

Abstract
The autosomal-dominant spinocerebellar ataxias (ADCA) are a heterogeneous group of neurodegenerative disorders with variable expression and phenotypic overlap. An accurate diagnosis relies on detection of a mutation in a specific causative gene, which is typically an abnormal number of CAG trinucleotide repeats. To streamline testing in a clinical setting, we converted our current panel of tests for the spinocerebellar ataxias (SCA) types SCA1, SCA2, SCA3, SCA6, and SCA7 from five independent amplification reactions analyzed by polyacrylamide gel electrophoresis (PAGE) to a single multiplex amplification reaction analyzed by capillary electrophoresis (CE). Multiplex amplification was facilitated by the use of chimeric primers; different lengths and fluorochromes distinguished the amplicons. During CE with commercially available molecular weight standards, the SCA amplicons migrated faster than predicted, thereby underestimating their length compared to that determined previously by PAGE. This was observed to varying degrees for each of the five loci, with the greatest size differential occurring in amplicons with greater (CAG)n. To determine accurate amplicon length, and therefore an accurate number of CAG repeats, a size correction formula was calculated for each locus. This multiplex semi-automated assay has been reliable during 1 year of use in a clinical setting during which 57 samples were tested and five positive samples were detected.
The autosomal-dominant spinocerebellar ataxias (ADCA) are a heterogeneous group of neurodegenerative disorders characterized by slowly progressive cerebellar dysfunction. Affected individuals have difficulty coordinating body movement, gait ataxia in particular, in addition to other associated findings. 1, 2 Over 15 distinct types of hereditary spinocerebellar ataxia (SCA) disorders have been described based on identification of different causative genes or chromosomal loci. 1 Because of variable expression and phenotypic overlap, the SCA disorders cannot be differentiated reliably on a clinical basis. Although algorithms may predict the likelihood of a specific type of SCA, 3, 4 an accurate diagnosis depends on molecular testing that detects a mutation in a specific causative gene. With few exceptions, mutations that cause SCA disorders are an abnormally large numbers of nucleotide-repeat motifs. The most common motif is a CAG trinucleotide repeat in the coding region of the respective genes, which on expansion causes SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 (Table 1) . 1, 5, 6, 7 Mutations involving expansion of other repeat motifs in coding and noncoding regions of additional genes are known to cause other types of SCA disease. 1, 8, 9
Table 1.
Molecular Basis of Five Spinocerebellar Ataxia Disorders
| Disease | Gene | Protein | Number of CAG repeats | Selected reference | ||
|---|---|---|---|---|---|---|
| Normal | Intermediate* | Abnormal | ||||
| SCA1 | SCA1 | Ataxin 1 | ≤36 | 37–44 | 44–83 | 17, 18 |
| SCA2 | SCA2 | Ataxin 2 | ≤31 | 30–34 | 35–∼400 | 19 |
| SCA3 | MJD | SCA3/MJD1 | ≤47 | 53, 54 | 55–86 | 20, 21 |
| SCA6 | CACNAIA | α1 Voltage-gated calcium channel | ≤20 | 20? | 21–33 | 22, 24 |
| SCA7 | SCA7 | Ataxin 7 | ≤19 | 28–35 | 36–>300 | 16, 23 |
Intermediate is used here to define two categories of alleles: those that are normal but may expand into the abnormal range upon transmission and those that show reduced penetrance. For simplicity, the table does not detail the rare alleles at the borders of the categories that are difficult to assign as normal, intermediate, or abnormal.
Typically, testing for the SCA disorders employs a panel that includes individual assays for the most prevalent types of SCA, for example SCA1, SCA2, SCA3, SCA6, and SCA7 in the U.S. population. The number of CAG repeats in each of the five causative genes is determined. The detection of an abnormally large number of CAG repeats in a gene means that the patient has, or is predisposed to develop, that type of SCA. Testing commonly employs radiolabeled primers to individually amplify the region containing the CAG-repeat motif from each of the five genes, polyacrylamide gel electrophoresis (PAGE) to resolve the amplicons by size, and autoradiography for visualization. To determine whether the amplicons have an abnormal number of repeats, their lengths are determined by co-migration with a DNA sequencing ladder. This method is labor intensive, requiring both the analysis of each locus individually and the precautions inherent in the use of radionuclides. Here, we detail for the first time, a rapid and precise method to assay SCA1, SCA2, MJD (SCA3), CACNA1A (SCA6), and SCA7 by multiplex amplification and capillary electrophoresis (CE).
