

Abstract
We developed a homogeneous format reverse transcription-polymerase chain reaction assay for quantitating hepatitis C virus (HCV) RNA based on the TaqMan principle, in which signal is generated by cleaving a target-specific probe during amplification. The test uses two probes, one specific for HCV and one specific for an internal control, containing fluorophores with different emission spectra. Titers are calculated in international units (IU)/ml by comparing the HCV signal generated by test samples to that generated by a set of external standards. Endpoint titration experiments demonstrated that samples containing 28 IU/ml give positive results 95% of the time. Based on these data, the limit of detection was set conservatively at 40 IU/ml. All HCV genotypes were amplified with equal efficiency and accurately quantitated: when equal quantities of RNA were tested, each genotype produced virtually identical fluorescent signals. The test exhibited a linear range extending from 64 to 4,180,000 IU/ml and excellent reproducibility, with coefficients of variation ranging from 21.6 to 30.4%, which implies that titers that differ by a factor of twofold (0.3 log10) are statistically significant (P = 0.005). The test did not react with other organisms likely to co-infect patients with hepatitis C and exhibited a specificity of 99% when evaluated on a set of samples from HCV seronegative blood donors. In interferon-treated patients, the patterns of viral load changes revealed by the TaqMan HCV quantitative test distinguished responders from nonresponders and responder-relapsers. These data indicate that the TaqMan quantitative HCV test provides an attractive alternative for measuring HCV viral load and should prove useful for prognosis and for monitoring the efficacy of antiviral treatments.
Nucleic acid amplification tests for HCV RNA have contributed to our understanding of the natural history of hepatitis C virus (HCV) infection and to the clinical management of chronic hepatitis C. Quantitative viral load tests are used for prognosis, 1, 2, 3, 4, 5, 6, 7, 8, 9 for evaluating new treatment regimens, 10 and for modifying treatment 11 or deciding whether to administer follow-up treatment. 12 A recent analysis of data from two multicenter studies indicates that pretreatment viral load, together with genotype, sex, age, and extent of fibrosis, can be used to decide whether patients should be treated with interferon plus ribavirin for 24 or 48 weeks. 13 Qualitative tests are used to diagnose infection 5, 14, 15, 16 and to detect a sustained response to antiviral therapy, 5, 14 which is associated with long-term clinical benefit. 17
The most sensitive and most reliable tests for HCV RNA are the qualitative AMPLICOR HCV and quantitative AMPLICOR HCV MONITOR tests (Roche Molecular Systems, Pleasanton, CA). 18, 19 These heterogeneous format tests use separate DNA hybridization procedures to detect products after a fixed number of amplification cycles and, thus, exhibit a trade-off between sensitivity and dynamic range. The quantitative test uses a limited number of amplification cycles so that high target concentrations do not reach saturation. This limits sensitivity because low concentration targets do not generate sufficient product to be detected. The qualitative test uses a large number of thermal cycles to maximize sensitivity. A quantitative assay could use a high number of amplification cycles to maximize sensitivity, but then high concentration samples would need to be diluted before testing to achieve accurate quantitation. Because viral load cannot always be estimated ahead of time, this approach would require that some samples be tested twice. At present, the most efficient strategy is to use a reasonably sensitive quantitative test for monitoring viral load and a highly sensitive qualitative test for diagnosis and test-of-cure.
A quantitative test that matches the sensitivity of the qualitative test while accurately quantitating high viral loads would be advantageous. Some chronic HCV patients have viral loads below 1000 copies/ml, 4, 7 and titers fall below this level in a substantial number of patients receiving interferon or combination interferon-ribavirin therapy. 9, 20 As has proven true for treatment of HIV-1, the likelihood of achieving a sustained response to therapy may increase with decreasing viral load, making it important to follow HCV-infected patients with the most sensitive test available.
Homogeneous reverse transcription-polymerase chain reaction (RT-PCR) tests based on the TaqMan 5′-nuclease assay format generate a signal by cleaving a target-specific probe during amplification. 21, 22 The probe contains a reporter fluorophore and a quencher dye that absorbs light emitted by the reporter. Cleavage of the probe physically separates the quencher from the reporter, enabling light emitted by the latter to be detected by a photomultiplier tube. Because amplification and detection are performed simultaneously, amplification products are measured during the exponential phase of DNA amplification regardless of the initial target concentration. Consequently, such tests combine exquisite sensitivity with an extremely broad dynamic range. In addition, they generate results more rapidly than the current heterogeneous test formats by eliminating the need to detect products after amplification is completed. Also, they eliminate the risk of carry-over contamination (ie, contamination with previously amplified products), because both amplification and detection are performed in a sealed reaction tube that is discarded at the end of the test. Furthermore, these assays yield quantitative data without requiring any extra effort by laboratory personnel. Thus, separate quantitative and qualitative assay formats will no longer be needed to achieve both sensitivity and dynamic range.
Indeed, several recently described TaqMan-based RT-PCR tests exhibit good sensitivity 23, 24, 25 and broad dynamic range. 23, 25, 26 We have developed a homogeneous format RT-PCR test for HCV, the TaqMan HCV quantitative test, that offers several advantages over the previously described assays. First, the test contains an internal control (IC), 27 which is added to each test sample before processing to monitor the integrity of sample processing and amplification and thereby eliminate false negative results. The IC is an RNA transcript with primer regions identical to those of the HCV target and a unique probe region. Second, the TaqMan HCV quantitative test uses standards calibrated in, and thus reports results in, international units (IU)/ml. The IU is defined by a standard solution prepared from a clinical specimen that, by international agreement, is defined as containing 100,000 IU/ml. 28 As other test developers adopt the IU/ml as a unit of measurement, comparisons of assay sensitivity, dynamic range, and precision will be possible.
