

Abstract
The fragile X syndrome is the most commonly inherited cause of mental retardation. Genetic diagnosis of this disease relies on the detection of triplet repeat expansion in the FMR1 gene on the X chromosome. Although the majority of disease in fragile X patients is due to mutations involving triplet repeat expansion, deletion of various portions of FMR1 has also been described in association with the fragile X syndrome. Here we describe a rare polymorphism in the noncoding region of FMR1 that mimics detection of a deletion in a commonly used assay for fragile X syndrome, which can result in misdiagnosis of the disease.
Fragile X mental retardation is the most common cause of inherited mental retardation, with an incidence of 1 in 4000 in males and 1 in 6000 in females. 1 This syndrome is characterized by mental retardation coupled with characteristic physical features, such as long facies, large ears, and macroorchidism. 2 At the molecular level, this disease is associated with expansion of a CGG repeat in the 5′ UTR of the FMR1 gene on the X chromosome. This expansion creates dynamic instability in the FMR1 gene, with the size of the repeat often increasing in size during female meiosis in succeeding generations. 3 When this expansion reaches a critical size, hypermethylation of both the repeat and the adjoining promoter region of FMR1 leads to the decreased transcription of FMR1. Although the vast majority of patients with fragile X disease show this pattern of expanded repeats, a small number of patients have been described in whom partial deletions of FMR1 result in a disease phenotype. 4
Molecular diagnosis of fragile X is based on the demonstration of expansion of the CGG repeat in patient samples. Traditionally, this diagnosis was made by cytogenetic detection of the fragile site after culture of patient cells in medium depleted of folic acid and thymidine. 5 After the discovery of the FMR1 gene and its role in disease, most clinical laboratories adopted DNA-based strategies to test for fragile X syndrome. These include methods based on both polymerase chain reaction (PCR) and Southern analysis of the FMR1 locus. In the genomic Southern hybridization method of direct mutation testing used in our laboratory, 6 genomic DNA isolated from peripheral white blood cells of the patient is digested with EcoRI and SacII (a methylation-sensitive enzyme). This digest is then probed with an FMR1 fragment (StB12.3) 6 to determine both the methylation status and relative length of the trinucleotide repeat. A normal male will show a single band of 2.8 kb, and a normal female will have an additional 5.2-kb band representing the inactive, methylated allele (Figure 1A) . Premutations (between 50 and 200 CGG repeats) and full mutations (>200 copies of the triplet) are detected by increases in the size of these bands.
Figure 1.
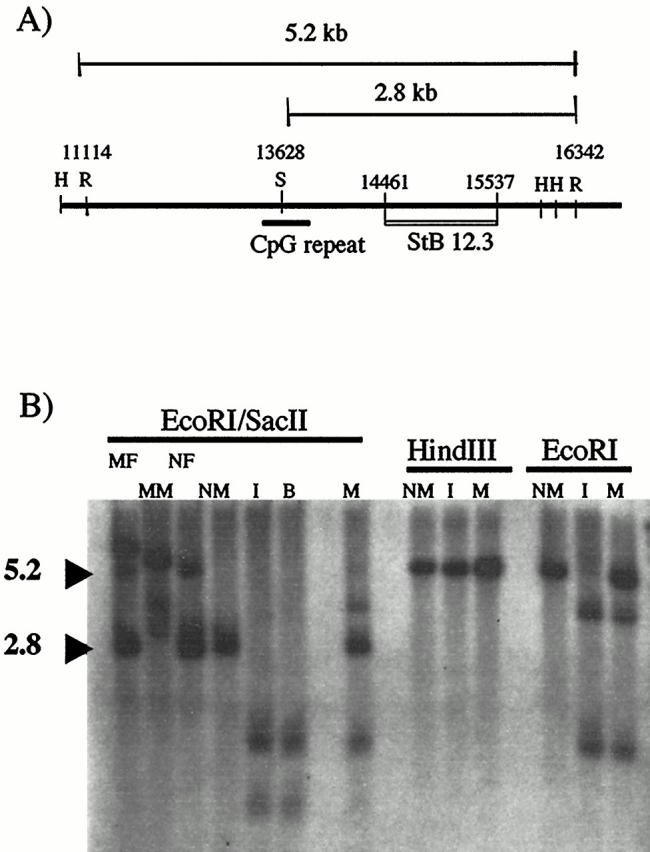
