

Abstract
The primary focus of this work was to determine the feasibility of obtaining representative expression array profiles from clinical core biopsies. For this purpose we performed six 16-gauge needle core biopsies and an excision biopsy on each of two different human xenografts, one from an Ewing’s sarcoma cell line and the second from neuroblastoma cell line grown in Beige-Scid mice. Three of the six core biopsies were processed separately and the remaining three were pooled and processed together. As the initial RNA material isolated from the core biopsies was not sufficient for microarray analysis, an amplification procedure using a modified Eberwine protocol was performed, and the amplified products applied onto a 6000-feature human cDNA microarray. Comparisons of the array results from core biopsies (amplified RNA) and surgical specimens (non-amplified RNA) showed maintenance of the expression profile as assessed by hierarchical clustering. Gene expression profiles obtained from microarray analysis clearly differentiated the Ewing’s sarcoma from the neuroblastoma with both core and excisional biopsies as starting material. Pooling the core biopsies did not improve the concordance with excisional biopsies. In conclusion, our results suggest that core biopsies can be used as a suitable and reliable material for the determination of tumor genetic profiles.
The recently described cDNA microarray technology allows researchers to monitor the expression of several thousand genes simultaneously and provides a format for the identification of new genes expressed in cancer states. 1, 2 Multiple studies have shown that cDNA microarrays are useful for characterizing human cancers and the resultant expression profiles are applicable in cancer diagnosis and prognosis. 3, 4, 5, 6
Most of these studies used total or poly(A) RNA from excised surgical specimens to obtain the minimal amount of RNA required for the preparation of the cDNA probe. To expand the utilization of cDNA microarrays to conditions in which starting material is the limiting factor such as material from fine-needle aspirates or core biopsies, RNA amplification approaches have been used. One such method, pioneered by Eberwine and colleagues, has been used to amplify RNA from single neuron. 7, 8
Needle core biopsy is a less invasive and less expensive alternative to surgical biopsy for the diagnosis of tumor lesions and provides similar diagnostic and molecular information. 9, 10, 11, 12 An added advantage of core biopsies, however, is that tumors can be serially sampled allowing for the monitoring of cellular changes after treatment.
The primary focus of this work was to determine the feasibility of obtaining representative expression array profiles from clinical core biopsies. As the RNA isolated from the core biopsies is not sufficient for standard microarray analyses, an amplification procedure using a modified Eberwine protocol was used. Comparisons of the array results from several core biopsies (using amplified RNA) and surgical specimens (using non-amplified RNA) from two human orthotopic xenografts (Ewing’s sarcoma and neuroblastoma), biopsied using identical instrumentation as for human needle biopsy procedures, showed maintenance of the tumor-specific gene expression profile, and concordance in identifying outliers. Gene expression profiles obtained from microarray analysis differentiated Ewing’s sarcoma from neuroblastoma with both core and surgical biopsies as starting material suggesting that core biopsies can be used as a suitable and reliable material for the determination of tumor genetic profiles.
Materials and Methods
Human Tumor Xenograft Models
Beige-Scid mice (Charles River Laboratories, Wilmington, MA) were housed under pathogen free conditions with a 12-hour light/12 hour-dark schedule, fed autoclaved standard chow and water ad libitum. Sites for orthotopic tumor implantation or injection was paralumbar musculature for the Ewing’s sarcoma xenograft (POB, LD, EWS, manuscript submitted) and intraadrenal for the neuroblastoma xenograft (SMS, KCNR, manuscript submitted). 13 The Ewing’s sarcoma and neuroblastoma xenografts shared many histological features, including high cellularity and small round blue cell morphology. Differences included occasional rosette formation and neuropil deposition in the neuoroblastoma xenograft (data not shown; respective manuscripts including histological analysis, submitted). All core and excisional biopsy procedures were collected post mortem following CO2 inhalation. Surgical sites were prepared by shaving skin and then cleansing using betadine scrub solution (E-Z Prep, Becton Dickinson, NJ) and 70% sterile alcohol. Animal care and use was in accordance with guidelines of the National Institutes of Health Animal Care and Use Committee (Committee, 1998 no. 185).
Tumor Samples and RNA Preparation
Six 16-gauge needle core biopsies and an excision biopsy were performed on each xenograft. Each specimen was snap frozen in liquid nitrogen and stored at −80°C. Phenol chloroform procedure (Trizol, Gibco, Grand Island, NY) was used to extract total RNA from each sample. Three core biopsies were processed separately while the other three were pooled together. Total RNA isolated from MCF7 breast cancer cell line cultured in RPMI 1640 medium (Gibco) supplemented with L-glutamine (2 mmol/L), 2% penicillin-streptomycin (10,000 U/ml) and 10% heat-inactivated fetal calf serum (FCS, Gibco) in an atmosphere of 95% air:5% CO2 at 37°C, served as a common reference. Eberwine’s RNA amplification procedure 8 with minor modifications was performed using total RNA from tumor specimen and MCF7 breast cancer cell line. Briefly, total RNA was reverse-transcribed by using a 63-nucleotide synthetic primer containing the T7RNA polymerase binding site (5′-GGC-CAG-TGA-ATT-GTA-ATA-CGA-CTC-ACT-ATA-GGG-AGG-CGG-(dT) 24–3′. Second strand cDNA synthesis (producing double-stranded cDNA) was performed with RNase H (Gibco-BRL), Escherichia coli DNA polymeraseI, and E. coli DNA ligase (Gibco-BRL). After cDNA was blunt-ended with T4 DNA polymerase (Gibco-BRL), it was purified and transcribed with T7 polymerase (T7 Megascript Kit 1334, Ambion), yielding amplified antisense RNA.
