

Abstract
A family of N-substituted glycine oligomers (peptoids) of defined length and sequence are shown to condense plasmid DNA into small particles, protect it from nuclease degradation, and efficiently mediate the transfection of several cell lines. The oligomers were discovered by screening a combinatorial library of cationic peptoids that varied in length, density of charge, side-chain shape, and hydrophobicity. Transfection activity and peptoid–DNA complex formation are shown to be highly dependent on the peptoid structure. The most active peptoid is a 36-mer that contains 12 cationic aminoethyl side chains. This molecule can be synthesized efficiently from readily available building blocks. The peptoid condenses plasmid DNA into uniform particles 50–100 nm in diameter and mediates the transfection of a number of cell lines with efficiencies greater than or comparable to DMRIE-C, Lipofectin, and Lipofectamine. Unlike many cationic lipids, peptoids are capable of working in the presence of serum.
Viral and nonviral gene transfer systems have been under intense investigation, as interest in the potential of gene therapy for the treatment and prevention of disease is greater than ever (1). Nonviral systems potentially offer many advantages over viral systems, such as ease of manufacture, safety, stability, lack of vector size limitations, low immunogenicity, and the modular attachment of targeting ligands (2). Most nonviral gene delivery systems are based on cationic compounds—either cationic lipids (2) or cationic polymers (3)—that spontaneously complex with a plasmid DNA vector by means of electrostatic interactions, yielding a condensed form of DNA that shows increased stability toward nucleases. Although cationic lipids have been quite successful at delivering genes in vitro, the success of these compounds in vivo has been modest, often because of their high toxicity and low transduction efficiency.
A wide variety of cationic polymers have been shown to mediate in vitro transfection, ranging from proteins [such as histones (4) and high mobility group (HMG) proteins (5)] and polypeptides [such as polylysine (3, 6), short synthetic peptides (7, 8), and helical amphiphilic peptides (9, 10)] to synthetic polymers [such as polyethyleneimine (11), cationic dendrimers (12, 13), and glucaramide polymers (14)]. Although the efficiencies of gene transfer vary with these systems, a large variety of cationic structures are effective. Unfortunately, it has been difficult to study systematically the effect of polycation structure on transfection activity.
To understand more fully the structure–activity relationship of these cationic polymer delivery systems, we examined a set of cationic N-substituted glycine oligomers (NSG peptoids) of defined length and sequence. Peptoids are a large family of synthetic oligomers that were originally developed for the combinatorial synthesis of compound libraries for drug discovery (15). Recent optimization of the solid-phase coupling chemistry has allowed the synthesis of long oligomers—up to 50 monomers in length—that are similar in size to many of the aforementioned polycationic transfection reagents. The synthesis of peptoid oligomers (16, 17) is substantially cheaper than the synthesis of peptides and allows the incorporation of diverse side-chain structures—giving flexibility to probe in detail the structural requirements of any active oligomer. Additionally, peptoids are stable to hydrolysis by proteases (18).
A diverse set of peptoid oligomers, composed of systematic variations in main-chain length, frequency of cationic side chains, overall hydrophobicity, and shape of side chain, were synthesized. Interestingly, only a small subset of peptoids were found to yield active oligomers. Many of the peptoids were capable of condensing DNA and protecting it from nuclease degradation, but only a repeating triplet motif (cationic-hydrophobic-hydrophobic) was found to have transfection activity. The most potent compound, a 36-mer, was shown to transfect many cell lines in the presence of fetal calf serum. Electron microscopy revealed that this 36-mer forms highly regular spherical structures (50–100 nm in diameter) when condensed with DNA.
MATERIALS AND METHODS
Peptoid Synthesis.
Solvents, amines, and other reagents were purchased from commercial sources and used without further purification. Peptoid oligomers were synthesized on 50 μmol of Rink amide resin (Nova Biochem) by a modification of previous methods (16, 17). Briefly, after removal of the first Fmoc group the following 90-min monomer addition cycle was performed by a robotic synthesizer and repeated until the desired length was obtained. The amino-resin was bromoacetylated by adding 830 μl of 1.2 M bromoacetic acid in N,N-dimethylformamide (DMF) and 200 μl of N,N′-diisopropylcarbodiimide (DIC). This solution was agitated for 40 min at 35°C, drained, and washed with DMF (three times with 2 ml each). Next, 0.85 ml of a 1 M solution of a primary amine in dimethyl sulfoxide was added to introduce the side chain. This solution was agitated for 40 min at 35°C, drained, and washed with DMF (four times with 2 ml each). After the last coupling the peptoid-resin was cleaved and lyophilized as previously described (17). The peptoids were dissolved in water at a concentration of 5.0 mM and stored at −20°C.