We evaluated the sensitivity, genotype reactivity, specificity, linear range, reproducibility, and precision of the TaqMan HCV test. We also evaluated the ability of the test to monitor changes in viral load in patients receiving interferon therapy.
Materials and Methods
Clinical Specimens
A panel of three HCV RNA-positive samples with HCV titers ranging from 4000 to 1,600,000 IU/ml was used to assess test precision. A set of 100 anti-HCV-negative plasma samples obtained from the Salzburg Blood Center (Salzburg, Austria) was used to evaluate test specificity. To demonstrate the test’s ability to monitor changes in viral load, serial samples from three patients with chronic HCV were tested. These patients had been treated with 5 million units of interferon (IFN)-α 3 times per week. For all samples, serum was separated from whole blood within 4 days of collection, aliquoted, and stored at or below −20°C.
RNA Transcripts
HCV RNA transcripts representing HCV genotypes 1a, 1b, 2a, 2b, 3a, 4, and 5 were generated by transcribing cloned HCV plasmid DNA and were purified by digestion with RNase-free DNase, extraction with phenol-chloroform, and chromatography on oligo-dT cellulose columns. The purified transcripts were quantified by measuring absorbance at 260 nm and were diluted to 100,000 copies/ml.
Microorganisms for Demonstrating Specificity
With the exception of the HIV and hepatitis B virus (HBV) samples and three bacterial isolates, all samples were obtained from the American Type Culture Collection (Manassas, VA). The HIV samples were high titer members of a cultured HIV-1 subtype panel consisting of purified viral particles in culture supernatant (provided by Prof. Gürtler, Ludwig Maximilian University, Munich, Germany); the viral titers of the panel members were determined by the COBAS AMPLICOR HIV-1 MONITOR Test, Version 1.5 (Roche Molecular Systems). The HBV sample was the EUROHEP HBV standard no.1, genotype A (adw2) with 8 × 108 copies/ml (supplied by Dr. K.-H. Heermann, Georg-August University, Goettingen, Germany). The Staphylococcus epidermis, S. aureus, and Propionibacterium acnes bacterial isolates were provided by the Roche Diagnostics Culture Collection Group (Penzberg, Germany).
Standardized Samples for Determining Limit of Detection
The WHO International Standard for HCV (96/790), which has an assigned concentration of 105 IU/ml, 28 was serially diluted in negative human plasma to a final concentration ranging from 12,000 to 1.5 IU/ml. A genotype 1b HCV RNA transcript was quantified by serially diluting the stock solution, performing multiple amplifications with the qualitative COBAS AMPLICOR HCV Test, and calculating the concentration from the fraction of negative reactions by application of Poisson’s law. 27, 29 The stock solution was serially diluted with HCV Specimen Diluent (Roche Molecular Systems) to concentrations of 800, 400, 200, 100, 50, 25, 12.5, 6.3, and 3.1 copies/ml, where a copy corresponds to an amplifiable molecule. Fifty-microliter aliquots of each dilution (equivalent to 20, 10, 5, 2.5, 1.25, 0.625, 0.3, 0.15, and 0.075 copies/PCR test) were amplified and detected.
An external HCV RNA quantitation standard with a nominal concentration of 8.3 × 106 IU/ml (as measured by the TaqMan HCV test) was serially diluted in anti-HCV negative human plasma to determine the linear range of the test.
Specimen Preparation
HCV viral RNA was extracted from 200 μl of serum or plasma with 400 μl of working HCV lysis buffer (68% w/v guanidine thiocyanate, 3% w/v dithiothreitol, <1% w/v glycogen in a Tris-HCl buffer) containing a known number of IC RNA molecules. After a 10-minute incubation at 60°C, the RNA was precipitated with isopropanol (600 μl), recovered by microcentrifugation at maximum speed (at least 12,500 × g) for 15 minutes at room temperature, washed with 1 ml 70% ethanol, and resuspended in 200 μl of HCV Specimen Diluent. Fifty microliters of the processed specimen were added to Working HCV Master Mix (prepared by adding HCV manganese solution to HCV Master Mix, Roche Molecular Systems) for the RT-PCR amplification reactions. The amplification reaction contained the HCV RNA recovered from 50 μl of serum or plasma.
Reverse Transcription, Amplification, and Detection
The test amplifies a 250-base target sequence that is defined by primers ST280 and ST778 and is located in a highly conserved 5′ untranslated region of the HCV genome. 30 DNA replication is detected with a dual-labeled fluorogenic hybridization probe that specifically anneals to the target DNA between the PCR primers. The primer sequences and the probe sequence are located in the most conserved domains within the 5′-untranslated region. 31 The probe contains a quencher and a fluorescent reporter, 6-carboxyfluorescein (FAM) for the HCV-specific probe and hexachloro-6-carboxyfluorescein (HEX) for the IC-specific probe. In the intact probe, the quencher absorbs fluorescence emitted by the reporter. The 5′ nuclease activity of the polymerase degrades the hybridization probe during replication, thereby releasing the reporter and producing an increase in fluorescent emission.
The reverse transcription and amplification reactions are performed with a modified version of the thermostable recombinant Thermus species ZO5 DNA polymerase, which was cloned, engineered, and expressed from thermophilic bacteria 32 using methods similar to those described previously. 33 In the presence of manganese and under the appropriate buffer conditions, ZO5 DNA polymerase has both reverse transcriptase and DNA polymerase activity, allowing both reverse transcription and PCR amplification to occur in the same reaction mixture. The Working HCV Master Mix is a tricine buffered solution containing glycerol, dimethylsulfoxide, potassium acetate, deoxynucleoside triphosphates (dNTPs including dATP, dCTP, dGTP, and dUTP), primer oligonucelotides, HCV-specific and IC-specific dual, fluorescently labeled probe oligonucleotides, ZO5 DNA polymerase, manganese acetate, AmpErase, ROX-labeled reference oligonucelotide (PE Corp., Norwalk, CT), and sodium azide.