Materials and Methods
Single SCA Gene Amplification and PAGE Analysis
Genomic DNA was purified from peripheral blood with the Puregene Human and Mammalian DNA Isolation kit (Gentra Systems, Inc., Minneapolis, MN). Segments of the genes SCA1, SCA2, MJD (SCA3), CACNA1A (SCA6), and SCA7 harboring the CAG-repeat region were amplified in five separate reactions in an ABI 9700 thermal cycler (Applied Biosystems, Foster City, CA). Reactions (20 μl) contained 2 μl of 10X reaction buffer (GeneAmp PCR Buffer II, Applied Biosystems); 200 μmol/L dNTPs (Amersham-Pharmacia Biotech, Piscataway, NJ); 0.2 pmol γ-32P-labeled forward primer; 1.5 units of polymerase (AmpliTaq, Applied Biosystems); 0.2 to 0.8 μg genomic DNA. For the SCA1, MJD (SCA3), and CACNA1A (SCA6) gene assays, 12 pmols of each primer were added; the SCA7 assay used 8 pmols of each primer; and the SCA2 assay used 1.3 pmols of SCA2A with 15 pmols of SCA2B. In addition, the SCA7 assay contained 5% dimethylsulfoxide (DMSO). Amplification of the SCA1, SCA2, MJD, and CACNA1A genes were performed for one cycle at 95°C for 5 minutes, and 28 cycles at 95°C for 1 minute, 61°C for 1 minute, 72°C for 1.5 minutes, with a final extension at 72°C for 5 minutes. Amplification of the SCA7 gene was performed for one cycle at 95°C for 5 minutes, and 35 cycles at 94°C for 45 seconds, 55°C for 75 seconds, and 72°C for 60 seconds, with a final extension at 72°C for 10 minutes. Samples were mixed with formamide-loading buffer (95% formamide, 20 mmol/L EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol) and electrophoresed through a denaturing polyacrylamide gel (GelMix6, Gibco/BRL) at 1900 V. The amplicons for each assay were electrophoresed on separate gels, adjacent to an M13 sequencing ladder, for a time sufficient to resolve the specific product. Amplicon lengths (excluding the trinucleotide-repeat region) are SCA1, 124 bp; SCA2, 59 bp; MJD,162 bp; CACNA1A, 102 bp; and SCA7, 277 bp.
Multiplex SCA Gene Amplification and CE Analysis
The multiplex assay was performed using the Expand Long Template PCR system (Roche Molecular Biochemicals, Indianapolis, IN). Chimeric primers (Table 2) were synthesized, labeled, and purified by high performance liquid chromatography (HPLC) (Applied Biosystems Inc., or Sigma-Genosys, The Woodlands, TX). A multiplex reaction of 20 μl consisted of SCA1 primers (1.5 pmol each), SCA2 primers (1.5 pmol each), MJD primers (4.0 pmol each), CACNAIA primers (1.5 pmol each), SCA7 primers (2.25 pmol each), 400 μmol/L dNTPs (Amersham-Pharmacia, Piscataway, NJ), 1X buffer #1 (1.75 mmol/L MgCl2), DMSO (4% v/v), Taq/Pwo polymerase (1.4 U), and 200 to 800 ng of genomic DNA purified from peripheral blood as described above. Reactions were performed in an ABI 9700 thermal cycler for 1 cycle at 95°C for 5 minutes, 32 cycles at 95°C for 1 minute, 60°C for 2 minutes (ramp 10% of default), 68°C for 1.5 minutes, and a final extension at 68°C for 10 minutes. One μl of a 1:5 dilution of the reaction products was added to 12 μl of formamide (HiDye formamide, Applied Biosystems Inc.) and 0.5 μl of GS500-ROX internal molecular weight standard (Applied Biosystems Inc.), denatured at 95°C for 2 minutes, and immediately placed on ice for a minimum of 3 minutes. Samples were injected into an ABI PRISM 310 Genetic analyzer (Applied Biosystems Inc.) with a 47 cm × 50 μm capillary containing Performance Optimized Polymer-4 (POP-4, Applied Biosystems Inc.) for 5 seconds with an injection kV of 15.0 and electrophoresed at 15 kV for 40 minutes at 65°C. Amplicon length was calculated by comparison with the GS500-ROX molecular weight standard by the Genescan program (Applied Biosystems Inc.) using the local Southern method.