Southern analysis of index patient. A: Patient DNA is digested with EcoRI (R) and SacII (S), then probed with the cloned fragment StB12.3. Normal males generate a 2.8-kb SacII/EcoRI fragment, whereas normal females generate an additional 5.2-kb EcoRI/EcoRI fragment from the methylated allele. Additional restriction sites used in this paper (HindIII (H)) are shown. B: Lanes 1–4 show the patterns seen after digestion with EcoRI/SacII in control patients with full mutations (MM, mutant male; MF, mutant female) as compared to normal controls (NM, normal male; NF, normal female). Both the index patient (I) and his brother (B) show a loss of this normal pattern and the appearance of smaller bands. The mother (M) has a more complex pattern. Digestion with HindIII produces identical bands in a normal male, the index patient, and his mother. In contrast, digestion with EcoRI alone demonstrates the presence of a new EcoRI site in both the index patient and his mother, when compared to the normal male control.
In the course of clinical testing in our laboratory, we discovered a male patient who appeared to have a deletion of the FMR1 gene, as evidenced by the absence of the 2.8-kb band described in the above assay. Here we describe the work-up of this patient and his family members and a novel polymorphism that produces this unusual result.
Materials and Methods
Fragile X Analysis
Patient DNA was isolated from anticoagulated peripheral blood with the “Puregene” DNA isolation procedure (Gentra Systems, Minneapolis, MN). Five micrograms of genomic DNA was digested with 50 U each of EcoRI and SacII at 37 degrees for 2 hours. The digested DNA was separated by electrophoresis on a 1.5% vertical agarose gel (Hoefer Scientific Instruments, San Francisco, CA) without ethidium bromide for 16 hours at 45 volts. Gels were subsequently stained with ethidium bromide (EtBr) and photographed to visualize DNA, followed by alkaline Southern transfer to nylon membranes (Zeta Probe; Biorad, Hercules, CA). Blots were probed with a [32P]dCTP-radiolabeled (redivue; Amersham Pharmacia Biotech, Piscataway, NJ) FMR1 probe (StB12.3) that hybridizes to the region from bp 14461 to 15537 in FMR1.
FMR1 PCR
The following PCR primers were designed to bracket regions of the StB12.3 probe fragment region of FMR1 (numbers indicate positions in the FMR1 sequence as denoted in GenBank, reference L29074): FMR1: CCTAAACATCATCTCCCAGCG (14373–14393); FMR2: TTAGACGCTGAAGCATGTGC (14775–14755); FMR3: GAGGGAAGGACTGGACTTGG (14153–14173); FMR4: CAGTTGCCATTGTGATTTGG (14604–14584); FMR5: GTAGTAAGAAGCGGTAGTCG (14562–14582); FMR6: CCAGCAGTGCATTGAAGAAG (14680–14700); FMR7: CAGCCTTCCTTCCACACGCA (15240–15220). Final primer concentrations were 0.2 μmol/L each. PCR was performed using 500 ng of genomic DNA with the following conditions: 30 cycles of 94°C for 45 seconds, 60°C for 1 minute, 72°C for 90 seconds, with a final extension of 72°C for 5 minutes. After PCR, 10 U of EcoRI was added directly to the PCR mixture. After a 15-minute incubation at 37°C, reactions were stopped by adding bromophenyl blue and visualized on a 3% agarose gel.
Sequence Analysis
Direct sequencing of PCR fragments amplified from primers FMR1 and FMR7 was performed by the Sanger method, using Big Dye fluorescent sequencing reagents and an ABI 373 analyzer (Applied Biosystems, Foster City, CA).