Fifty microliters of extracted serum RNA or diluted standard RNA were added to MicroAmp Reaction Plate (PE Corp.) tubes containing 50 μl of reaction mix. The samples were amplified and detected with a PE Applied Biosystems Prism 7700 Sequence Detection System (PE Corp.) using the following thermal cycling parameters: 50°C for 4 minutes (inactivation of any previously amplified DNA by AmpErase), 30 minutes at 61°C (reverse transcription), 2 PCR cycles of 15 seconds at 95°C and 50 seconds at 58°C, and 48 PCR cycles of 15 seconds at 91°C and 50 seconds at 58°C. The ABI Prism 7700 Sequence Detector System measured fluorescent emissions, which increase in direct proportion to the increase of amplified product, continuously during the PCR amplification.
HCV RNA Quantitation
HCV viral RNA was quantitated by using a set of five external HCV RNA quantitation standards, which have been calibrated against the HCV WHO Standard 96/790 and assigned concentration values in IU/ml. The standards are non-infectious, 639 nucleotides in length, in vitro-transcribed RNA molecules that contain the 5′ non-coding region of HCV and have primer and probe binding sites identical to those of the HCV target sequence. These standards are reverse transcribed, amplified, and detected in separate reactions that are run in parallel with the samples being tested. The IC could not be used as a quantitation standard because the Prism 7700 software is not designed to calculate concentration by comparing the signals generated by the target and an internal standard.
The five standards cover a range of 4 logs to enable generation of a standard curve by the ABI Prism 7700 Sequence Detection System over approximately 1 × 103 to 1 × 107 IU/ml. The standard curve is generated automatically by plotting the threshold cycle (CT, first cycle with a reporter fluorescence above the baseline) versus log10(N), where N is the initial concentration of the standard in IU/ml, and calculating the best-fit line by linear regression analysis. The HCV RNA level in each specimen is determined by locating its CT on the standard curve. Samples that had a CT corresponding to a concentration below 103 IU/ml were reported as “HCV RNA detected but below 103 IU/ml”. Samples that had a CT corresponding to a concentration above 107 IU/ml were reported as “HCV RNA detected, greater than 107 IU/ml”. Samples that had a CT corresponding to a concentration below 40 IU/ml were reported as “HCV RNA not detected”, provided the IC gave a positive result. Any samples with no detectable HCV RNA concentration and a negative IC result were reported as invalid.
Results
Limit of Detection
The limit of detection was determined using two serial dilution series prepared in HCV-negative plasma, one prepared from the WHO International Standard for HCV (96/790) and one prepared from a genotype 1b transcript. The WHO International Standard samples and the genotype 1b transcripts yielded positive results in 100% of replicate reactions at concentrations greater than or equal to 47 IU/ml and 100 copies/ml, respectively (Table 1) . Below these concentrations, the detection failure rate increased as the concentration decreased (Table 1) . A detailed probit analysis indicated that the limit of detection (the concentration at which 95% or more of the replicates tested gave a positive response) of the test was 28 IU/ml for the WHO International Standard and 57 copies/ml for the genotype 1b transcript. A concentration of 57 copies/ml corresponds to 2.9 copies/amplification reaction. This result is consistent with Poisson statistics, which predicts that 5.8% of replicate reactions will yield negative results for a mean input concentration of 2.9 copies/ml. This agreement is not surprising, since transcript copy number was determined by limiting dilution and is thus a measure of amplifiable molecules (see Materials and Methods).
Table 1.
Limit of Detection
| WHO International Standard (96/790) | Genotype 1b RNA transcript | ||
|---|---|---|---|
| Actual input (IU/ml) | % positive (number positive) | Actual input (copies/ml)* | % positive (number positive) |
| 188 | 100.0 (8) | 400 | 100.0 (8) |
| 94 | 100.0 (8) | 200 | 100.0 (8) |
| 47 | 100.0 (8) | 100 | 100.0 (8) |
| 24 | 87.5 (7) | 50 | 87.5 (7) |
| 12 | 75.0 (6) | 25 | 50.0 (4) |
| 6 | 37.5 (3) | 12.5 | 37.5 (3) |
| 3 | 12.5 (1) | 6.3 | 12.5 (1) |
| 1.5 | <12.5 (0) | 3.1 | <12.5 (0) |
Eight replicate tests were performed for each dilution of the WHO International Standard for HCV (96/790) and a genotype 1b transcript. A test result was considered positive if the HCV-specific signal had a CT < 48. A sample was considered negative if the HCV-specific signal had a CT ≥ 48 and the IC-specific signal had a CT < 48.
The input concentration of the transcript was determined by limiting dilution (see Materials and Methods).
Concentrations above 188 IU/ml and 400 copies/ml also yielded positive results for all eight replicates.
HCV Genotype Reactivity
HCV transcripts representing genotypes 1a, 1b, 2a, 2b, 3a, 4, and 5 were quantified by OD260 measurement and diluted in HCV-negative plasma so that each was tested at a concentration of 100,000 copies/ml. Four replicates of each genotype were tested with the TaqMan HCV quantitative test. Each genotype yielded positive results (CT < 48). Furthermore, all of the genotypes tested were amplified with comparable efficiency because average CT values differed by less than one amplification cycle, which corresponds to a coefficient of variation (CV) of 1.56% (Figure 1) .
Figure 1.