Preparation and Hybridization of Fluorescent-Labeled cDNA
The cDNA probes were prepared from amplified RNA or total RNA as described elsewhere. 7 Briefly, we used 3 μg of amplified RNA or 50 μg of total RNA for Cy3 labeling, and 3 μg of amplified RNA or 100 μg of total RNA for Cy5-labeling. Cy-dye incorporation was achieved in a reverse-transcription reaction using 6 μg of random hexamers (for amplified RNA) or 2 μg oligodT (for total RNA) primer in the presence of Cy3 or Cy5-labeled dUTP (Amersham, Piscataway, NJ) and 400 units of Superscript II reverse-transcriptase enzyme (Gibco-BRL). After probe purification using Centricon-30 microconcentrator (Amicon), the two separated probes were combined, mixed with hybridization solution, denatured and hybridized in a humidified chamber at 65°C for 16 hours. The slides were then rinsed by submersion and agitation for 2 minutes in 2X standard saline citrate (SSC) with 0.1% sodium dodecyl sulfate (SDS), followed by 1X SCC, 0.2 × SCC and 0.05X SCC and then dried. To exclude labeling biases, each experiment was repeated after having labeled each RNA-target with the reciprocal fluorochrome.
Scanning and Data Processing
Following hybridization, arrays were scanned using a 10-μm resolution GenePix 4000 scanner (Axon Instruments, Inc., Foster City, CA) at variable photomultiplier tube (PMT) voltage to obtain maximal signal intensity with <1% probe saturation. Resulting TIFF images for each fluorescent were analyzed with GenePix software version 3.0 (Axon Instruments, Inc., Foster City, CA). The data files generated by GenePix v3.0 were entered into a web-based database maintained by the Bioinformatics and Molecular Analysis Section of the Center for Information Technology, National Cancer Institute, Bethesda, MD. To study the gene expression profiles, an average linkage hierarchical cluster analysis using a correlation metric of similarity for clustering genes was performed as described by Eisen and colleagues. 14 A metric multidimensional scaling for analyzing and visualizing the correlation among expression profiles of samples was also performed. 15
Results
Core Biopsies Can Be Used to Obtain a Representative Gene Expression Profile of Tumors
To determine the feasibility of obtaining representative expression array profiles from clinical core biopsies, we performed six 16-gauge needle core biopsies and an excision biopsy on each of two different human xenografts, one from an Ewing’s sarcoma cell line and the second from neuroblastoma cell line grown in Beige-Scid mice at orthotopically relevant sites. Three of the six core biopsies were processed separately and the remaining three were pooled and processed together. To assess potential bias introduced by the RNA amplification procedure, a sample of total RNA from the excisional biopsies was also amplified and analyzed separately.
We first asked whether amplified RNA from the core biopsies would give expression profiles similar to those obtained from excisional specimens. To accomplish this, we compared the gene expression profiles generated from each core, pooled cores, and excisional biopsy for the tumor xenografts using the unsupervised hierarchical clustering technique of Eisen and co-workers. 14 As shown in Figure 1 , expression profiles from the core and excisional biopsies of each tumor xenograft clustered together and appeared highly reproducible. This suggests that microarray profiles from core biopsies are reliable, and are able to distinguish an Ewing sarcoma from a neuroblastoma.
Figure 1.