EcoRI Screening of Patient Samples
Stored genomic DNA from previous samples submitted to our laboratory for analysis were anonymously screened using the PCR/EcoRI digestion protocol described above.
Results
Fragile X Analysis of Index Patient Sample by Southern Blot Analysis
Figure 1B shows the results of Southern blot analysis of the index patient and family members, using the StB12.3 probe described above. After the EcoRI/SacII digestion used for routine clinical testing in our laboratory, the index patient (I) showed a loss of the 2.8-kb fragment expected in male subjects, suggesting a deletion within FMR1 in the area of the StB12.3 probe. (The smaller bands seen in Figure 1B were not seen in the original clinical gel. Presumably these shorter fragments were eluted off the gel, because of the longer electrophoretic times used in clinical testing to provide adequate separation of the premutation fragments.) Further analysis of family members showed that the patient’s brother (B) also had a loss of the 2.8-kb fragment. The mother (M) of the index patient showed a more complex abnormal pattern, with two additional bands seen in addition to the expected 2.8-kb and 5.2-kb fragments.
Because deletions in the FMR1 gene are rare and have variable fragile X phenotypes, we decided to use additional restriction enzymes that flank the StB12.3 probe site to determine the extent and location of the deletion. Surprisingly, digestion with HindIII (Figure 1B) and BglII (data not shown) produced a fragment in the index patient and his mother that was identical to that seen in normal control samples, indicating that there was no large-scale deletion in this area of FMR1. This observation suggested that the “deletion” of the 2.8-kb band seen in the index patient might be due to a novel EcoRI site in FMR1. Subsequent analysis showed that digestion with EcoRI alone produced bands in both the index patient and his mother that were not seen in normal control patients (Figure 1B) .
Restriction Fragment Length Polymorphism Analysis of the StB12.3 Fragment
Analysis of the published sequence of the FMR1 gene revealed six potential sites in the StB12.3 fragment where a single base change would introduce a new EcoRI site. We constructed a series of PCR primers to span these areas, amplified each region, and digested the resulting PCR fragments with EcoRI. PCR fragments generated from control DNA, using primers FMR1 and FMR7, did not cut with EcoRI (Figure 2 , lane 2). In contrast, DNA from the index patient generated a PCR fragment that was cleaved by EcoRI into two smaller fragments (Figure 2 , lane 4). Genomic DNA isolated from the mother of this patient showed that she was heterozygous for this mutation (lane 6), and a brother also carried the mutation (data not shown). Sequence analysis of this region confirmed that an A → G transition at position 14744 had produced a new EcoRI site in both the index patient and his mother (Figure 3) . No other novel EcoRI sites were detected using the other FMR primer combinations.
Figure 2.