Efficiency of amplification of different HCV genotypes. HCV transcripts representing genotypes 1a, 1b, 2a, 2b, 3a, 4, and 5 were diluted to 100,000 copies/ml and tested with the TaqMan HCV quantitative Test. The CT for four replicates of each genotype were averaged to determine the mean CT and SD.
Specificity
The analytical specificity of the TaqMan HCV test was demonstrated by analyzing microbes and viruses that could potentially co-infect HCV-positive individuals as well as skin flora that could contaminate blood during collection. The organisms were tested at an approximate concentration of 105 organisms/ml (equivalent to 5 × 103 organisms/PCR) in normal human plasma. All of the following organisms gave positive IC signals and negative HCV signals (CT of 48): adenovirus types 2, 3, and 7; Chlamydia trachomatis; coxsackievirus B1; cytomegalovirus; echovirus 1; hepatitis A; hepatitis B; herpes simplex types 1 and 2; human herpesvirus 6; HIV-1, Group M subtypes A-E and G and Group O; P. acnes; S. aureus; S. epidermis; and varicella zoster (data not shown).
In addition, cross-reactivity was assessed by testing a set of 100 samples obtained from HCV-seronegative blood donors. Four of the specimens gave invalid results (the IC had a CT of 48). All but one of the remaining 96 samples gave positive IC signals and negative HCV signals (CT of 48) for a specificity of 99% (data not shown). This specimen gave a weak positive result (<100 IU/ml) when initially tested and yielded a negative result when retested. It was also negative when tested in the qualitative COBAS AMPLICOR HCV, Version 2.0 test. These observations suggest that the original test aliquot inadvertently became cross-contaminated with one of the external standards during loading of samples into the amplification tray wells.
Linear Range
The linear range was determined by analysis of two identical dilution series that were independently prepared in HCV-negative plasma from one of the external HCV RNA quantitation standards, which had a nominal concentration of 8.3 × 106 IU/ml. The quantitation standard was diluted with HCV-negative plasma to generate a set of samples with concentrations ranging from 64 to 4,180,000 IU/ml plasma. Each dilution series was tested in duplicate. For each dilution, the log10 measured concentration was calculated for both replicates and averaged to determine the mean log10, which was compared to the log10 input concentration. Linear regression analysis showed that the linear range of the test was 64 to 4,180,000 IU/ml. Both dilution series yielded similar regression lines: the slopes were 0.997 and 0.913, the intercepts were 0.044 and 0.436, and the R2 values were 0.993 and 0.997, respectively (Figure 2) . Because the slopes approached 1.0 and the intercept approached 0.0, the measured concentration within the linear range was, on average, close to the nominal concentration. For each dilution series, the mean log10 measured concentration was within 8% of the log10 nominal concentration with the exception of the 128 and 255 IU/ml dilutions in series 2, which were within 15% and 12% of the nominal values, respectively. When the log10 values from both dilution series were averaged together, the overall mean log10 measured concentration was within 6% of the nominal concentration with the exception of the 255 IU/ml dilution, which was within 9% of the nominal value.
Figure 2.

Linear range for the TaqMan HCV test. Two independent dilution series were prepared by diluting an external HCV RNA quantitation standard in HCV-negative plasma. Each dilution in each series was tested in duplicate. At each input, RNA concentration, the mean of the log10 calculated RNA concentrations (circles and squares), and the SD were determined. Linear regression analysis was performed on the mean log10 values for each dilution series (solid and dashed lines and associated equations).
Precision Study
A panel of three clinical plasma samples was tested each day over a 10-day period. Each day, the samples were tested on two different instruments. For each instrument run, three replicate aliquots of each sample were carried though extraction, amplification, and detection. Thus, the precision reported here includes the variability contributed by all of the test processes. Eighteen tests (3 replicates per sample per instrument × 3 samples × 2 instruments) were performed each day, for a total of 180 tests over the 10-day period. Each instrument run also included four replicates of each of the five RNA quantitation standards and one negative control. All 80 negative control tests gave negative results. Both instruments exhibited similar within-run precision; the individual daily within-run CVs ranged from 1.3 to 47.0% for the first instrument and 2.6 to 43.9% for the second instrument over the 10 days (data not shown). Most of the variability was due to variation between replicates, with the within-run CVs ranging from 17.0 to 26.6% (Table 2) . The total variation, which includes between-run and between-day variation, was only slightly larger, with CVs ranging from 21.6 to 30.4% (Table 2) .
Table 2.
Precision of the TaqMan HCV Test
| Variable | Input IU/ml | ||
|---|---|---|---|
| 4000 | 80,000 | 1,600,000 | |
| Total number of replicates | 60 | 60 | 60 |
| Mean calculated RNA conc. (IU/mL) | 3,101 | 98,199 | 3,010,026 |
| Minimum value | 1,325 | 46,611 | 1,127,390 |
| Maximum value | 5,600 | 155,064 | 4,644,024 |
| Within-run | |||
| Standard deviation | 926 | 18,087 | 511,429 |
| Coefficient of variation | 26.6% | 18.4% | 17.0% |
| Total | |||
| Standard deviation | 941 | 22,928 | 649,456 |
| Coefficient of variation | 30.4% | 23.4% | 21.6% |
These calculations was performed as described in the National Committee for Clinical Laboratory Standards guideline EP5-T2 for within-run and total precision. Total precision includes between-run (instrument) variation and between-day variation.
The minimum measured concentrations differed from the nominal concentration by factors of 0.33, 0.58, and 0.70 for the 4000, 80,000, and 1,600,000 IU/ml samples, respectively (Table 2) . The maximum measured concentrations differed from the nominal concentration by factors of 1.40, 1.94, and 2.90 for the 4000, 80,000, and 1,600,000 IU/ml samples, respectively (Table 2) . These results demonstrate that a particular test result will estimate the actual concentration within a factor of 3 (0.5 log10). Furthermore, an overall CV of 30% implies that results that differ by 90%, corresponding to an approximately twofold (0.3 log10) change in viral titer, are statistically significant (P = 0.005).