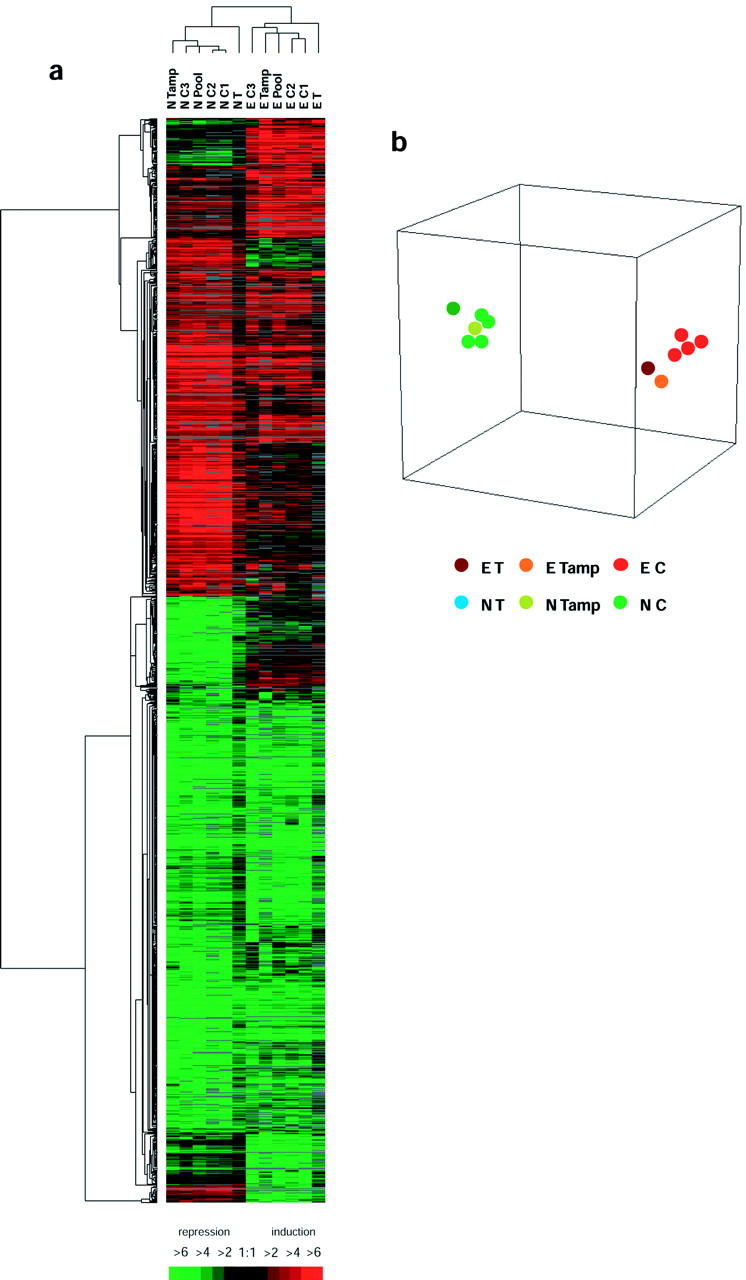
a: Hierarchical cluster analysis of all genes with a ratio greater than 2.0 or less than 0.5 compared with a reference cell line (MCF7) in at least 25% of arrays. Each row represents a single gene and each column represents the average of two reciprocal experiments. Red bars indicate genes over-expressed in tumor xenografts and green bars indicate genes that are under-expressed in tumor xenografts as compared to MCF7 breast cancer cell line. Black bars indicate genes with approximately equivalent expression levels and gray bars indicate missing or filter-excluded data. E, Ewing’s sarcoma; N, neuroblastoma; T, tumor (total RNA); C, core biopsy (amplified RNA); Tamp, tumor (amplified RNA). b: Multidimensional-scaling plot showing the correlation among expression profiles of different samples. Each colored spot represents an excisional or a core biopsy experiment.
Core Biopsies Are Representative of the Entire Tumor
Two factors that may contribute to the variability of the system are sampling errors from obtaining core biopsies, and potential bias from the T7 RNA amplification procedure. Representative scatter plots indicating the level of similarity between the cDNA microarray results comparing different biopsies are shown in Figure 2 . When profiles from two core biopsies from the same tumor were assessed (Figure 2f) , their Pearson correlation coefficient was very high (r = 0.87). When amplified RNA was used from the parent excisional biopsy (Figure 2c and 2d) , the correlation with the profile from the resultant core was also high (r = 0.85, 0.91). In contrast, core biopsies from different tumors, as expected, gave distinctly different expression profiles (Figure 2h , r = 0.37). These data suggest that when amplified RNA is used as template, array profiles from sequential core biopsies of an individual tumor are reproducible, that they are highly representative of the profiles from the parent tumor, and that cores from different tumors can be distinguished from one another.
Figure 2.

Representative scatter plots indicating the level of similarity between the cDNA microarray results comparing different protocols. a and b: Comparison of all targets between excisional biopsy using total RNA and core biopsy using amplified RNA for Ewing’s sarcoma and neuroblastoma, respectively. c and d: Comparison of all targets between excisional and core biopsies both using amplified RNA for Ewing’s sarcoma and neuroblastoma respectively. e and f: Comparison between two excisional (non-amplified) and two core biopsies (amplified) from the same tumor, respectively. g and h: Comparison between two excisional and two core biopsies from different tumor types. The correlation coefficient for each experiment appears in bold.
When we compared the results from the excisional biopsies (total RNA) with those from the resultant core biopsies (amplified RNA) we also observed excellent correlation (Figure 2a and 2b) , albeit with slightly lower correlation coefficients (r = 0.77, 0.78). This reduced correlation appears to be due to minor, but global differences in the Cy5/Cy3 ratios. These data suggest that although total and amplified RNA show small differences in expression ratios of individual genes, the composite expression profiles remain intact, and that comparisons between core biopsies using the same amplification technique can be used to distinguish tumor types.
We also assessed the variance of the ratios for the genes represented in the array. The coefficients of variance (C.V.) were calculated for genes showing either a ratio of ≥ 1.5, ≥ 2.0, ≥ 2.5, and ≥ 3.0 from experiments using either total RNA or amplified RNA. Our results show that the higher the ratio cut-off, the lower the C.V., and the C.V.’s of the core biopsies are slightly but consistently larger than that for the non-amplified excisional biopsies (Figure 3) .
Figure 3.