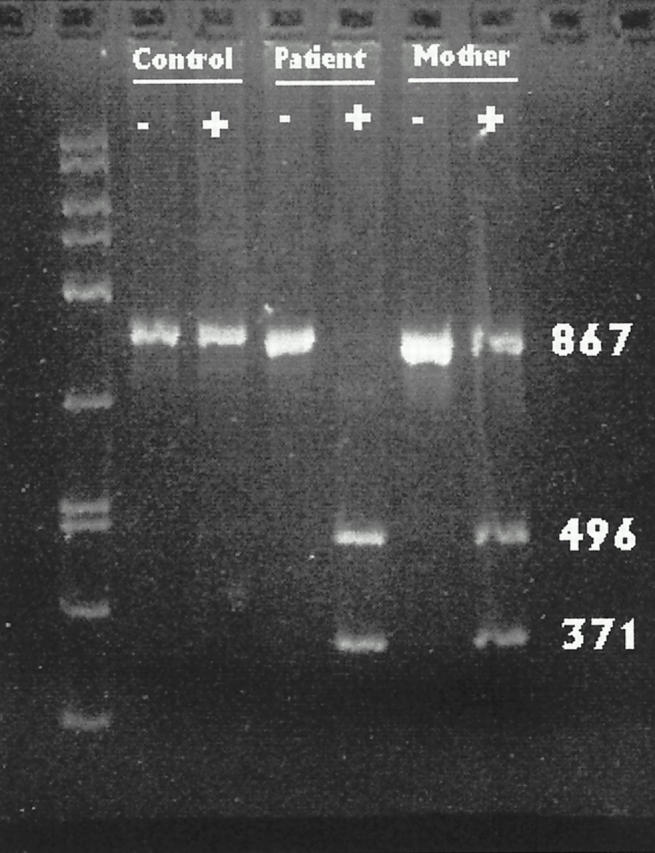
PCR-restriction fragment length polymorphism analysis for an EcoRI site. Genomic DNA was amplified using primers FMR1 and FMR7 to produce a 867-bp fragment spanning bases 14373 to 15240 in the FMR1 gene. Lanes marked + have been digested with EcoRI. PCR using control DNA from a normal subject produces a fragment that does not cut with EcoRI. Digestion of the PCR product from the index patient’s DNA produces fragments of 371 and 496 bp. DNA from the mother shows that she is heterozygous for the EcoRI site.
Figure 3.
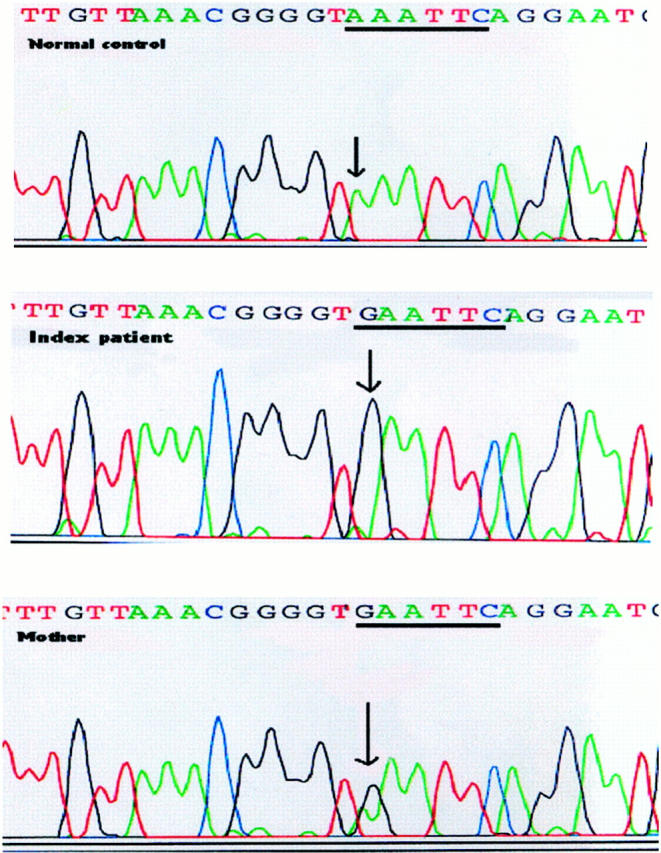
Sequence analysis of patient DNA. The PCR fragment amplified using primers FMR5 and FMR7 was sequenced from both the index patient and his mother. Patient DNA shows an A → G transition at bp 14744. The mother is heterozygous for this mutation.
Frequency of Polymorphism
Because this polymorphism has not previously been reported, we used the PCR/EcoRI assay to rapidly screen DNA samples from our patient stores to estimate the prevalence of this polymorphism in the general population. Seventy-two patient samples (40 male, 32 female, 104 chromosomes) were analyzed by PCR restriction fragment length polymorphism analysis. No additional examples of this polymorphism were found, suggesting that the frequency of this polymorphism in the general population is less than 1%. In addition, a retrospective review of more than 900 fragile X assays performed in our laboratory over the past 5 years with the EcoRI-SacII genomic Southern hybridization protocol revealed no evidence of similar “pseudodeletions” in past testing.