Table 2.
Chimeric Primers for Multiplex Amplification of Five Spinocerebellar Ataxia Genes
| Name* | Fluorochrome | Sequences (5′ → 3′) | Amplicon (bp) |
|---|---|---|---|
| SCA1F | gcg gtcccaa aag ggtcagt AAC TGG AAA TGT GGA CGT AC | ||
| SCA1R | 6-FAM | ggtcccaaaagggtcagtCAA CAT GGG CAG TCT GAG | 162 + (CAG)n |
| SCA2F | aaaagggtcagt GGG CCC CTC ACC ATG TCG | ||
| SCA2R | HEX | caaaagggtcagtCGG GCT TGC GGA CAT TGG | 84 + (CAG)n |
| SCA3F | NED | gcggtcccaaaagggtcagt CCA GTG ACT TTG ATT CG | |
| SCA3R | gcggtcccaaaagggtcagt TGG CCT TTC ACA TGG ATG TGA A | 201 + (CAG)n | |
| SCA6F | caaaagggtcagt CAG GTG TCC TAT TCC CCT GTG ATC C | ||
| SCA6R | NED | aaagggtcagtTGG GTA CCT CCG AGG GCC GCT GGT G | 126 + (CAG)n |
| SCA7F | HEX | gcggtcccaaaagggtcagtTGT TAC ATT GTA GGA GCG GAA | |
| SCA7R | gtcccaaaagggtcagtCAC GAC TGT CCC AGC ATC ACT T | 314 + (CAG)n |
R, reverse; F, forward primers for each SCA locus.
Upper case, SCA locus-specific primer sequence; lower case, sequence from M13mp18 (gcggtcccaaaagggtcagt). The SCA locus-specific sequences are the same as those utilized in the PAGE assay and were described previously. 27 28 29 30 31
Length of unique sequence given for each amplicon.
Results
Multiplex Assay Development and Optimization
To streamline SCA testing and shorten turn-around time, we sought to convert our current panel of tests for SCA1, SCA2, SCA3, SCA6, and SCA7 from five independent amplification reactions analyzed by PAGE to a single multiplex amplification reaction analyzed by CE. For multiplex amplification, our strategy was to employ chimeric primers, which were shown previously to facilitate optimization of multiplex polymerase chain reaction (PCR). 10 New primers were synthesized with 10 to 20 bases of an M13 universal sequence added to the 5′ end of each SCA primer that was used in our existing individual SCA assays (Table 2) . The number of M13 bases that was added varied to approximate a melting temperature (Tm) of ∼80°C and a similar GC content between primer pairs and also among the five loci. Fluorescent labels were selected based on normal allele size ranges for each locus, making sure that loci with overlapping size ranges were given different labels (Figure 1) . For example, the primers for CACNAIA (SCA6) and MJD (SCA3) were both labeled with NED because the size of their amplicons do not overlap. Similarly, both the amplicons of SCA2 and SCA7 were labeled with HEX because their sizes do not overlap, with the possible exception of rare instances of large expansions in neonatal/juvenile onset cases (Table 1) .
Figure 1.