Assessment of coefficients of variance between genes from excisional (total RNA) and core biopsies (amplified RNA). Average of C. V. for the expression ratios are greater for amplified RNA from core biopsies than for non-amplified RNA from excisional specimens.
Are Core Biopsies Reliable for the Detection of Outliers?
An important use of microarray technology is the identification of genes that are potentially differentially expressed. We asked whether sampling from core biopsies would reliably identify outliers. Outliers were defined arbitrarily as genes producing array spots that exhibit Cy5/Cy3 ratios greater than 2.0 or less than 0.5. To test this, we assessed the percentage of concordance between the outlier lists from the non-amplified total RNA and the core biopsy amplified RNAs. When total RNA from a given excisional biopsy was compared to total RNA from a different excisional biopsy of the same tumor, the outlier concordance was between 49 to 68%. This baseline reproducibility of the system is consistent with previous reports. 7 When excisional biopsy total RNA was compared to individual cores (amplified RNA) from the same tumor, the concordance rate was similar (41 to 79%). In the neuroblastoma experiments, the level of concordance was higher when amplified core biopsy was compared to amplified excisional biopsy (77 to 86%). Thus core biopsies have a similar robustness to that of larger excisional biopsies in their ability to identify outlier genes. Pooling the core biopsies did not significantly improve the concordance with the excisional specimens. Thus, microarray analysis of core biopsies are representative of their native tumors, and can be used to identify differentially expressed genes as part of a gene discovery program.
Ewing’s sarcomas and neuroblastomas are occasionally misdiagnosed because of their histological similarities as small blue round cell tumors. The Ewing’s sarcoma and neuroblastoma xenografts shared many histological features, including high cellularity and small round blue cell morphology. Our results suggest that there are distinct differences in gene expression that can distinguish between the two tumor types. Tables 1 and 2 list the differences in gene expressions that segregate Ewing’s sarcoma from neuroblastoma. As expected, N-myc is significantly elevated in the neuroblastoma as compared to the Ewing’s sarcoma (7.2-fold), and c-myc is expressed predominantly in the Ewing’s sarcoma (3.7-fold). 16, 17, 18, 19 Interestingly, several genes were highly differentially expressed between the Ewing’s and the neuroblastoma xenografts. The tumor suppressor, BRCA2, and the transcription factor, ETV4, an E1A enhancer binding protein, were elevated by greater than 35-fold in the Ewing’s xenograft, whereas the GATA3, GATA binding protein, was increased by ninefold in the neuroblastoma tumor. Thus the expression profiling uncovered potential new diagnostic markers distinguishing Ewing’s sarcoma from neuroblastoma. However, these potential markers will require validation in a clinical investigation with an adequate sample number.
Table 1.
Genes Expressed at Higher Levels in the Ewing’s Sarcoma Cell Line
| Ratio* | Map | Gene | IMAGE no. | Ratio | Map | Gene | IMAGE no. |
|---|---|---|---|---|---|---|---|
| 42.60 | 13 | BRCA2 | 429238 | 2.82 | 12q23–q24 | EPS8 | 665304 |
| 37.17 | 22q12.3–q13.2 | SLC16A3 | 502151 | 2.78 | 1 | PEF | 137353 |
| 33.15 | 17q21 | ETV4 | 430297 | 2.78 | 15q21–q22.2 | B2M | 324872 |
| 12.90 | 11p15.5 | IGF2 | 245330 | 2.76 | 1p32–p36 | PABPC4 | 842820 |
| 10.65 | 11p15.5 | IGF2 | 245330 | 2.74 | 2p14–p13 | UGP2 | 486436 |
| 10.57 | 1p36.13–p36.12 | ID3 | 756405 | 2.73 | 1p31–p22 | CYR61 | 486700 |
| 8.65 | 13q14.3 | LCP1 | 344589 | 2.71 | 11q13 | GSTP1 | 774710 |
| 8.27 | 4q28–q32 | ANXA5 | 786680 | 2.71 | 4p16 | CTBP1 | 347702 |
| 8.03 | 12p12.3–p12.1 | MGST1 | 768443 | 2.67 | 6q22.1–22.3 | RPS5P1 | 376217 |
| 7.62 | 1q21 | S100A4 | 472180 | 2.65 | 1q32 | MCP | 796994 |