N-terminally modified peptoids (e.g., AMCA-NpeNpeNae; see Tables 1 and 2 for nomenclature) were obtained by treating the fully elongated peptoid-resin with 1.4 ml of Fmoc-aminohexanoic acid (0.4 M)/hydroxybenzotriazole (0.4 M) in DMF and 133 μl of DIC. The reaction mixture was incubated at 35°C for 2 hr, drained, and washed with DMF (three times with 2 ml each). The Fmoc group was removed with 20% piperidine in DMF. The free primary amino group was then acylated with 1 ml of 0.2 M aminomethylcoumarin-succinimide (Molecular Probes) in DMF for 1 hr at 35°C, followed by washing with DMF (three times with 2 ml each). The peptoid-lipid conjugate was prepared as described elsewhere (C. Huang, T.U., J.M., S. Lee, J.D.H., J. Escobedo, F.E.C., R. Radhakrishnan, V.D., and R.N.Z., unpublished work).
Table 1.
Cationic peptoid oligomers
| Designator | Molecular weight
|
Designator | Molecular weight
|
Designator | Molecular weight
|
|||
|---|---|---|---|---|---|---|---|---|
| Calc | Found | Calc | Found | Calc | Found | |||
| (Nae)18 | 1819.0 | ND | (NchmNchmNae)12 | 4895.9 | 4894.0 | (NgalNgalNae)12 | 6479.2 | 6479.4 |
| (Nae)36 | 3621.0 | ND | (NbnNbnNae)12 | 4750.7 | 4748.0 | (Ngal)12(NpeNpeNae)12 | 7717.8 | 7715.1 |
| (NspeNspeNae)6 | 2552.2 | 2550.5 | (NpeNpeNae)12 | 5087.4 | 5086.3 | (NaeNpeNpe)12 | 5087.4 | 5085.1 |
| (NspeNspeNae)8 | 3397.3 | 3395.7 | (NppNppNae)12 | 5424.1 | 5421.1 | AMCA(NpeNpeNae)12 | 5415.8 | 5412.7 |
| (NspeNspeNae)12 | 5087.4 | 5085.8 | (NhpeNhpeNae)12 | 5471.4 | 5468.5 | DMPE(NpeNpeNae)12 | 5763.3 | 5760.9 |
| (NspeNspeNae)16 | 6775.8 | 6773.9 | (NcpeNcpeNae)12 | 5914.0 | 5910.9 | (NaeNpeNpe)12 | 5087.4 | 5086.3 |
| (NrpeNrpeNae)12 | 5087.4 | 5085.3 | (NmpeNmpeNae)12 | 5808.1 | 5805.6 | (NpeNaeNpe)12 | 5087.4 | 5084.7 |
| (NmeNmeNae)12 | 3981.7 | 3980.1 | (NieNieNae)12 | 6023.9 | 6021.2 | (NaeNmpeNmpe)12 | 5808.1 | 5805.0 |
AMCA, N-(7-amino-4-methylcoumarin-3-acetyl)-6-aminohexanoyl; DMPE, N-(dimirystoylphosphatidylethyl)glycine; see Table 2 for structures and names of Nae, etc. ND, not determined.
Table 2.
N-substituted glycine side chains and designators

The indicated bond is the site of attachment to the main-chain glycine nitrogen. Nae, N-(2-aminoethyl)glycine; Napr, N-(3-aminopropyl)glycine; Nme, N-(2-methoxyethyl)glycine; Nbn, N-benzylglycine; Nspe, (S)-N-(1-phenylethyl)glycine; Nchm, N-cyclohexylmethylglycine; Npe, N-(2-phenylethyl)glycine; Npp, N-(3-phenylpropyl)glycine; Nag, N-(6-aminogalactosyl)glycine; Nie, N-(2-(3′-indolylethyl)glycine; Nmpe, N-(2-(p-methoxyphenylethyl))glycine; Ncpe, N-(2-(p-chlorophenylethyl)glycine; Nhpe, N-[2-(p-hydroxyphenylethyl)]glycine.