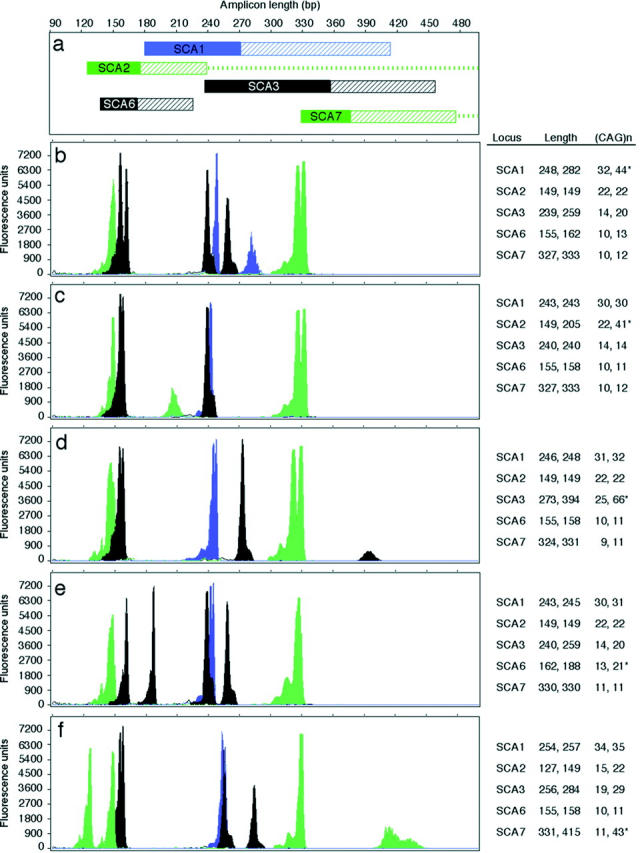
Capillary electrophoresis of multiplexed SCA loci. a: Schematic showing the range of amplicon lengths for each locus. Colors indicate fluorochromes: green, HEX; blue, 6-FAM; black, NED; solid bar, range of normal sized alleles; hatched bar, range of abnormally expanded alleles; dashes, the exceptionally large expansions observed in affected neonates/children. b–f: Electropherograms showing the test results for an affected subject with an abnormal CAG-repeat expansion. The table adjacent to each panel gives the amplicon length and number of CAG repeats for each allele of the samples tested (*, abnormal (CAG)n). b: Abnormal test result for SCA1. c: Abnormal test result for SCA2. d: Abnormal test result for SCA3 (MJD gene). e: Abnormal test result for SCA6 (CACNAIA gene). f: Abnormal test result for SCA7.
To facilitate efficient amplification of both normal and abnormal alleles, assay parameters and polymerases that favored the amplification of long templates were used (Materials and Methods). Assay optimization experiments were performed with a normal control subject and a patient with an expanded allele of known CAG-repeat length for each locus. Assay parameters were adjusted to optimize multiplex amplification and CE analysis. For example, the addition of DMSO was required for robust amplification of SCA7, but too much inhibited amplification of MJD (SCA3); 4% DMSO was optimal. Differences in amplification efficiencies and in fluorescence intensities of the three dyes resulted in varying peak amplitudes of fluorescence units in the electropherograms. Therefore, primer concentrations were adjusted to yield peaks of comparable amplitude to ensure detection of alleles of varied length.
Examples of the results of the optimized assay are shown in Figure 1 . Heterozygotes with normal alleles differing by one or two CAG repeat were detectable (Figure 1 , panel c, CACNA1A and SCA7). Expanded alleles, in the size ranges tested (Figure 2) , were detected consistently; drop out of expanded alleles was never observed. Somatic mosaicism and stuttering, observed routinely in PAGE assays, was also observed in the CE electropherograms (Figure 1 , panels d and f, MJD and SCA7). However, the dominant peak could be clearly assigned based on amplitude or peak area. Amplicon molecular weights calculated by Genescan based on the GS500-ROX internal molecular weight standard were underestimated compared to the manual PAGE assay.
Figure 2.