Quantitation of HCV RNA in Clinical Specimens
The ability of the TaqMan HCV test to monitor changes in HCV RNA titer was assessed in sera from three patients receiving interferon therapy. The first patient initially responded to interferon and then relapsed (Figure 3A) . The viral load decreased 6,500,000 IU/ml to 30 IU/ml during first 5 months of IFN-α treatment. The load increased to 3,200,000 between months 5 and 8 of treatment and then decreased to 680 IU/ml, at which time therapy was terminated. The viral load rebounded to pretreatment levels after cessation of therapy. These changes paralleled changes in alanine amino-transferase (ALT) concentration.
Figure 3.
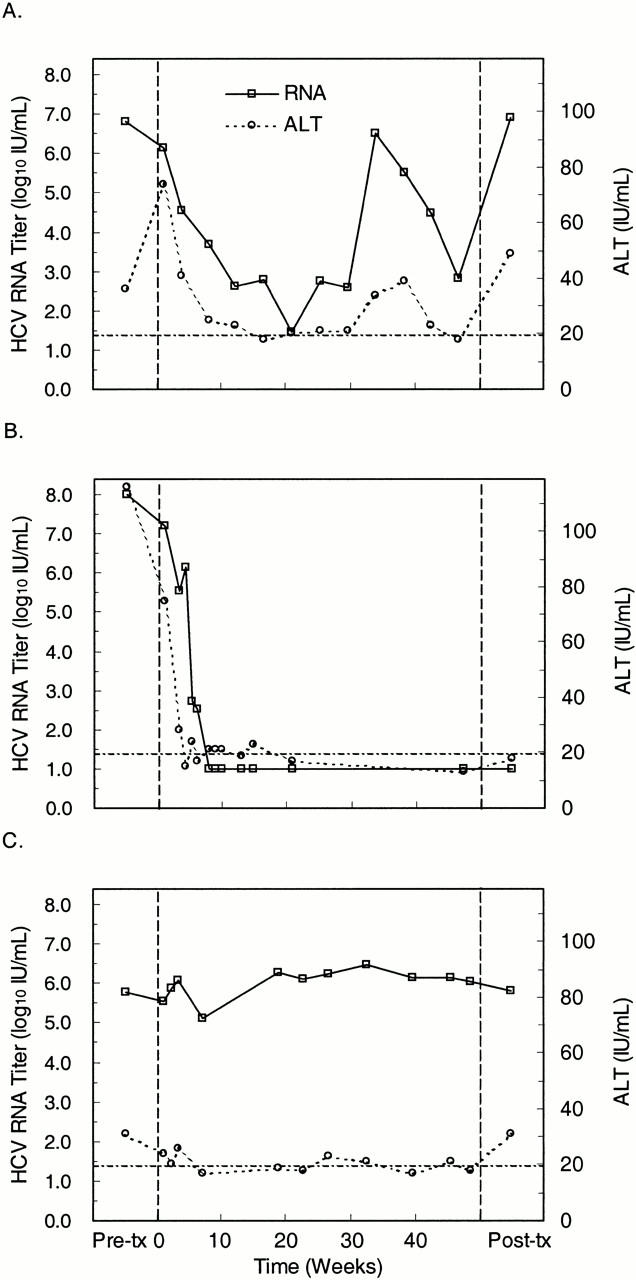
Quantitation of HCV RNA (left axis) and serum ALT (right axis) levels in a responder-relapser (A), responder (B), and nonresponder (C) to interferon therapy. Baseline HCV RNA and ALT levels (first data point) were measured 12 (A), 4 (B), and 65 (C) weeks before the start of interferon therapy (day 0, vertical dashed line). RNA and ALT levels were periodically monitored starting at 1 week after initiating therapy (second data point). The posttreatment RNA levels (last data point) were measured at 25 (A), 4 (B) or 38 (C) weeks after completion of interferon therapy at week 48 (vertical dashed line). The dotted-dashed line indicates both the limit of detection for the HCV RNA test and the upper limit of the normal ALT range. Samples that tested negative for HCV RNA were assigned an arbitrary value (10 IU/ml) below the limit of detection.
The second patient exhibited a complete virological and biochemical response. Viral load steadily decreased, ultimately dropping below the limit of detection and remaining undetectable after termination of therapy (Figure 3B) . Again, the change in viral load paralleled the change in ALT concentration.
The third patient was a nonresponder whose viral load varied minimally during the course of treatment (Figure 3C) . This patient had a slightly elevated ALT level at baseline that decreased during therapy and returned to the baseline level after therapy was terminated. Thus, for this patient, viral load provided a better indication of non-responsiveness to interferon therapy.
The patterns of viral load changes revealed by the TaqMan HCV quantitative test in these patients are similar to those seen when interferon-treated patients are monitored with heterogeneous format tests such as COBAS AMPLICOR HCV MONITOR. This, suggests that the TaqMan HCV quantitative test reliably monitors viral load changes.
