships among different breast lesions from a single tissue sample, to trace the tumor development history, and to gain an understanding of the molecular pathways to breast cancer.
Because a large number of genes may be involved in breast cancer development and progression, it is necessary to cover most, if not all, chromosomal regions when the individual lesions are analyzed. In this way, the mechanisms underlying breast cancer development may be studied in a comprehensive way. However, the amount of DNA isolated from microdissection is usually small. With the conventional single-locus-based genotyping procedure, this is only enough for analysis with one or very few loci. To address this issue, we have developed a multiplex genotyping approach. With this approach, the polymorphic sequences at a large number of marker loci can be amplified to analyzable amounts simultaneously by a three-round polymerase chain reaction (PCR) protocol. The allelic sequences amplified from different loci are then resolved by denaturing gradient gel electrophoresis (DGGE),9, 10, 11, 12, 13 which is capable of separating DNA fragments differing by as little as 1 bp. With this multiplex genotyping system, we were able to determine the genotypes of individual lesions isolated from paraffin-embedded 5-μm breast tissue sections at 28 genetic marker loci distributed on 18 chromosomal arms in an efficient way. The results allowed us to detect loss of heterozygosity (LOH), a common indication of tumor suppressor gene inactivation, in different proliferative lesions.
Materials and Methods
Isolation of Cells of Individual Lesions from Paraffin-Embedded Breast Tissue
Breast tissue specimens used in this study were from pathological specimens. Duplicate 5-μm sections were prepared from paraffin-embedded tissue blocks. After H&E staining, one coverslipped slide was used as the reference slide and subjected to histological analysis. The lesions were marked by a surgical pathologist (H. F.) with extensive experience in breast tissue evaluation. The other slide, with no coverslip (sample slide), was overlaid on the reference slide and the tissue sections on both slides were aligned. The selected lesions on the sample slide were isolated by scraping with a 271/2-gauge syringe needle (Figure 1) . The material isolated from each lesion was placed into a 0.5-μl tube containing 5 μl of lysis buffer (200 mmol/L KOH and 50 mmol/L dithiothreitol). After 10 minutes’ incubation at 65°C, the lysate was neutralized with 5 μl of neutralization buffer (200 mmol/L HCl, 900 mmol/L Tris-HCl, pH 8.3, and 300 mmol/L KCl).14
Figure 1.
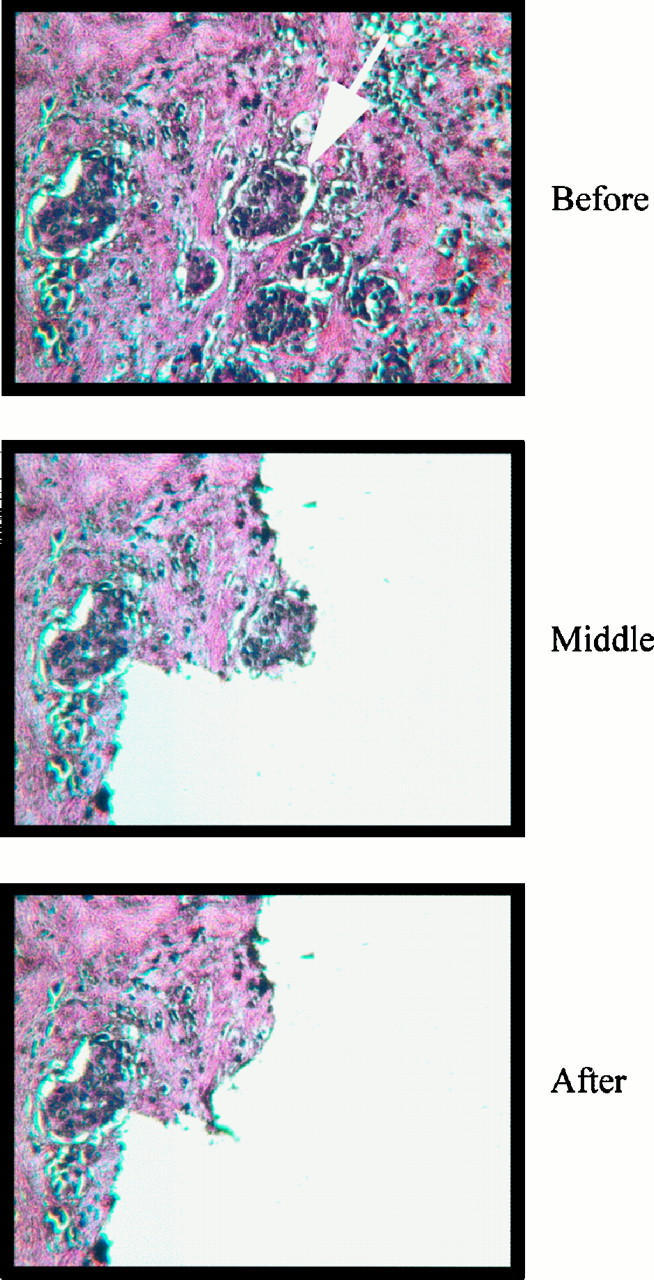
Microdissection of a small group of cells containing ∼30 nuclei in a breast tissue section. For large areas, cells may be microdissected directly with a fine needle (271/2-gauge). For small groups of cells as indicated by the arrow above, an adjacent area was cleared first (middle) before the desired cell group was dissected.