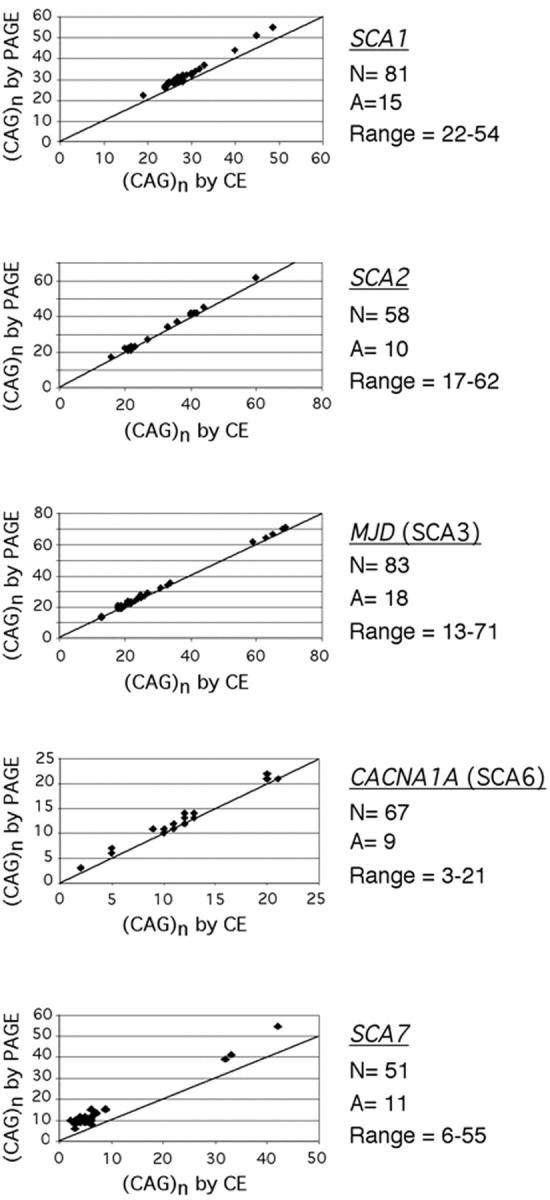
Comparision of SCA amplicon lengths as determined by PAGE versus CE. The graph for each locus documents that CE results in an underestimation of length, albeit to different degrees. The diagonal line represents the expected results if the two methods gave equivalent lengths. N, the total number of alleles tested; A, the number of alleles that had different (CAG)n; range, the range of (CAG)n tested. This assay had high precision of ± 1 repeat for normal alleles and ± 2 repeats for abnormal alleles based on data from our laboratory and from data reported by 25 other laboratories on samples provided for proficiency testing (MGLA 1999 and MGLB 2000 surveys, College of American Pathologists, Northfield, IL). In addition, several normal alleles were cloned and sequenced to confirm the number of CAG repeats; this was not possible for abnormal alleles due to their instability in Escherichia coli. The formulae for calculation of (CAG)n from amplicons sized by CE for each locus are: number of (CAG)n in SCA1 = [CE product size (bp) − 162]/3 × 1.0678 + 1.115; number of (CAG)n in SCA2 = [CE product size (bp) − 84]/3 × 1.0121 + 0.5342; number of (CAG)n in SCA3/MJD = [CE product size (bp) − 201]/3 × 1.0184 + 0.7062; number of (CAG)n in SCA6/CACNAIA = [CE product size (bp) − 126]/3 × 1.0075 + 0.5572; number of (CAG)n in SCA7 = [CE product size (bp) − 314]/3 × 1.1211 + 5.0958.
To analyze the apparent discrepancy in amplicon size, samples were assayed retrospectively by the multiplexed CE assay. Allele lengths were compared to those determined previously by the PAGE assay. During CE, both normal and expanded alleles for each locus migrated faster than predicted (Figure 2) . The degree of anomalous migration was different at each locus. Amplicons with greater numbers of CAG repeats tended to migrate faster, which was most pronounced at SCA1 and SCA7 (Figure 2) . Inter-assay variability, calculated as a 95% confidence interval, of 10 replicate assays of a normal and an expanded allele at each SCA locus, was ± 2 bp (data not shown).