| 7.17 | 1p36 | CDW52 | 301723 | 2.60 | 15q22.1–q22.33 | MAP2K1 | 309258 |
| 7.12 | 5q31.3–q32 | SPARC | 324597 | 2.59 | 1p13.3 | GSTM4 | 840990 |
| 6.64 | 3p21.3 | GPX1 | 625473 | 2.58 | 20q13.31 | PCK1 | 742082 |
| 6.58 | 6 | RAB32 | 472186 | 2.52 | 12q23–q24.1 | TXNRD1 | 789376 |
| 5.77 | 11p15.5 | IGF2 | 296448 | 2.51 | 5q12.2–q13.3 | GTF2H2 | 345525 |
| 5.58 | 11p15.5 | IGF2 | 296448 | 2.46 | 9q13–q21 | CDC20 | 754999 |
| 5.41 | 13q14.3 | LCP1 | 712280 | 2.43 | 6q22–q23 | LAMA2 | 471642 |
| 5.20 | 1q22 | IFI16 | 824602 | 2.42 | 2 | BRE | 739993 |
| 5.15 | 22q13.1 | LGALS1 | 376168 | 2.41 | 19p13.3–p13.2 | ICAM1 | 293413 |
| 5.10 | 8q24 | PLEC1 | 781362 | 2.39 | 2p22–p21 | CAD | 274638 |
| 4.83 | 15 | LOC56851 | 110503 | 2.36 | 19p13.3 | BSG | 756533 |
| 4.72 | Unknown | PTPN9 | 342927 | 2.35 | 8 | ADE2H1 | 273546 |
| 4.54 | 16q22.1 | CBFB | 322494 | 2.34 | 22q13.2 | ST13 | 210887 |
| 4.49 | 1p36.3–p36.2 | ENO1 | 512247 | 2.30 | 11q13 | GSTP1 | 136235 |
| 4.32 | 4q13–q21 | IL8 | 549933 | 2.30 | 7p22 | PDGFA | 435470 |
| 4.26 | Xq26 | FGF13 | 785796 | 2.30 | 1p36.1 | CDC42 | 859586 |
| 3.93 | 22q11.21 | UFD1L | 80708 | 2.30 | 5q12–q13 | FOXD1 | 382564 |
| 3.87 | 19p13.1–p12 | PRKCL1 | 153010 | 2.29 | Unknown | EST | 32231 |
| 3.82 | 9 | SR-BP1 | 324210 | 2.29 | 15q26.1 | IQGAP1 | 898148 |
| 3.79 | 8q24.12–q24.13 | MYC | 812965 | 2.29 | 17 | RPL27 | 272185 |
| 3.75 | 19 | ISYNA1 | 809508 | 2.27 | 6q27 | RPS6KA2 | 22711 |
| 3.75 | 11q22.2–q22.3 | CASP4 | 470160 | 2.27 | 13q31.2–q32.3 | STK24 | 773137 |
| 3.70 | 16q13 | MT1G | 202535 | 2.27 | 12q22–q23 | IGF1 | 287327 |
| 3.68 | 8q24.12–q24.13 | MYC | 812965 | 2.23 | 1p31 | PDE4B | 788136 |
| 3.63 | 6q23.3 | CITED2 | 491565 | 2.22 | 3p21 | CTNNB1 | 774754 |
| 3.63 | 10p15.3–p15.2 | PFKP | 950682 | 2.22 | 19p13.1 | MYO9B | 279085 |
| 3.62 | 6q26 | IGF2R | 79712 | 2.20 | 1p34 | AK2 | 626880 |
| 3.61 | Xp11.3–p11.23 | TIMP1 | 771755 | 2.19 | 22q13.2 | ST13 | 210887 |
| 3.60 | 6p21.3 | HLA-DRA | 726209 | 2.17 | 18p11.31–p11.21 | YES1 | 204634 |
| 3.59 | 17 | ITGA3 | 755402 | 2.16 | 17q25 | CSNK1D | 302527 |
| 3.58 | 11q22.3 | MMP1 | 624924 | 2.16 | 8p12–p11 | DUSP4 | 756596 |
| 3.54 | 8 | E2F5 | 809828 | 2.15 | 4q24 | NFKB1 | 789357 |
| 3.52 | 11p15.4 | LDHA | 897567 | 2.12 | 7 | SYPL | 770444 |
| 3.50 | 17 | EST | 768299 | 2.11 | 11 | PRO1073 | 257287 |
| 3.42 | 15q24.3 | BCL2A1 | 814478 | 2.10 | 10q22 | CAMK2G | 366154 |
| 3.39 | 16q22.1 | CBFB | 624754 | 2.10 | 6q21 | CCNC | 647007 |
| 3.29 | 15q24.3 | BCL2A1 | 814478 | 2.10 | 2q36–q37 | INPP5D | 826405 |
| 3.29 | 8 | KIAA0143 | 530310 | 2.08 | 7p12–q21 | PHKG1 | 612224 |
| 3.28 | 13q12–q13 | ARPC2 | 162208 | 2.07 | 11q23 | CD3E | 1130062 |
| 3.25 | 1p34 | AK2 | 45464 | 2.07 | 1p34.1 | HDAC1 | 548736 |
| 3.24 | 15 | LOC56851 | 110503 | 2.06 | Xq26 | ARHGEF6 | 687990 |
| 3.21 | 4 | KIAA0746 | 809374 | 2.06 | 17q11.2 | MAP2K3 | 45641 |
| 3.20 | 2q37 | COL6A3 | 138991 | 2.05 | 12p12.3 | ARHGDIB | 841332 |
| 3.17 | 4q34–q35 | FAT | 503119 | 2.05 | 3 | EST | 137984 |
| 3.10 | 1p34 | LAPTM5 | 753313 | 2.05 | 6p21.3 | DAXX | 841498 |
| 2.98 | 6p21.3 | HLA-A | 853906 | 2.05 | 17q25 | BIRC5 | 796694 |
| 2.98 | 3p21.2 | IMPDH2 | 292008 | 2.05 | 11q23.2–q23.3 | H2AFX | 114416 |
| 2.94 | 7q11.23 | LIMK1 | 1086811 | 2.05 | 22 | SIVA | 501643 |
| 2.93 | 17q25 | H3F3B | 950574 | 2.04 | 20p12.1–p11.23 | JAG1 | 141815 |
| 2.92 | 10q22–q23 | ANXA11 | 810117 | 2.04 | 20q12 | SDC4 | 504763 |
| 2.89 | 16p13.3 | NME4 | 203003 | 2.04 | 8q24 | RAD21 | 383190 |
| 2.88 | 4 | UGT2B10 | 293742 | 2.03 | 6q23 | SGK | 840776 |
| 2.88 | 20pter–p12 | PRNP | 812048 | 2.03 | 8 | ADE2H1 | 273546 |
| 2.87 | 9q33–q34.1 | ENG | 307887 | 2.02 | 19p13.3 | KHSRP | 123400 |
| 2.84 | 10q25.3 | PGAM1 | 486108 | 2.01 | 6pter–q12 | NMOR2 | 824024 |
| 2.84 | 11p15.5 | LSP1 | 1323933 | 2.01 | 1q21–q25 | TAGLN2 | 45544 |
| 2.83 | 17 | NMT1 | 756480 | 2.00 | 12p13 | CCND2 | 366412 |
| 2.82 | 5q14–q21 | PAM | 140806 | 2.00 | 10q22–q23 | ATP5C1 | 845519 |
Relative gene expression ratios of Ewing’s sarcoma compared to neuroblastoma.