Multiplex PCR Amplification
A panel of 28 genetic markers distributed on 18 chromosomal arms were chosen (Table 1) . For each marker locus, three primers were designed (Figure 2) . One of the primers, primer O, was a regular primer with a ∼20-base locus-specific sequence. The other two, primers, R and C, each contained 20-base locus-specific sequences at their 3′ portions and a 20-base nongenomic sequence (either tail 1 or tail 2) that was universal for all loci at its 5′ portion. Tail 1 was AT-rich and tail 2 was GC-rich to facilitate DGGE separation. The C primer was an internal (nested) primer with respect to the O primer and was used in the second round of amplification to enhance the yield and specificity. Two universal primers, T1 and T2, were also synthesized. T1 was identical to tail 1 with an additional 5-base AT-rich sequence at its 5′ end. T2 was identical to tail 2 in its 3′ portion and contained a 20-base GC-rich portion at its 5′ end for attaching more GC-rich sequence to the final PCR products. (Because of the large number of primers, the primer sequences are not included in this publication, but will be provided on request).
Table 1.
Genotypes of Lesions from the Two Patients
| Number | Locus | Location | Patient 1 | Patient 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NT | DH | ADH | DCIS | NT | DCIS | IC-1 | IC-2 | |||
| 1 | AT3 | 1q32 | ○ | ○ | ○ | ○ | ||||
| 2 | DAG1 | 3p21 | ○ | ○ | ○ | ○ | • | |||
| 3 | AGTR1 | 3q21-q25 | • | |||||||
| 4 | SRDA | 5p15 | ○ | ○ | ○ | ○ | ||||
| 5 | DRD1 | 5q35.1 | ○ | ○ | ○ | ○ | ||||
| 6 | OTF | 6p21.3 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 7 | CA2 | 8q22 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 8 | ABO | 9q34.1 | ||||||||
| 9 | WT1 | 11p13 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 10 | A2M | 12p13.3-p12.3 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 11 | CF7 | 13q34 | ○ | ○ | ○ | ○ | • | |||
| 12 | GALNS | 16q24 | • | • | • | |||||
| 13 | TP53 | 17p13.1 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 14 | VNT | 17q11 | ○ | ○ | ○ | ○ | ||||
| 15 | HSD | 17q11-q21 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 16 | BOX | 17q21.1 | ○ | ○ | ○ | ○ | ||||
| 17 | KRT9 | 17q21.1-q21.2 | ○ | ○ | ○ | ○ | • | |||
| 18 | COL1A1 | 17q21.3-q22 | ○ | ○ | ○ | ○ | • | |||
| 19 | NME | 17q21-q22 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 20 | P4HB | 17q25 | ○ | ○ | ○ | ○ | ||||
| 21 | BCL2 | 18q21 | ○ | ○ | ○ | ○ | • | |||
| 22 | INSR | 19p13.3 | ○ | ○ | ○ | ○ | ||||
| 23 | HRC | 19p13.2-q13.3 | ○ | ○ | ○ | ○ | ||||
| 24 | C3 | 19p13.3-p13.2 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 25 | CKM | 19q13.3 | ||||||||
| 26 | GNAS1 | 20q13.2-13.3 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| 27 | CHRNA4 | 20q13.2-q13.3 | ○ | ○ | ○ | ○ | ||||
| 28 | CBS | 21q22.3 | ||||||||
, Heterozygous; ○, homozygous; •, LOH; , allelic reduction.