To adjust for anomalous migration of amplicons during CE, a size correction formula was calculated by regression analysis of a pair-wise comparison of (CAG)n values determined by PAGE versus CE (Figure 2 legend). Because the precision of the PAGE assay (Table 2) was lower than the CE assay, multiple samples with the same number of CAG repeats as determined by PAGE were included in the analyses. To facilitate correction of amplicon length and minimize calculation errors in the clinical laboratory, a spreadsheet was generated in Excel (Microsoft, Redmond, WA) that converted raw amplicon length to number of CAG repeats based on the specific correction formula for each SCA. Fractional CAG-repeat numbers that were greater than 0.5 were rounded up to the next whole number. An advantage of the CE assay is that amplicons from a patient sample can be mixed with those from an abnormal control and re-injected to confirm allele sizes. For example, we routinely re-inject a mixture of any patient sample that has between 18 to 22 CAG repeats at CACNA1AI (SCA6) with the abnormal control (genotype 13, 21) to confirm that the patient’s repeat is ≥ 21 and therefore in the abnormal range. Before placing the multiplex/CE assay on-line in the clinical laboratory, 12 clinical samples were tested prospectively by both the PAGE and multiplex/CE assays with consistent results. To date, we have used this assay to test 57 patient samples referred for diagnostic or carrier testing of SCA1, SCA2, SCA3, SCA6, and SCA7. Five samples were positive, including one SCA1 case, two SCA2 cases, and two SCA3 cases.
Discussion
We have developed a rapid and precise semi-automated assay that employs multiplex PCR and CE for the molecular diagnosis of the spinocerebellar ataxias types 1, 2, 3, 6, and 7. Relatively minimal optimization was required for the multiplex reaction, presumably due to the use of chimeric primers. The observation that the SCA amplicons migrated faster than anticipated during CE, resulting in an underestimation of their lengths, was not unexpected. Capillary electrophoresis of fluorescent-labeled amplicons of the trinucleotide-repeat disorders Huntington disease (CAG)n and fragile X mental retardation (CGG)n, resulted in faster migration and subsequent underestimation of size. 11, 12, 13, 14 Faster migration of SCA, HD, and FMR1 amplicons compared to the internal size standards could be attributed to significant differences in base composition (repeat-containing versus non-repeat-containing), particularly GC-rich sequences. 13 Other factors contributing to sizing discrepancies could be different sieving effects of cross-linked PAGE compared to CE linear polymer, electro-osmotic effect, or extent of denaturation. 12, 13, 14
In trinucleotide-repeat disorders, an underestimation of amplicon size could result in a misdiagnosis, particularly for alleles that are near the threshold of the normal, intermediate, or abnormal size ranges. Routine clinical use of CE for diagnosis of at least some trinucleotide-repeat disorders requires a method that corrects the sizing discrepancy. For Huntington disease, an allele-specific molecular weight ladder was assembled using gene fragments that varied in CAG-repeat number, thereby minimizing sequence differences between amplicons and size markers. 13, 15 This approach could not be used in the SCA assay because it consists of amplicons of five loci that migrate differentially during CE. For fragile X mental retardation, a commercially-available molecular weight ladder was used and amplicons lengths were multiplied by a migration factor to give an accurate length. 11 This approach was applied successfully to the multiplex SCA assay, in part due to the high precision of the instrument in calculating amplicon length and the low inter-assay variability.
This assay has proved reliable in a clinical setting for 1 year. There is no evidence that factors, such as capillary age or replacement, have affected the correction formulae. Analysis of normal and abnormal controls showed no change in amplicon length as calculated by Genescan using the GS500-ROX molecular weight standard over this time. It is reasonable to expect that our assay for the diagnosis of SCA types 1, 2, 3, 6, and 7 could be extended to include some additional SCA loci. Up to 15 loci have been multiplexed successfully 10 and other unique fluorochromes are available and being developed, depending on the instrument. It is important to note that our assay is not expected to detect the very large CAG expansions in infantile/juvenile onset SCA2 or SCA7. The electrophoresis ends before such large fragments would be detected, assuming that they would amplify sufficiently in this assay. Therefore, additional testing is needed for at-risk SCA2 and SCA7 infants and juveniles. 16 This multiplex semi-automated assay greatly improves testing efficiency and turn-around time thereby facilitating the diagnosis of spinocerebellar ataxia in the clinical setting. It will also be invaluable in the research setting for screening cohorts with autosomal-dominant spinocerebellar ataxia and/or sporadic ataxia to determine prevalence of specific SCA types.