Discussion
The TaqMan HCV quantitative test provides a rapid, extremely sensitive, specific method for measuring HCV viral load over a very broad dynamic range. Results are available within 2.5 hours of initiating amplification instead of the 4 to 6 hours for heterogeneous format tests, in which products are quantified in a separate detection assay performed on completion of amplification. The 28 IU/ml limit of detection for TaqMan HCV quantitative test exceeds the 50 IU/ml limit of detection for the qualitative COBAS AMPLICOR HCV Test, Version 2.0, which is currently the most sensitive, commercially available, heterogeneous format test 18 (Lee et al, manuscript submitted). The TaqMan HCV quantitative test exhibited a linear response over a 65,000-fold (4.8 log10) concentration range; in contrast, the quantitative COBAS AMPLICOR HCV MONITOR Test, Version 2.0 is linear over a 1400-fold (3.2 log10) concentration range (Lee et al, manuscript submitted). These improved performance characteristics are a direct consequence of the assay’s homogeneous format, in which amplification products are detected as they are formed (see Introduction). Furthermore, high sensitivity was achieved without compromising specificity; the test exhibited a specificity of 99% when performed on HCV seronegative blood donors and did not cross-react with other organisms that could co-infect HCV-infected individuals.
The TaqMan HCV quantitative test appeared to be more sensitive than other recently described homogeneous format HCV PCR tests, reliably detecting (95% probability of a positive result) a synthetic RNA transcript at a concentration of 57 copies/ml, which is equivalent to 2.9 copies/amplification reaction. The other tests detected synthetic RNA transcripts at 330 copies/ml, 23 2000 copies/ml, 26 and 10 copies/amplification reaction. 25 One test reported a 50 copy/ml detection limit, but did not specify whether RNA transcripts or clinical samples were used and did not show any data. 24 These comparisons must be considered indirect for two reasons. First, none of the other studies demonstrated the probability of a positive result was at least 95% at the stated limit of detection. Second, each study used a different method, ie, endpoint titration (this study), radiolabeling, 23 or A260, 25, 26 to determine the concentration of the synthetic HCV RNA transcripts used to assess sensitivity. Unfortunately, we cannot compare sensitivities in IU/ml because the other studies did not evaluate sensitivity using the WHO standard or another standard calibrated in IU/ml. The specificity of the TaqMan HCV quantitative test was similar to the value reported for two of the other homogeneous format tests, 99.3% (272/274) 24 and 100% (50/50). 25 The remaining two studies did not evaluate specificity. 23, 26
The 4.8 log10 dynamic range of the TaqMan HCV quantitative test was comparable to that of the other homogeneous format tests, which were linear over a 5 log10 23, 26 and 7 log10 25 range; linearity was not evaluated for one of the assays. 24 It is possible that the TaqMan HCV quantitative test would have exhibited an even broader dynamic range had we included higher concentration samples in the evaluation. The broad dynamic range makes these tests well suited for managing patients throughout the course of treatment. The ability to discriminate between high and low baseline viral titers (with a cutoff of approximately 105 to 106 copies/ml) is required to assess the likelihood that a patient will respond to antiviral therapy. 4, 6, 7, 8 At the same time, a very sensitive test is needed to assess the response to therapy, because viral load drops dramatically in those who respond to treatment. 4, 7, 9, 20
The TaqMan HCV quantitative test should accurately measure viral load regardless of the infecting genotype, since it reacted equally well with RNA transcripts representing HCV genotypes 1a, 1b, 2a, 2b, 3a, 4, and 5. One of the recently described homogenous format HCV assays reacted equally well with genotypes 1a, 1b, 2a, and 2b transcripts, 25 whereas another tended to underestimate viral load in patients infected with genotypes 2 and 4. 23 The remaining two tests detected all genotypes that were tested, but the accuracy of the viral load measurement was not evaluated. 24, 26 It will be important to evaluate the genotype reactivity of the TaqMan HCV quantitative test on clinical specimens, as short, synthetic RNA transcripts may behave differently than full-length viral RNA. 23, 34
The TaqMan HCV qualitative test exhibited good reproducibility, having total CVs ranging from 21.6 to 30.4%, with lower titer specimens exhibiting higher values. Given this level of variation, a greater than threefold (0.5 log10) difference between two samples will be statistically significant (P < 0.005). The other recently described homogeneous format HCV tests exhibited similar reproducibility. The assay developed by Mercier et al 24 exhibited CVs ranging from 8.7 to 74.7% (as calculated from the results in Table 1 of Ref. 24 ). Kawai et al did not evaluate assay precision. 26 Takeuchi et al 25 calculated assay variation from log-transformed titers (although they calculated mean values from the nontransformed titers), obtaining between-run CVs between 0.9 and 4.7% (see Table 4 of Ref. 25 ). The corresponding between-run CVs calculated from the nontransformed titers reported in Table 4 25 ranged from 13.8 to 45.9%, which is similar to the overall CVs reported here. Martell et al reported a between-run CV of 6.2% based on the variation of the CT values. 23 Because CT values are proportional to the log of the viral RNA titer, this is equivalent to calculating the variance from log-transformed data. Thus, the Martell et al assay exhibited slightly higher between-run variation than the Takeuchi et al assay. The overall variation exhibited by these two assays may be greater than the between-run variation. Because their experimental design and method for calculating variances were not specified, we do not know whether the between-run variation for these assays includes within-run variation. If it does not, the overall variation would be higher, because it would include the between-run variation and the within-run variation, which is similar in magnitude to the between-run variation (see Table 3 of Ref. 25 ).
The TaqMan HCV quantitative test detected changes in viral load during IFN therapy that paralleled changes in serum ALT levels, as did one of the other homogeneous format HCV tests. 25 Similar viral load responses have been documented with heterogeneous format tests. Thus, a TaqMan-based quantitative test for HCV could provide a more rapid and sensitive alternative for monitoring patients receiving antiviral therapy.