Figure 2.

Schematic demonstration of the three-round multiplex PCR amplification. Three loci are shown. Specific sequences are indicated as either dash- or line-filled bars. Primers are represented by different arrows. Polymorphic sites are indicated by letters. Non-genomic sequences of tails 1 and 2 in the specific primers and the universal primers T1 and T2 are shown as either black (GC-rich) or hollow (AT-rich) bars. Note that with DGGE, fragments amplified from different loci do not have to be of different lengths for separation.
About one-third of the material microdissected from each lesion was used for analysis. A three-round multiplex PCR procedure described in our previous publication15 and modified in the present study was used for amplifying the sequences at the 28 loci. Briefly, all PCR samples contained 1× PCR buffer (100 mmol/L Tris-HCl pH 8.3, 50 mmol/L KCl, 1.5 mmol/L MgCl2, and 0.1 mg/ml gelatin), the four dNTPs (100 μmol/L each), and 1 unit of Taq DNA polymerase in a final volume of 50 μl. PCR was performed on a DNA Thermal Cycler 480 (Perkin Elmer, Norwalk, CT). In the first round, R and O primers (50 nmol/L each) for all loci were used. Each PCR cycle consisted of 1 minute at 94°C for denaturation and 3 minutes at 55°C, followed by 5 minutes ramping from 55°C to 70°C, for annealing and extension. Thirty-five cycles were performed. Amplification with the R primers attached tail 1 sequence to all target fragments. In the second round, a 2-μl aliquot from each first-round product was reamplified. All R primers were replaced by only one primer, T1 (0.2 μmol/L), which was identical to tail 1. All O primers were replaced by the corresponding C primers (20 nmol/L each). Amplification with C primers attached the other universal tail, tail 2, to all target sequences. The PCR profile used in the first round was used in the second round. Fifteen cycles were performed. In the third round, only two primers (0.2 μmol/L each), T1 and T2, were used to amplify all PCR products to analyzable amounts. The PCR profile for the third round was 1 minute at 94°C for denaturation and 1 minute at 60°C for annealing and extension. Thirty-five cycles were performed. To minimize the amount of heteroduplexes, additional enzyme (0.5 units) and primers (to final concentration of 0.5 μmol/L for each) were added to each sample before the last PCR cycle in the third round. The PCR cycle consisted of 2 minutes at 95°C for denaturation, 1 minute at 60°C for annealing, and 10 minutes at 72°C for extension, as described previously.16
Separation of the Allelic PCR Products Amplified from Different Loci
The allelic PCR products amplified from different loci in the multiplex PCR were separated by DGGE with 10% polyacrylamide gels containing a denaturing gradient of 40 to 75% (100% denaturing gel contains 7 mol/L urea and 40% formamide). The samples were electrophoresed at 115 volts and 60°C for 15 hours. The gels were stained with SYBR2 Green (Molecular Probes, Eugene, OR) and visualized under UV illumination.
Results
Marker Selection
A panel of 28 genetic markers distributed on 18 chromosomal arms (Table 1) were selected either from our previous publication15 or from the Genome Database (GDB, Johns Hopkins University, Baltimore, MD). All markers except one were single nucleotide polymorphisms (SNPs); the exception was a 10-base insertion/deletion. Other than some markers on chromosome 17, most markers were close to the telomeres. The reason for such a selection was to maximize LOH detection, because markers close to the telomeres are likely to be involved in LOH caused by whole chromosome loss, non-interstitial chromosomal segment deletion, and mitotic recombination. Several markers in different regions on chromosome 17 were chosen because several tumor suppressor genes may be present along this chromosome.17, 18, 19, 20, 21, 22, 23, 24 PCR primers were synthesized for the three-round multiplex amplification as described in Materials and Methods. The lengths of the specific sequences in the final PCR products ranged from 56 to 121 bp, plus the 65-bp nongenomic sequences from the universal tails and universal primers. This length range is especially suitable for this analysis because DNA in archived tissue is usually highly degraded. Because the selected markers were all biallelic, 56 possible bands were expected from the 28 SNPs. Although it is possible to resolve 56 well-spaced bands in one gel lane as shown in Figure 3 , not all bands from the 28 loci were well-spaced. Therefore, the 28 markers were subdivided into two groups of 17 and 11, respectively. Allelic bands from two pairs of markers (HRC and KRT9, CBS and GNAS1) could not be well resolved in the denaturing range used for resolving most markers. They could be resolved with a slightly changed denaturing range and were therefore kept in the system. The polymorphic sequences at all 28 marker loci were coamplified in the first round of amplification using about one-third of the material microdissected from each lesion. The two groups of markers were amplified separately in the second and the third rounds by using the corresponding primers.