Conclusions
We have described a novel polymorphism in the FMR1 gene, where an A → G transition produces a new EcoRI site. For the diagnostic method discussed here, the presence of this polymorphism results in cleavage of the normal unmethylated 2.8-kb fragment into two smaller fragments of 1.6 and 1.2 kb. These smaller fragments may run off the gel under the conditions used for clinical testing, as gels are run for prolonged periods on high-percentage agarose to optimize separation of the larger fragments necessary for detection of premutations. The loss of the normal 2.8-kb band or the presence of smaller fragments generated by this new EcoRI site can give the appearance of a deletion in FMR1, as was seen in this case.
It is not clear whether this polymorphism has functional significance for the mental retardation phenotype. The mutation is located outside both the coding and promoter regions of FMR1 and does not appear to produce a new splice site. However, it is interesting to note that the mother shows preferential methylation of the mutant allele, as evidenced by the pattern seen with Southern analysis after the EcoRI/SacII digestion. The presence of four bands in the mother’s sample is consistent with heterozygosity for the mutation, and comparison of the fragments generated from the maternal allele without the polymorphism shows that the 5.2-kb fragment (methylated allele) is much less intense than the 2.8-kb fragment (unmethylated allele), suggesting a skewed pattern of X chromosome inactivation. However, it is not clear whether this shift in methylation is associated with the polymorphism or is a separate phenomenon.
For the purposes of clinical diagnostic testing, this patient did not have a triplet repeat expansion typically associated with clinical symptoms of the fragile X syndrome. The final report for the patient described in this study was “Triplet repeat expansion not detected,” and consultation with the ordering physician was provided to describe the results more fully. Although the direct phenotypic influence(s) of this unusual polymorphism with respect to the fragile X syndrome is unknown, from a diagnostic standpoint, this newly described polymorphism can affect the ability of laboratories to correctly diagnose clinical samples. Laboratories that use the method of detection described here could incorrectly identify the patient as having a deletion of part of the FMR1 gene. Because the deletions of FMR1 that have been described show variable association with the mental retardation phenotype, misclassification of a patient as carrying a FMR1 deletion could result in the misdiagnosis of a genetic basis for the patient’s mental retardation.
Address reprint requests to Dr. Barbara A. Zehnbauer, Molecular Diagnostics Laboratory, Box 8118, 660 S. Euclid Ave., Washington University School of Medicine, St. Louis, MO 63110. E-mail: zehnbauer_b@kids.wustl.edu.
References
- 1.Turner G, Webb T, Wake S, Robinson H: Prevalence of fragile X syndrome. Am J Med Genet 1996, 64:196-197 [DOI] [PubMed] [Google Scholar]
- 2.de Vries BBA, Halley DJ, Oostra BA, Niermeijer MF: The fragile X syndrome. J Med Genet 1998, 35:579-589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL: Instability of a 550-base pair DNA fragment and abnormal methylation in fragile X syndrome. Science 1991, 252:1097-1102 [DOI] [PubMed] [Google Scholar]
- 4.Hammond LS, Macias MM, Tarleton JC, Pai GS: Fragile X syndrome and deletions in FMR1: new case and review of the literature. Am J Med Genet 1997, 72:430-434 [PubMed] [Google Scholar]
- 5.Sutherland GR: Heritable fragile sites on human chromosome. I. Factors affecting expression in lymphocyte culture Am J Hum Genet 1979, 31:125-135 [PMC free article] [PubMed] [Google Scholar]
- 6.Rousseau F, Heitz D, Biancalana V, Blumenfeld S, Kretz C, Boue J, Tommerup N, Van Der Hagen C, DeLozier-Blanchet C, Croquette M-F, Gilgenkrantz S, Jalbert P, Voelckel M-A, Oberle I, Mandel J-L: Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med 1991, 325:1673-1681 [DOI] [PubMed] [Google Scholar]