Individual peptoid oligomers were analyzed by reverse-phase HPLC on C4 columns (Vydac, 5 μm, 300 Å, 1 × 150 mm) on a Magic 2002 system (Michrom Bioresources, Auburn, CA). A linear gradient of 5–95% solvent B in solvent A in 40 min was used at a flow rate of 100 μl/min [solvent A = 0.1% trifluoroacetic acid (TFA) in water; solvent B = 0.1% TFA in acetonitrile] at a column temperature of 60°C. Preparative HPLC was performed on a Delta-Pak C4 column (15 μm, 300 Å) on a Waters Prep LC3000 system using the same solvent system. Peaks were eluted with a linear gradient of 20–70% B in 40 min at a flow rate of 50 ml/min. Electrospray mass spectrometry was performed on a Platform II instrument (Micromass, Beverly, MA) at a cone voltage of 30 V.
Amine Submonomers.
N-t-BOC-1,2-diaminoethane and N-t-BOC-1,3-diaminopropane were prepared as described (19). 1,2,3,4-O-Diisopropyliden-6-amino-6-deoxy-α-d-galactose, O-tert-butyltyramine and Nin-t-Boc-tryptamine were prepared as described elsewhere (T.U., E. Beausoleil, R. Goldsmith, and R.N.Z., unpublished work).
Plasmids and Cell Lines.
NIH 3T3, HT1080, and COS cells were obtained from the American Type Culture Collection. 293 cells were obtained from Microbix (Toronto). HT1080, COS, and 293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; JRH Biosciences, Lenexa, KS) containing 10% fetal calf serum (FCS) (Summit Biotechnology, Ft. Collins, CO). NIH 3T3 cells were grown in DMEM containing 10% calf serum (HyClone).
The plasmid pCMVkmLuc was constructed by inserting the luc gene from pSP-luc+ (Promega) into the mammalian expression vector pCMVkm2. pCMVkm2 was derived from the plasmid pCMV6c (20) by replacing the simian virus 40 polyadenylation signal with the bovine growth hormone polyadenylation signal from pcDNA-3 (Invitrogen). Finally, the expression cassette, containing the luc gene driven by the cytomegalovirus promoter, was moved into the plasmid pHHS, which contains a kanamycin-resistance gene (21).
DNA Binding and Nuclease Protection.
The concentration of negative charges on DNA is calculated by using 3.03 nmol of phosphate per 1 μg of DNA. The formula weight of each peptoid is calculated as the semi-trifluoroacetate salt, and the peptoid concentration is determined on the basis of the lyophilized weight. Amino groups on peptoids are formally considered to be fully protonated to obtain the number of positive charges on the peptoid of interest when the +/− charge ratio is calculated.
DNA binding and nuclease protection studies are typically performed by incubating 3 μg of plasmid DNA with the appropriate amount of peptoid in 30 μl of water. After a 30-min incubation, complexes are divided into three parts. The first fraction (7.5 μl) is mixed with 2.5 μl of a sample buffer containing 40% sucrose, 0.25% bromophenol blue, 200 mM Tris⋅HCl at pH 7.8, and 5 mM EDTA (22) prior to electrophoresis. The second 7.5-μl fraction is mixed with 2.5 μl of an identical sample buffer which also contains 5% SDS. The third fraction is incubated with 1 unit of DNase (Boehringer Mannheim) in a 10 mM MgCl2 buffer for 15 min at 37°C. This fraction is then mixed with sample buffer containing SDS, and electrophoresed. For serum protection studies, complexes are mixed at a 1:1 ratio with serum from BALB/c mice, incubated at 37°C for 15 min, mixed with sample buffer containing SDS, and subjected to agarose gel electrophoresis.
Physical Characterization of Peptoid DNA Complexes.
Transmission electron microscopy of negative-stained complexes was performed with a Zeiss 10C transmission electron microscope. The sample solution was placed on a grid precoated with carbon and Formvar, and excess liquid was removed by blotting with a tissue after 5 min. To stain the sample, 4% aqueous uranyl acetate was placed on the grid for 2–5 sec and then removed by blotting, and the grid was allowed to dry. Particle size of the complex was measured by dynamic light scattering using a N4 Plus instrument (Coulter).