Figure 3.

Multiplex genotype determination at 28 marker loci (listed in Table 1 ) for the lesions microdissected from breast tissues of the two patients. NT, normal tissue; DH, ductal hyperplasia; ADH, atypical ductal hyperplasia; DCIS, ductal carcinoma in situ; IC, invasive carcinoma; M, mixture of PCR products separately amplified from the 28 loci in corresponding heterozygous DNA samples used as molecular markers. Bands representing different alleles are distinguished by “−1” and “−2.” Alleles for the loci with LOH are indicated by boxed numbers. The confirming panel is for confirmation of the results from the loci showing LOH in patient 2. The loci in this panel were separately amplified by using aliquots from the first-round PCR products. The PCR products from different loci for each samples were mixed, respectively, before being loaded onto the DGGE gel. Only results from tumor 2 are shown.
During PCR, heteroduplex DNA may be generated due to annealing between the DNA strands of different allelic sequences. These heteroduplexes may be detected as gel bands and thus complicate the multiplex analysis. We showed that heteroduplexes could be eliminated or reduced to insignificant amounts by adding a long PCR cycle at the end of the multiplex amplification with additional primers and DNA polymerase.16 This allowed us to obtain clean results from the multiplex analysis by DGGE (Figure 3) .
To confirm the results of multiplex genotyping, all loci showing LOH in patient 2 were retyped with two different approaches. With the first approach, the same sets of samples typed by the PCR-DGGE method were prepared by microdissection. In the first round, the samples were amplified with the same conditions used in the three-round multiplex amplification. Aliquots from the PCR products were reamplified separately with nested primers for each locus. For the loci that were not natural restriction fragment length polymorphisms (RFLPs), primers with single bases mismatching to their templates, which were next to or near the polymorphic sites, were used to convert polymorphic sites into RFLPs. All final PCR products were digested with corresponding enzymes. Three sets of duplicated samples were typed in this way. Results from one set are shown in Figure 4 . All results were consistent with the PCR-DGGE results.
Figure 4.

Confirmation of the LOH results from PCR-DGGE by restriction enzyme digestion. One of the three duplicates is shown for the samples from patient 2. For each locus, samples in the lanes from left to right are normal tissue, DCIS, and IC. PCR products were digested by corresponding restriction enzymes. Allelic bands are indicated by arrows.
Another concern about the typing results from PCR-DGGE is whether the multiple rounds of amplification would generate artifacts. To address this issue, aliquots from the first-round PCR products were reamplified separately with the primers used in the second round of the three-round multiplex amplification. The resulting PCR products of different loci for each lesion were pooled and subjected to DGGE analysis. The results were also consistent with those obtained from the multiplex analysis (confirming panel, Figure 3 ), indicating that the multiplex approach is reliable.
Genetic Alterations in Breast Tissues from Two Patients
Using samples microdissected from six separate lesions and the normal tissue in two patients, the multiplex genotyping system allowed us to determine the corresponding 8 × 28 = 224 genotypes very efficiently (Table 1) . From patient 1, normal tissue (NT), DH, ADH, and DCIS were isolated and analyzed. One locus, GALNS on chromosome 16q24, was shown to be associated with LOH in both ADH and DCIS but not in DH. This result is consistent with the notion that ADH and DCIS are more advanced lesions than DH. It is also consistent with the previous observations that inactivation of a tumor suppressor gene on 16q is involved in the progression of proliferative breast lesions.25, 26, 27, 28, 29, 30, 31, 32
From patient 2, NT, DCIS, and two foci of IC were isolated. LOH was observed in DCIS at the marker loci on six chromosomal arms, 3p (DAG1, 3p21), 3q (AGTR1, 3q21-q25), 13q (CF7, 13q34), 16q (GALNS, 16q24), 17q (KRT9, 17q21.1-q21.2; COL1A1, 17q21.3-q22), and 18q (BCL2, 18q21). LOH on these chromosomal arms has been observed in many other breast cancer studies.18, 19, 20, 21, 22, 23, 24, 28, 30, 32, 33, 34, 35, 36, 37, 38, 39, 40 Allelic reduction was observed at the marker loci on 3q, 16q, and 17q for both IC samples. Reduction rather than loss is likely related to the fact that small groups of infiltrating cancer cells are difficult to separate from stromal and inflammatory cells. Interestingly, allele loss or reduction affected different alleles for the marker CF7 on chromosome 13q. The mechanism of this phenomenon and the impact to breast cancer is not clear. Neither allelic loss nor allelic reduction was observed for the markers at 3p and 18q in the IC samples. A possible explanation is that the DCIS and IC samples isolated from this patient progressed from the same founder cell through inactivation of a set of tumor suppressor genes including those on chromosomes 3q, 16q, and 17q, but diverged before the DCIS stage. Gaining additional genetic alterations on chromosome 3p and 18q was not sufficient to convert the DCIS into IC, whereas the two foci of IC may have developed by gaining other genetic alterations in chromosomal regions that were not covered or not informative in the present analysis. These results suggest that at the molecular level, even within the same breast tissue, lesions evolve differently.