Map, chromosomal location.
IMAGE, International Molecular Analysis of Genomics and their Expression.
Table 2.
Genes Expressed at Higher Levels in the Neuroblastoma Cell Line
| Ratio* | Map | Gene | IMAGE no. | Ratio | Map | Gene | IMAGE no. |
|---|---|---|---|---|---|---|---|
| 9.06 | 10p15 | GATA3 | 214068 | 2.54 | 11q13 | CCND1 | 841641 |
| 7.39 | 5q | ISL1 | 362795 | 2.52 | Unknown | EST | 503064 |
| 7.19 | 2p24.1 | MYCN | 298309 | 2.50 | 4q11–q12 | KIT | 265060 |
| 6.68 | 2p25 | ODC1 | 545502 | 2.48 | 7q11.23 | GTF2IP1 | 548957 |
| 6.48 | 6q21 | CD24 | 204335 | 2.47 | 1 | CELSR2 | 175103 |
| 6.20 | 6p21.3 | HSPA1A | 265267 | 2.47 | 20p13 | CENPB | 809720 |
| 5.59 | 17 | EST | 430186 | 2.47 | 18 | EST | 364506 |
| 5.54 | 10 | INA | 784876 | 2.45 | Xp22.2–p22.1 | PDHA1 | 489212 |
| 5.38 | 6p21.3 | HSPA1A | 265267 | 2.43 | 3q27–q28 | SIAT1 | 194295 |
| 5.23 | 6q21 | CD24 | 163189 | 2.37 | 6p21.3 | ZNF184 | 814014 |
| 5.08 | 20q13.3 | KCNQ2 | 179534 | 2.35 | 5q21–q23 | CAMK4 | 814780 |
| 4.55 | 16p13.2 | EMP2 | 109863 | 2.34 | 8 | KIAA1249 | 291537 |
| 4.40 | 2 | IGFBP5 | 45542 | 2.33 | 10q26 | OAT | 783696 |
| 4.34 | 3p21.3 | MST1R | 612616 | 2.33 | 20pter–p12 | PCNA | 789182 |
| 3.98 | Unknown | EST | 530662 | 2.32 | 5 | PPIC | 487437 |
| 3.90 | 1 | RGS4 | 429349 | 2.30 | 7q22 | TRIP6 | 811108 |
| 3.85 | 12q13 | KRT8 | 897781 | 2.30 | 14q32.32 | AKT1 | 785669 |
| 3.64 | 1p22 | D1S155E | 839623 | 2.25 | 11q13 | UCP2 | 236034 |
| 3.62 | 6p22.3 | SOX4 | 366815 | 2.20 | 2q31 | COL3A1 | 122159 |
| 3.56 | 4q34–q35 | FACL2 | 82734 | 2.20 | 16q22 | ATP6D | 384078 |
| 3.55 | 2p25 | ODC1 | 796646 | 2.19 | 17p13.2–p13.3 | MYO1B | 840474 |
| 3.52 | 19 | EIF3S4 | 857319 | 2.17 | 9q22 | C9ORF3 | 768292 |
| 3.36 | 20q12 | NCOA3 | 502333 | 2.15 | 12p13 | CD9 | 727251 |
| 3.32 | 2 | EST | 768324 | 2.14 | 16q13–16q21 | MMP15 | 784589 |
| 3.22 | 22q12.3 | TIMP3 | 75410 | 2.14 | 11 | IFITM1 | 509641 |
| 3.19 | 3q13.1–q13.2 | GAP43 | 44563 | 2.10 | 17q | CBX1 | 786084 |
| 3.17 | 20p13 | CDC25B | 786067 | 2.09 | Xq26 | FHL1 | 813266 |
| 3.10 | 6q27 | PDCD2 | 303183 | 2.08 | 18 | EST | 141234 |
| 2.97 | 6p21.3 | HSPA1L | 50615 | 2.08 | 3q12–q13 | MOX2 | 51363 |
| 2.89 | Xp11.4–p11.3 | MAOA | 359661 | 2.08 | 16p13.3 | SRM300 | 489453 |
| 2.82 | 19p13.1 | IFI30 | 724506 | 2.07 | 1q31–q32 | PTPRC | 239287 |
| 2.80 | 18q21.3 | BCL2 | 342181 | 2.04 | Xq11 | TM4SF2 | 307471 |
| 2.79 | 7q22 | CUTL1 | 701751 | 2.02 | 20q11.2–q12 | NNAT | 139681 |
| 2.78 | 20p13 | CDC25B | 786067 | 2.02 | 17p13.3 | YWHAE | 266106 |
| 2.71 | 11 | PIG11 | 365972 | 2.02 | 11p11.2–p11.11 | ACP2 | 70332 |
| 2.69 | 11q13 | CCND1 | 841641 | 2.02 | 4 | CCNI | 248295 |
| 2.64 | Unknown | EST | 488574 | 2.01 | 15q25 | NTRK3 | 35356 |
| 2.55 | 8p21 | NEFL | 28422 | 2.00 | 9q34.3 | PTGES | 491213 |
Relative gene expression ratios of neuroblastoma compared to Ewing’s sarcoma.