Like the other homogeneous format tests described to date, the TaqMan HCV quantitative test utilizes external standards to construct a standard curve that is used to calculate viral titers in test samples. This approach has two limitations. First, the concentration range of the external standards, which range from 103 to 107 IU/ml, limits the dynamic range. Even though we demonstrated that the TaqMan HCV quantitative test exhibits a linear response for concentrations as low as 64 IU/ml, samples giving a positive result below 103 IU/ml are reported as “HCV RNA detected but below 103 IU/ml” in routine use. Likewise, samples giving a result above 107 IU/ml are reported as “HCV RNA detected, greater than 107 IU/ml.”
The second limitation is that the test does not control for sample-to-sample differences in sample processing and amplification efficiency. The IC detects samples in which sample preparation or amplification fails, thus preventing false negative results. But if amplification is partly inhibited or there is a partial loss of nucleic acid during sample processing, the CT for the sample will be higher than it would have been under ideal conditions and will consequently yield an artificially low RNA reading on the standard curve. This problem can be avoided by using the IC as an internal quantitation standard and comparing the CT of target to the CT of the internal standard. The COBAS TaqMan HCV assay now being developed by Roche Molecular Systems utilizes this format, which further increases assay throughput by eliminating the need to run external standards.
In summary, the TaqMan quantitative HCV test offers better sensitivity, greater dynamic range, and higher throughput than heterogeneous format tests. It is more sensitive than other homogeneous format tests, and at least as reproducible. The TaqMan HCV quantitative test is a prototype test and is not being commercialized. Instead, the technology will be enhanced and further standardized, ultimately resulting in the COBAS TaqMan HCV test. Given the properties of the prototype test, the COBAS TaqMan HCV test should prove useful for prognosis and for monitoring the efficacy of antiviral treatments.
Acknowledgments
For assistance we thank Monika Auer, Gordan Graf, Martina Klemmer, and Bettina Reithmeier of Roche Molecular Systems. For advice we thank Christoph Berding and Peter Ebert of Roche Molecular Systems. For samples we thank Maria Martell, Silvia Sauleda, and Juan Esteban from Hospital General Universitari Vall d’Hebron, Barcelona, Spain. For helpful suggestions and continuing support of this work we thank Thomas White, Elizabeth Dragon, and Knut Bartl of Roche Molecular Systems.
Address reprint requests to Dr. Maurice J. Rosenstraus, Roche Molecular Systems, 1080 Route 202, Somerville, NJ 08876. E-mail: maurice.rosenstraus@roche.com.
References
- 1.Berenguer M, Wright TL: Hepatitis C and liver transplantation. Gut 1999, 45:159-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charlton M, Seaberg E, Wiesner R, Everhart J, Zetterman R, Lake J, Detre K, Hoofnagle J: Predictors of patient and graft survival following liver transplantation for hepatitis C. Hepatology 1998, 28:823-830 [DOI] [PubMed] [Google Scholar]
- 3.Fried MW: Clinical applications of hepatitis C virus genotyping and quantitation. Clinics Liver Dis 1997, 1:631-645 [DOI] [PubMed] [Google Scholar]
- 4.Ichijo T, Matsumoto A, Kobayashi M, Furihata K, Tanaka E: Quantitative measurement of HCV RNA in the serum: a comparison of three assays based on different principles. J Gastroenterol Hepatol 1997, 12:500-506 [DOI] [PubMed] [Google Scholar]
- 5.: NIH Consensus Statement Online: Management of Hepatitis C. 1997, 15 (http://odp.od.nih.gov/consensus/cons/105/105_statement.htm):1-41 [PubMed] [Google Scholar]
- 6.Reichard O, Norkrans G, Fryden A, Braconier JH, Sonnerborg A, Weiland O: Comparison of 3 quantitative HCV RNA assays: accuracy of baseline viral load to predict treatment outcome in chronic hepatitis C. Scand J Infect Dis 1998, 30:441-446 [DOI] [PubMed] [Google Scholar]
- 7.Shiratori Y, Kato N, Yokosuka O, Hashimoto E, Hayashi N, Nakamura A, Asada M, Kuroda H, Ohkubo H, Arakawa Y, Iwama A, Omata M: Quantitative assays for hepatitis C virus in serum as predictors of the long-term response to interferon. J Hepatol 1997, 27:437-444 [DOI] [PubMed] [Google Scholar]
- 8.Soffredini R, Rumi MG, Del Ninno E, Parravicini ML, Russo A, Colombo M: Serum levels of hepatitis C virus RNA predict non-response to interferon therapy: comparison of two commercial assays. J Viral Hepatitis 1999, 6:65-71 [DOI] [PubMed] [Google Scholar]
- 9.Zeuzem S, Lee JH, Franke A, Ruster B, Prummer O, Herrmann G, Roth WK: Quantification of the initial decline of serum hepatitis C virus RNA and response to interferon alfa. Hepatology 1998, 27:1149-1156 [DOI] [PubMed] [Google Scholar]
- 10.Rosenstraus M, Gutekunst K, Dale B: Utility of hepatitis virus nucleic acid assays in therapeutic drug trials. Schinazi RF Sommadossi JP Thomas HC eds. Therapies for Viral Hepatitis. 1998, :115-127 International Medical Press Ltd. London [Google Scholar]
- 11.Bonetti P, Chemello L, Antona C, Breda A, Brosolo P, Casarin P, Crivellaro C, Dona G, Martinelli S, Rinaldi R, Zennaro V, Santonastaso M, Urban F, Pontisso P, Alberti A: Treatment of chronic hepatitis C with interferon-alpha by monitoring the response according to viraemia. J Viral Hepat 1997, 4:107-112 [DOI] [PubMed] [Google Scholar]