Discussion
For a systematic genetic analysis of breast cancer development, it is necessary to include a large number of genetic markers for both the invasive tumor and its purported precursors. However, this approach is limited by the amount of material that can be obtained from microscopic precursor lesions. Microdissection, together with the multiplex genotyping approach used in the present study, was shown to be promising for analysis with a large number of markers in a nonradioactive way. We showed that only about one-third of the material from each of DH, ADH, and DCIS in a 5-μm tissue section was sufficient for the multiplex analysis at 28 loci. More material may be obtained from ICs, although contamination may be a problem with some infiltrating patterns. Therefore, material obtained from each tissue section should be sufficient for the analysis with ∼84 markers, or very likely with ∼100 loci. Because the thickness of a 5-μm tissue section is about the size of a nucleus, and it is possible to prepare several successive sections containing the same set of lesions from each tissue block, we have virtually demonstrated the feasibility of analyzing breast lesions at several hundred loci. If 600 well-spaced markers (5 cM each) are included in the analysis, the human genome could be well covered. Although 600 markers cannot be all informative for a given case, analysis of a sufficient number of cases should allow one to locate a large number of candidate tumor suppressor genes to small chromosomal regions.
All but one of the genetic markers used in the present study were SNPs. A major advantage of using SNP-based markers is the ability to incorporate a large number of markers into a multiplex format. However, because allelic sequences at an SNP locus differ by only 1 bp, the alleles of each locus need to be discriminated with a highly sensitive approach. In the present study, DGGE was used for this purpose. With DGGE, not only can the allelic sequences at each marker locus be well separated, but the allelic sequences can be separated based on sequence differences rather than on the sizes of the amplified fragments. This is especially important for genetic analysis of archived tissues, in which DNA is usually highly degraded and is present as short fragments. Because DGGE separation does not depend on the lengths of the sequences, short sequences, as long as they are different, can be used for the analysis.
The observation that lesions at different proliferative stages are present in cancer-containing breast tissue and the results from long-term epidemiological studies have led to the hypothesis that breast cancer progresses through successive stages in a stepwise manner. Recent reports of genetic analyses of individual breast lesions isolated by microdissection indicate that genetic alterations detected from early (less morphologically advanced) lesions are generally fewer than those from later (more advanced) lesions. In many tumors, later lesions often contain all genetic changes found in the early lesions plus additional genetic alterations that are not seen in the early lesions.40, 41, 42 Such a correlation was also shown on a genomic scale by comparative genome hybridization.43, 44 These observations support the hypotheses that the less advanced lesions are the precursors of more advanced lesions and that development of early lesions is a necessary step for the development of the later lesions, at least in a significant percentage of patients.
However, several lines of data challenge the above notion. Deng et al38 showed that cells from a breast carcinoma and those in the adjacent histologically normal terminal ductal-lobular units (TDLU) contained the same set of genetic alterations, suggesting that the histologically normal TDLU cells may be immediate precursors of cancer cells. Furthermore, in the studies discussed above40, 41, 42, 43, 44 showing the later lesions containing all genetic alterations found in the early lesion, exceptions were also noted. In particular, early lesions had genetic alterations that were not found in more advanced lesions from the same species. Discrepancies were also found between ICs and metastatic carcinomas.44 In the present study, Patient 2 was such an example. LOH on chromosomal arms 3p and 18q was detected in DCIS but not in two separate IC samples. Thus, at least some of the earlier lesions are not immediate precursors of the later lesions found in the same patient, and some of the earlier changes may not be necessary for the development of later lesions.