Map, chromosomal location.
IMAGE, International Molecular Analysis of Genomics and their Expression.
Discussion
The recently described cDNA microarray technology allows researchers to monitor the expression of several thousand genes simultaneously. The power of this technique is that the comprehensive expression profile of a tumor cannot only be used to discover new genes involved in a disease process, but also to develop molecular fingerprints of a tumor that can be used for diagnosis and for correlation with clinical outcomes. The successful molecular classification of diverse tumors on the basis of gene expression profile indicates that cDNA microarray technology is potentially a powerful tool for the development of personalized treatment. 6, 20 However, one of its restrictions for broader clinical utilization is the need of large amount of RNA required for its utilization, in the range of 50 to 100 μg of total RNA. This limitation may be resolved by the application of different amplification approaches such as the T7-based Eberwine’s procedure.
The purpose of this study was to determine the feasibility of obtaining representative expression array profiles from limited amount of tissue such as core biopsies applying T7-based amplification procedure. Needle core biopsy is an established, highly accurate method for the diagnosis of many tumor lesions and one of its advantages is to be a less invasive and less expensive alternative to surgical biopsy. Our study shows that gene expression profiles from a core biopsy were able to distinguish two histologically similar tumor phenotypes. These profiles appeared to be highly reproducible across several core biopsies suggesting that core biopsies can provide reliable and reproducible gene expression profiles, which can be used to distinguish between tumor types. The xenograft systems used in these studies were selected for their close histology and biology to Ewing’s sarcoma and neuroblastoma, respectively. Historically tumor xenograft models have used heterotopic (ie, subcutaneous) injection of human cancer cell lines into mice. Under these heterotopic conditions it is common to find highly cellular tumors relatively devoid of normal stroma or vasculature. These heterotopic xenografts have limited value in the study of cancer biology. Orthotopic tumor injection have been shown to influence several facets of tumor biology, including proliferation rate, invasion, metastases, and even chemosensitivity and furthermore have significantly enhanced the relevance of most tumor xenograft systems studied. 21, 22, 23 Our orthotopic models included chest wall (thoracolumbar) injections of the Ewing’s sarcoma cell line (similar to Askin’s-type Ewing’s sarcoma) and intraadrenal injection of neuroblastoma cell lines. In both cases the biology, histology, and metastatic behavior of the xenografts was highly representative of the human disease (respective manuscipts submitted). Most important to this microarray study the tumor xenografts demonstrated intratumoral heterogeneity including normal stromal and vascular invasion and distinct areas of necrosis and differentiation. This intratumor heterogeneity contributed to the relevance and interpretation of the gene expression profiles generated from whole tumor and needle biopsy. It should be noted that the core biopsy technique used for sampling xenograft tumors uses identical instrumentation as used in patient biopsy procedures. However, both xenograft tumors were biopsied without imaging techniques using a percutaneous approach. The addition of tumor imaging may allow biopsies to be collected from specific areas of a tumor rather than randomly as was done here.
Though the use of comprehensive gene expression profiling can be accomplished using core biopsies, our observations suggest that when ascertaining the expression levels of individual genes, the variance of the results for each gene is significant especially using amplified RNA (Figure 3) . Thus, microarray results for individual genes should be used with caution in clinical settings requiring precise quantitation, except for where small number of genes are highly expressed or where the genes are highly differentially expressed.
Acknowledgments
We thank Soek-Ying Neo and Olga Aprelikova for critical review of the manuscript.
Address reprint requests to Edison T. Liu, M.D., Executive Director, Genome Institute of Singapore, 1 Research Link, IMA Building, 04–01, Singapore 117604. E-mail: gisliue@nus.edu.sg.