- 12.Tong MJ, Blatt LM, Tong LT, Sayadzadeh K, Conrad A: Long-term retreatment in chronic hepatitis C patients who were non-responders to an initial course of interferon-alpha 2b. J Viral Hepat 1998, 5:323-331 [DOI] [PubMed] [Google Scholar]
- 13.Poynard T, McHutchinson J, Goodman Z, Ling MH, Albrecht J, : the ALGOVIRC Project Group: Is an “a la carte” combination interferon alpha-2b plus ribavirin regimen possible for the first line treatment in patients with chronic hepatitis C? Hepatology 2000, 31:211-218 [DOI] [PubMed] [Google Scholar]
- 14.Dhumeaux D, Doffoel M, Galmiche JP: A French consensus conference on hepatitis C: screening and treatment. J Hepatol 1997, 27:941-944 [DOI] [PubMed] [Google Scholar]
- 15.Pawlotsky JM, Lonjon I, Hezode C, Raynard B, Darthuy F, Remire J, Soussy CJ, Dhumeaux D: What strategy should be used for diagnosis of hepatitis C virus infection in clinical laboratories? Hepatology 1998, 27:1700-1702 [DOI] [PubMed] [Google Scholar]
- 16.Schneeberger PM, Keur I, van der Vliet W, van Hoek K, Boswijk H, van Loon AM, van Dijk WC, Kauffmann RH, Quint W, van Doorn LJ: Hepatitis C virus infections in dialysis centers in the Netherlands: a national survey by serological and molecular methods. J Clin Microbiol 1998, 36:1711-1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau DTY, Kleiner DE, Ghany MG, Park Y, Schmid P, Hoofnagle JH: 10-Year follow-up after interferon-alpha therapy for chronic hepatitis C. Hepatology 1998, 28:1121-1127 [DOI] [PubMed] [Google Scholar]
- 18.Doglio A, Laffont C, Caroli-Bosc FX, Rochet P, Lefebvre J: Second generation of the automated Cobas Amplicor HCV assay improves sensitivity of hepatitis C virus RNA detection and yields results that are more clinically relevant. J Clin Microbiol 1999, 37:1567-1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lelie PN, Cuypers HTM, van Drimmelen AAJ, Quint WGV: Quality assessment of hepatitis C virus nucleic acid amplification methods. Infusionsther Transfusionsmed 1998, 25:102-110 [Google Scholar]
- 20.Yasui K, Okanoue T, Murakami Y, Itoh Y, Minami M, Sakamoto S, Sakamoto M, Nishioji K: Dynamics of hepatitis C viremia following interferon-alpha administration. J Infect Dis 1998, 177:1475-1479 [DOI] [PubMed] [Google Scholar]
- 21.Holland PM, Abramson RD, Watson R, Gelfand DH: Detection of specific polymerase chain reaction product by utilizing the 5′-3′ exonuclease activity of Thermus aquaticus. Proc Nat Acad Sci USA 1991, 88:7276-7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Flood SJA, Marmaro J, Giusti W, Deetz K: Oligonucleotides with fluourescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybrization. PCR Methods Applic 1995, 4:357-362 [DOI] [PubMed] [Google Scholar]
- 23.Martell M, Gomez J, Esteban JI, Sauleda S, Quer J, Cabot B, Esteban R, Guardia J: High-throughput real-time reverse transcription-PCR quantitation of hepatitis C virus RNA. J Clin Microbiol 1999, 37:327-332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercier B, Burlot L, Ferec C: Simultaneous screening for HBV DNA and HCV RNA genomes in blood donations using a novel TaqMan PCR assay. J Virol Methods 1999, 77:1-9 [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi T, Katsume A, Tanaka T, Abe A, Inoue K, Tsukiyama-Kohara K, Kawaguchi R, Tanaka S, Kohara M: Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology 1999, 116:636-642 [DOI] [PubMed] [Google Scholar]
- 26.Kawai S, Yokosuka O, Kanda T, Imazeki F, Maru Y, Saisho H: Quantification of hepatitis C virus by TaqMan PCR: comparison with HCV Amplicor Monitor assay. J Med Virol 1999, 58:121-126 [DOI] [PubMed] [Google Scholar]
- 27.Rosenstraus M, Wang Z, Chang SY, DeBonville D, Spadoro JP: An internal control for routine diagnostic PCR: design, properties and effect on clinical performance. J Clin Microbiol , 36:191-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saldanha J, Lelie N, Heath A, : WHO Collaborative Study Group: Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCV RNA. Vox Sang 1999, 76:149-158 [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Chang SY, MacMullen G, Huang D, Kwok S, Spadoro J: Use of internal control quantitative standards to quantify target nucleic acid sequences by PCR. Program and abstracts of the American Association for Clinical Chemistry San Diego Conference: The Genetic Revolution. 17–19November1994. San Diego, CA
- 30.Young K, Resnick R, Myers T: Detection of hepatitis C virus RNA by a combined reverse-transcriptase-polymerase chain reaction assay. J Clin Microbiol 1993, 31:882-886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bukh J, Purcell RH, Miller RH: Sequence analysis of the 5′ noncoding region of hepatitis C virus. Proc Natl Acad Sci USA 1992, 89:4942-4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degryse E, Glansdorff N, Pierard A: A comparative analysis of extreme thermophilic bacteria belonging to the genus Thermus. Arch Microbiol 1978, 117:189-196 [DOI] [PubMed] [Google Scholar]
- 33.Lawyer FC, Stoffel S, Saiki RK, Chang SY, Landre PA, Abramson RD, Gelfand DH: High-level expression, purification, and enzymatic characterization of full-length thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity. PCR Methods Applic 1993, 2:275-287 [DOI] [PubMed] [Google Scholar]
- 34.Mellor J, Hawkins A, Simmonds P: Genotype dependence of hepatitis C virus load measurement in commercially available quantitative assays. J Clin Microbiol 1999, 37:2525-2532 [DOI] [PMC free article] [PubMed] [Google Scholar]