In summary, multiplex genotype analysis of microdissected lesions at a large number of loci provides a powerful tool for the systematic genetic analysis of breast cancer and for understanding the genetic factors involved in the evolution of cancer. With genetic markers covering the entire genome, it is possible to correlate morphologically distinct lesions with patterns of genetic abnormalities in a comprehensive way.
Acknowledgments
We thank Dr. David P. Beck for his comments on the manuscript.
Address reprint requests to Honghua Li, Coriell Institute for Medical Research, 401 Haddon Avenue, Camden, NJ 08103. E-mail: holi@umdnj.edu.
Footnotes
Supported in part by National Institutes of Health grant CA77363, an institutional grant from the Coriell Institute, the Emlen Stokes Chair in Genetics (to H. L.), and Department of Defense grant DAMD17–94-J-4177 (to H. F.).
Dr. Lin’s current address: Department of Cellular and Molecular Physiology, Pennsylvania State University College of Medicine, Hershey, PA 17033.
References
- 1.Bodian CA, Perzin KH, Lattes R, Hoffmann P, Abernathy TG: Prognostic significance of benign proliferative breast disease. Cancer 1993, 71:3896-3807 [DOI] [PubMed] [Google Scholar]
- 2.Bodian CA, Perzin KH, Lattes R: Lobular neoplasia: long term risk of breast cancer and relation to other factors. Cancer 1996, 78:1024-1034 [DOI] [PubMed] [Google Scholar]
- 3.Dupont WD, Page DL: Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985, 312:146-151 [DOI] [PubMed] [Google Scholar]
- 4.Page DL, Dupont WD: Histopathologic risk factors for breast cancer in women with benign breast disease. Semin Surg Oncol 1988, 4:213-217 [DOI] [PubMed] [Google Scholar]
- 5.Page DL, Dupont WD: Histologic indicators of breast cancer risk. Am Coll Surg Bull 1991, 76:16-23 [Google Scholar]
- 6.Page DL, Dupont WD: Indicators of increased breast cancer risk in humans. J Cell Biochem (suppl) 1992, 16G:175-182 [DOI] [PubMed] [Google Scholar]
- 7.Page DL, Dupont WD, Jensen RA: Papillary apocrine change of the breast: associations with atypical hyperplasia and risk of breast cancer. Cancer Epidemiol Biomarkers Prev 1996, 5:29-32 [PubMed] [Google Scholar]
- 8.Radford DM, Fair K, Thompson AM, Ritter JH, Holt M, Steinbrueck T, Wallace M, Wells SA, Jr, Donis-Keller HR: Allelic loss on a chromosome 17 in ductal carcinoma in situ of the breast. Cancer Res 1993, 53:2947-2949 [PubMed] [Google Scholar]
- 9.Fischer SG, Lerman LS: Separation of random fragments of DNA according to properties of their sequences. Proc Natl Acad Sci USA 1980, 77:4420-4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer SG, Lerman LS: DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: correspondence with melting theory. Proc Natl Acad Sci USA 1983, 80:1579-1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers RM, Fischer SG, Maniatis T, Lerman LS: Modification of the melting properties of duplex DNA by attachment of a GC-rich DNA sequence as determined by denaturing gradient gel electrophoresis. Nucleic Acids Res 1985, 13:3111-3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myers RM, Fischer SG, Lerman LS, Maniatis T: Nearly all single base substitutions in DNA fragments joined to a GC-clamp can be detected by denaturing gradient gel electrophoresis. Nucleic Acids Res 1985, 13:3131-3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheffield VC, Cox DR, Lerman LS, Myers RM: Attachment of a 40-base-pair G + C-rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chain reaction results in improved detection of single-base changes. Proc Natl Acad Sci USA 1989, 86:232-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui X, Li H, Goradia TM, Lange K, Kazazian HH, Galas D, Arnheim N: Single