Address reprint requests to Karen Stephens, Ph.D., University of Washington, 1959 NE Pacific St., Room I-204, Medical Genetics 357720, Seattle, WA 98195-7720. E-mail: millie@u.washington.edu.
Footnotes
Supported in part by Department of Defense Neurofibromatosis Research Program grant DAMD17–00-1–0542 (to K.S.).
References
- 1.Bird TD: Ataxia overview. GeneClinics: Clinical Genetic Information Resource [database online]. University of Washington, Seattle, updated 07/25/00, www.geneclinics.org/profiles/ataxias
- 2.Klockgether T, Wullner U, Spauschus A, Evert B: The molecular biology of the autosomal-dominant cerebellar ataxias. Mov Disord 2000, 15:604-612 [DOI] [PubMed] [Google Scholar]
- 3.Schelhaas HJ, Ippel PF, Beemer FA, Hageman G: Similarities and differences in the phenotype, genotype, and pathogenesis of different spinocerebellar ataxias. Eur J Neurol 2000, 7:309-314 [DOI] [PubMed] [Google Scholar]
- 4.Tan EK, Ashizawa T: Genetic testing in spinocerebellar ataxias: defining a clinical role. Arch Neurol 2001, 58:191-195 [DOI] [PubMed] [Google Scholar]
- 5.Stephens K, Tait JF, Jacobs DS, Garg U: Molecular Genetic Testing. Jacobs DS DeMott WR Oxley DK eds. Laboratory Test Handbook. 2001, :pp 701-718 Lexi-Comp Inc., Cleveland [Google Scholar]
- 6.Potter NT, Nance MA; for the Ataxia Mol Diagnostic Testing Group [Allingham-Hawkins DJ, Bellissimo D, Bridge P, Graham L, Brown C, Garber A, Leonard D, Rennert H, Margolis R, McIntosh N, Muralidharan K, Popovich B, Anoe K, Ray P, Richards CS, Gunaratne P, Schaefer F, Seltzer W, Sims K, Xin W, Snow K, Stephens K, Wick M]: Genetic testing for ataxia in North America. Mol Diagn 2000, 5:91–99 [DOI] [PubMed]
- 7.Stevanin G, Dürr A, Brice A: Clinical and molecular advances in autosomal-dominant cerebellar ataxias: from genotype to phenotype and physiopathology. Eur J Hum Genet 2000, 8:4-18 [DOI] [PubMed] [Google Scholar]
- 8.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T: Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 2000, 26:191-194 [DOI] [PubMed] [Google Scholar]
- 9.Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP: An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 1999, 21:379-384 [DOI] [PubMed] [Google Scholar]
- 10.Shuber AP, Grondin JV, Klinger KW: A simplified procedure for developing multiplex PCRs. Genome Res 1995, 5:488-493 [DOI] [PubMed] [Google Scholar]
- 11.Larsen LA, Gronskov K, Norgaard-Pedersen B, Brondum-Nielsen K, Hasholt L, Vuust J: High-throughput analysis of fragile X (CGG)n alleles in the normal and premutation range by PCR amplification and automated capillary electrophoresis. Hum Genet 1997, 100:564-568 [DOI] [PubMed] [Google Scholar]
- 12.Le H, Fung D, Trent RJ: Applications of capillary electrophoresis in DNA mutation analysis of genetic disorders. Mol Pathol 1997, 50:261-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams LC, Hegde MR, Herrera G, Stapleton PM, Love DR: Comparative semi-automated analysis of (CAG) repeats in the Huntington disease gene: use of internal standards. Mol Cell Probes 1999, 13:283-289 [DOI] [PubMed] [Google Scholar]
- 14.Rosenblum BB, Oaks F, Menchen S, Johnson B: Improved single-strand DNA sizing accuracy in capillary electrophoresis. Nucleic Acids Res 1997, 25:3925-3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruland O, Almqvist EW, Goldberg YP, Boman H, Hayden MR: Accurate determination of the number of CAG repeats in the Huntington disease gene using a sequence-specific internal DNA standard. Clin Genet 1999, 55:198-202 [DOI] [PubMed] [Google Scholar]