Footnotes
Supported in part by the “Fond National de Recherche Scientifique,” Belgium, Grant Ext 260, V6/5/2-ILF, 14773 to Christos Sotiriou.
References
- 1.Schena M, Shalon D, Davis RW, Brown PO: Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 20:270:467–470 [DOI] [PubMed]
- 2.DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM: Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet 1996, 14:457-460 [DOI] [PubMed] [Google Scholar]
- 3.Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, van de Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO: Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 2000, 24:227-235 [DOI] [PubMed] [Google Scholar]
- 4.Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, Wilfond B, Borg A, Trent J: Gene-expression profiles in hereditary breast cancer. N Engl J Med 2001, 344:539-548 [DOI] [PubMed] [Google Scholar]
- 5.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D: Molecular portraits of human breast tumours. Nature 2000, 406:747-752 [DOI] [PubMed] [Google Scholar]
- 6.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Staudt LM: Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403:503-511 [DOI] [PubMed] [Google Scholar]
- 7.Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM: High-fidelity mRNA amplification for gene profiling. Nature Biotechnol 2000, 18:457-459 [DOI] [PubMed] [Google Scholar]
- 8.Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P: Analysis of gene expression in single live neurons. Proc Natl Acad Sci USA 1992, 89:3010-3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Florentine BD, Cobb CJ, Frankel K, Greaves T, Martin SE: Core needle biopsy: a useful adjunct to fine-needle aspiration in select patients with palpable breast lesions. Cancer 1997, 81:33-39 [DOI] [PubMed] [Google Scholar]
- 10.Koscick RL, Petersilge CA, Makley JT, Abdul-Karim FW: CT-guided fine needle aspiration and needle core biopsy of skeletal lesions: complementary diagnostic techniques. Acta Cytol 1998, 42:697-702 [DOI] [PubMed] [Google Scholar]
- 11.Willman JH, White K, Coffin CM: Pediatric core needle biopsy: strengths and limitations in evaluation of masses. Pediatr Dev Pathol 2001, 3:46-52 [DOI] [PubMed] [Google Scholar]
- 12.McManus AP, Gusterson BA, Pinkerton CR, Shipley JM: Diagnosis of Ewing’s sarcoma and related tumours by detection of chromosome 22q12 translocations using fluorescence in situ hybridization on tumour touch imprints. J Pathol 1995, 176:137-142 [DOI] [PubMed] [Google Scholar]
- 13.Reynolds CP, Biedler JL, Spengler BA, Reynolds DA, Ross RA, Frenkel EP, Smith RG: Characterization of human neuroblastoma cell lines established before and after therapy. J Natl Cancer Inst 1986, 76:375-387 [PubMed] [Google Scholar]
- 14.Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 1998, 95:14863-14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tenenbaum JB, de Silva V, Langford JC: A global geometric framework for nonlinear dimensionality reduction. Science 2000, 290:2319-2323 [DOI] [PubMed] [Google Scholar]
- 16.Sollazzo MR, Benassi MS, Magagnoli G, Gamberi G, Molendini L, Ragazzini P, Merli M, Ferrari C, Balladelli A, Picci P: Increased c-myc oncogene expression in Ewing’s sarcoma: correlation with Ki67 proliferation index. Tumori 1999, 85:167-173 [DOI] [PubMed] [Google Scholar]
- 17.Winters JL, Geil JD, O’Connor WN: Immunohistology, cytogenetics, and molecular studies of small round cell tumors of childhood: a review. Ann Clin Lab Sci 1995, 25:66-78 [PubMed] [Google Scholar]
- 18.Michon J, Delattre O, Zucker JM, Peter M, Delonlay P, Luciani S, Mosseri V, Vielh P, Neuenschwander S, Thomas G: Prospective evaluation of N-myc amplification and deletion of the short arm of chromosome 1 in neuroblastoma tumours: a single institution study. Prog Clin Biol Res 1994, 385:11-17 [PubMed] [Google Scholar]
- 19.Breit S, Schwab M: Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J Neurosci Res 1989, 24:21-28 [DOI] [PubMed] [Google Scholar]
- 20.Bubendorf L, Kolmer M, Kononen J, Koivisto P, Mousses S, Chen Y, Mahlamaki E, Schraml P, Moch H, Willi N, Elkahloun AG, Pretlow TG, Gasser TC, Mihatsch MJ, Sauter G, Kallioniemi OP: Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst 1999, 20:1758-1764 [DOI] [PubMed] [Google Scholar]
- 21.Singh RK, Tsan R, Radinsky R: Influence of the host microenvironment on the clonal selection of human colon carcinoma cells during primary tumor growth and metastasis. Clin Exp Metastasis 1997, 15:140-150 [DOI] [PubMed] [Google Scholar]
- 22.Fidler IJ, Wilmanns C, Staroselsky A, Radinsky R, Dong Z, Fan D: Modulation of tumor cell response to chemotherapy by the organ environment. Cancer Metastasis Rev 1994, 13:209-222 [DOI] [PubMed] [Google Scholar]
- 23.Togo S, Shimada H, Kubota T, Moossa AR, Hoffman RM: Host organ specifically determines cancer progression. Cancer Res 1995, 55:681-684 [PubMed] [Google Scholar]