sperm typing: determination of genetic distance between the Gγ-globin and parathyroid hormone loci using the polymerase chain reaction and allele specific oligomers. Proc Natl Acad Sci USA 1989, 86:9389-9393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Z, Cui X, Li H: Multiplex genotype determination at a large number of gene loci. Proc Natl Acad Sci USA 1996, 93:2582-2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui X, Li H: Discriminating between allelic and interlocus differences among human immunoglobulin VH4 sequences by analyzing single spermatozoa. Hum Genet 1997, 100:96-100 [DOI] [PubMed] [Google Scholar]
- 17.Coles C, Thompson AM, Elder PA, Cohen BB, Mackenzie IM, Cranston G, Chetty U, Mackay J, Macdonald M, Nakamura Y, Hoyheim B, Steel CM: Evidence implicating at least two genes on chromosome 17p in breast carcinogenesis. Lancet 1990, 336:761-763 [DOI] [PubMed] [Google Scholar]
- 18.Lindblom A, Skoog L, Andersen TI, Rotstein S, Nordenskjold M, Larsson C: Four separate regions on chromosome 17 show loss of heterozygosity in familial breast carcinomas. Hum Genet 1993, 91:6-12 [DOI] [PubMed] [Google Scholar]
- 19.Cropp CS, Champeme MH, Lidereau R, Callahan R: Identification of three regions on chromosome 17q in primary human breast carcinomas which are frequently deleted. Cancer Res 1993, 53:5617-5619 [PubMed] [Google Scholar]
- 20.Cornelis RS, Devilee P, van Vliet M, Kuipers-Dijkshoorn N, Kersenmaeker A, Bardoel A, Khan PM, Cornelisse CJ: Allele loss patterns on chromosome 17q in 109 breast carcinomas indicate at least two distinct target regions. Oncogene 1993, 8:781-785 [PubMed] [Google Scholar]
- 21.Saito H, Inazawa J, Saito S, Kasumi F, Koi S, Sagae S, Kudo R, Saito J, Noda K, Nakamura Y: Detailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2-cM region on 17q21.3 often and commonly deleted in tumors. Cancer Res 1993, 53:3382-3385 [PubMed] [Google Scholar]
- 22.Kirchweger R, Zeillinger R, Schneeberger C, Speiser P, Louason G, Theillet C: Patterns of allele losses suggest the existence of five distinct regions of LOH on chromosome 17 in breast cancer. Int J Cancer 1994, 56:193-199 [DOI] [PubMed] [Google Scholar]
- 23.Nagai MA, Medeiros AC, Brentani MM, Brentani RR, Marques LA, Mazoyer S, Mulligan LM: Five distinct deleted regions on chromosome 17 defining different subsets of human primary breast tumors. Oncology 1995, 52:448-453 [DOI] [PubMed] [Google Scholar]
- 24.Phelan CM, Borg A, Cuny M, Crichton DN, Baldersson T, Andersen TI, Caligo MA, Lidereau R, Lindblom A, Seitz S, Kelsell D, Hamann U, Rio P, Thorlacius S, Papp J, Olah E, Ponder B, Bignon YJ, Scherneck S, Barkardottir R, Borresen-Dale AL, Eyfjord J, Theillet C, Thompson AM, Larsson C: Consortium study on 1280 breast carcinomas: allelic loss on chromosome 17 targets subregions associated with family history and clinical parameters. Cancer Res 1998, 58:1004-1012 [PubMed] [Google Scholar]
- 25.Dutrillaux B, Gerbault-Seureau M, Zafrani B: Characterization of chromosomal anomalies in human breast cancer: a comparison of 30 paradiploid cases with few chromosome changes. Cancer Genet Cytogenet 1990, 49:203-217 [DOI] [PubMed] [Google Scholar]
- 26.Hainsworth PJ, Raphael KL, Stillwell RG, Bennett RC, Garson OM: Cytogenetic features of twenty-six primary breast cancers. Cancer Genet Cytogenet 1991, 53:205-218 [DOI] [PubMed] [Google Scholar]
- 27.Cornelisse CJ, Kuipers-Dijkshoorn N, van Vliet M, Hermans J, Devilee P: Fractional allelic imbalance in human breast cancer increases with tetraploidization and chromosome loss. Int J Cancer 1992, 50:544-548 [DOI] [PubMed] [Google Scholar]