- 16.Hsieh M, Lin SJ, Chen JF, Lin HM, Hsiao KM, Li SY, Li C, Tsai CJ: Identification of the spinocerebellar ataxia type 7 mutation in Taiwan: application of PCR-based Southern blot. J Neurol 2000, 247:623-629 [DOI] [PubMed] [Google Scholar]
- 17.Brandt V, Zoghbi HY: Spinocerebellar ataxia type 1. GeneClinics: Clinical Genetic Information Resource [database online]. University of Washington, Seattle, updated 01/26/01, www.geneclinics.org/profiles/sca1
- 18.Orr HT: The ins and outs of a polyglutamine neurodegenerative disease: spinocerebellar ataxia type 1 (SCA1). Neurobiol Dis 2000, 7:129-134 [DOI] [PubMed] [Google Scholar]
- 19.Pulst S-M: Spinocerebellar ataxia type 2. GeneClinics: Clinical Genetic Information Resource [database online]. University of Washington, Seattle, updated 01/19/01, www.geneclinics.org/profiles/sca2
- 20.Subramony SH, McDaniel DO, Smith S, Vig PJS: Spinocerebellar ataxia type 3. GeneClinics: Clinical Genetic Information Resource [database online]. University of Washington, Seattle
- 21.van Alfen N, Sinke RJ, Zwarts MJ, Gabreels-Festen A, Praamstra P, Kremer BP, Horstink MW: Intermediate CAG-repeat lengths (53,54) for MJD/SCA3 are associated with an abnormal phenotype. Ann Neurol 2001, 49:805-807 [DOI] [PubMed] [Google Scholar]
- 22.Gomez CM: Spinocerebellar ataxia type 6. GeneClinics: Clinical Genetic Information Resource [database online]. University of Washington, Seattle, updated 07/24/00, www.geneclinics.org/profiles/sca6
- 23.Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, Zoghbi HY: Molecular and clinical studies in SCA7 define a broad clinical spectrum and the infantile phenotype. Neurology 1998, 51:1081-1086 [DOI] [PubMed] [Google Scholar]
- 24.Matsuyama Z, Wakamori M, Mori Y, Kawakami H, Nakamura S, Imoto K: Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. J Neurosci 1999, 19:RC14 (1–5) [DOI] [PMC free article] [PubMed]
- 25.Babovic-Vuksanovic D, Snow K, Patterson MC, Michels VV: Spinocerebellar ataxia type 2 (SCA2) in an infant with extreme CAG-repeat expansion. Am J Med Genet 1998, 79:383-387 [PubMed] [Google Scholar]
- 26.Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, Holmberg M: Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG-repeat length on the clinical manifestation. Hum Mol Genet 1998, 7:171-176 [DOI] [PubMed] [Google Scholar]
- 27.Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY: Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993, 7:513-520 [DOI] [PubMed] [Google Scholar]
- 28.Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A, DeJong P, Rouleau GA, Auburger G, Korenberg JR, Figueroa C, Sahba S: Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996, 14:269-276 [DOI] [PubMed] [Google Scholar]
- 29.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, Kimura J, Narumiya S, Kakizuka A: CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:213–215, 221–228 [DOI] [PubMed]
- 30.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC: Autosomal-dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α 1A-voltage-dependent calcium channel. Nat Genet , 15:62-69 [DOI] [PubMed] [Google Scholar]
- 31.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A: Cloning of the SCA7 gene reveals a highly unstable CAG-repeat expansion. Nat Genet , 17:65-70 [DOI] [PubMed] [Google Scholar]