- 28.Aldaz CM, Chen T, Sahin A, Cunningham J, Bondy M: Comparative allelotype of in situ and invasive human breast cancer: high frequency of microsatellite instability in lobular breast carcinomas. Cancer Res 1995, 55:3976-3981 [PubMed] [Google Scholar]
- 29.Iida A, Isobe R, Yoshimoto M, Kasumi F, Nakamura Y, Emi M: Localization of a breast cancer tumour-suppressor gene to a 3-cM interval within chromosomal region 16q22. Br J Cancer 1997, 75:264-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen T, Sahin A, Aldaz CM: Deletion map of chromosome 16q in ductal carcinoma in situ of the breast: refining a putative tumor suppressor gene region. Cancer Res 1996, 56:5605-5609 [PubMed] [Google Scholar]
- 31.Radford DM, Fair KL, Phillips NJ, Ritter JH, Steinbrueck T, Holt MS, Donis-Keller H: Allelotyping of ductal carcinoma in situ of the breast: deletion of loci on 8p, 13q, 16q, 17p and 17q. Cancer Res 1995, 55:3399-3405 [PubMed] [Google Scholar]
- 32.O’Connell P, Pekkel V, Fuqua SA, Osborne CK, Clark GM, Allred DC: Analysis of loss of heterozygosity in 399 premalignant breast lesions at 15 genetic loci. J Natl Cancer Inst 1998, 90:697-703 [DOI] [PubMed] [Google Scholar]
- 33.Sato T, Tanigami A, Yamakawa K, Akiyama F, Kasumi F, Sakamoto G, Nakamura Y: Allelotype of breast cancer: cumulative allele losses promote tumor progression in primary breast cancer. Cancer Res 1990, 50:7184-7189 [PubMed] [Google Scholar]
- 34.Devilee P, van Vliet M, van Sloun P, Kuipers Dijkshoorn N, Hermans J, Pearson PL, Cornelisse CJ: Allelotype of human breast carcinoma: a second major site for loss of heterozygosity is on chromosome 6q. Oncogene 1991, 6:1705-1711 [PubMed] [Google Scholar]
- 35.Sato T, Akiyama F, Sakamoto G, Kasumi F, Nakamura Y: Accumulation of genetic alterations and progression of primary breast cancer. Cancer Res 1991, 51:5794-5799 [PubMed] [Google Scholar]
- 36.Andersen TI, Gaustad A, Ottestad L, Farrants GW, Nesland JM, Tveit KM, Borresen AL: Genetic alterations of the tumour suppressor gene regions 3p, 11p, 13q, 17p, and 17q in human breast carcinomas. Genes Chromosomes Cancer 1992, 4:113-121 [DOI] [PubMed] [Google Scholar]
- 37.Chen LC, Kurisu W, Ljung BM, Goldman ES, Moore D, 2d,, Smith HS: Heterogeneity for allelic loss in human breast cancer. J Natl Cancer Inst 1992, 84:506-510 [DOI] [PubMed] [Google Scholar]
- 38.Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS: Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 1996, 274:2057-2059 [DOI] [PubMed] [Google Scholar]
- 39.Dillon EK, de Boer WB, Papadimitriou JM, Turbett GR: Microsatellite instability and loss of heterozygosity in mammary carcinoma and its probable precursors. Br J Cancer 1997, 76:156-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhuang Z, Lininger RA, Man YG, Albuquerque A, Merino MJ, Tavassoli FA: Identical clonality of both components of mammary carcinosarcoma with differential loss of heterozygosity. Mod Pathol 1997, 10:354-362 [PubMed] [Google Scholar]
- 41.Radford DM, Phillips NJ, Fair KL, Ritter JH, Holt M, Donis-Keller H: Allelic loss and the progression of breast cancer. Cancer Res 1995, 55:5180-5183 [PubMed] [Google Scholar]
- 42.O’Connell P, Pekkel V, Fuqua S, Osborne CK, Allred DC: Molecular genetic studies of early breast cancer evolution. Breast Cancer Res Treat 1994, 32:5-12 [DOI] [PubMed] [Google Scholar]
- 43.Kuukasjarvi T, Tanner M, Pennanen S, Karhu R, Kallioniemi OP, Isola J: Genetic changes in intraductal breast cancer detected by comparative genomic hybridization. Am J Pathol 1997, 150:1465-1471 [PMC free article] [PubMed] [Google Scholar]
- 44.Kuukasjarvi T, Karhu R, Tanner M, Kahkonen M, Schaffer A, Nupponen N, Pennanen S, Kallioniemi A, Kallioniemi OP, Isola J: Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res 1997, 57:1597-1604 [PubMed] [Google Scholar]