Transfections.
Twenty-four hours before transfection, cells were plated in DMEM containing 10% (vol/vol) FCS at 2 × 105 cells per well of a 6-well dish. The following day the medium was removed from each well and replaced with 2 ml of fresh DMEM containing 10% (vol/vol) FCS or Opti-MEM (GIBCO/BRL). To form transfection complexes, peptoid was added to Opti-MEM at the appropriate concentration and allowed to incubate for 5 min, and DNA was added to a final DNA concentration of 10 μg/ml. After a 5-min incubation, 100 μl of this solution was added to each well. The cationic lipids Lipofectin, Lipofectamine, and DMRIE-C were obtained from GIBCO/BRL. These lipids were used according to the manufacturer’s protocol. Poly(l-lysine) (Mr 23,800) (Sigma) and DNA, at a 1:1 (wt/wt) ratio, were incubated in Opti-MEM for 15 min before they were added to cells. Polylysine-mediated transfections were performed in either DMEM containing 10% FCS or DMEM containing 10% FCS and 100 mM chloroquine (Sigma).
Analysis of Luciferase Activity.
After transfection, cells were grown for 48–72 hr before lysis. Firefly luciferase content of the lysate was quantified by comparing to a known standard using the Promega luciferase assay reagent on an automated luminometer (Dynatech ML2250). Total protein content of the supernatant was determined by BCA assay (Pierce BCA protein assay reagent).
RESULTS AND DISCUSSION
Synthesis.
The reported DNA transfer activities of polylysine and other cationic peptides suggest that DNA transfer can be achieved with relatively short oligomers (7–10). Little effort has been made to systematically optimize the structures of these compounds. Because peptoids are protease resistant (18) and can have potent biological activity (15), we undertook a systematic investigation of the DNA transfer properties of a diverse library of cationic peptoid oligomers. A number of parameters were varied, including the oligomer length, periodicity of cationic residues, overall hydrophobicity, and side-chain shape. In total, we synthesized and characterized 67 oligomers for this study. In this paper, we describe the characterization of a representative subset of 24 oligomers (Table 1). The structure and nomenclature of the peptoid side chains are shown in Table 2.
Peptoids were synthesized on a robotic synthesizer (23) by a modification of the solid-phase submonomer method (16). Multiple parallel synthesis of dozens of oligomers of defined length and sequence was achieved from very low-cost starting materials. Each 90-min monomer-addition cycle generates an N-substituted glycine monomer in two steps from bromoacetic acid and a variety of primary amines. The commercial availability of more than one thousand amines and the high-yield coupling chemistry mean that an incredibly wide variety of sequence-specific oligomers can be generated and evaluated for DNA transfer activity. Furthermore, a structure–activity relationship can be generated for active oligomers because precise and subtle changes in the structure can be easily made.
The purity of most crude 36-mers was found to be 60–75%, indicating that the coupling yields per monomer addition cycle were in excess of 98%. Crude preparations were used for the screening, but active molecules were purified by preparative C4 reverse-phase HPLC and retested. Electrospray mass spectrometry confirmed the structures of all compounds (Table 2 and Fig. 1).
Figure 1.

Analysis of the most active cationic peptoid (NaeNpeNpe)12 by reverse-phase HPLC, showing the crude (i) and purified (ii) products, and by electrospray mass spectrometry of the crude product (Inset).
Formation of Peptoid–DNA Complexes.
Gel shift assay. All of the peptoid-based polymers synthesized for this study would be predicted to complex spontaneously with plasmid DNA owing to an extensive network of electrostatic interactions. Binding of a cationic peptoid to plasmid DNA results in neutralization of negative charges in the phosphate backbone of DNA. This interaction can be measured by the inability of these large electroneutral complexes to migrate toward the cathode during agarose gel electrophoresis. The ability of peptoids to complex with DNA was characterized by incubating peptoids with plasmid DNA at a 3:1 (+/−) charge ratio and analyzing the complexes by agarose gel electrophoresis (Fig. 2A). The majority of peptoids tested in this and other experiments (data not shown) are capable of completely retarding the migration of DNA into the gel. In this experiment, the single exception is peptoid (NieNieNae)12, which fails to retard the migration of DNA into the gel. Thus, the electrostatic complexation with DNA is very tolerant to changes in the specific sequence of side-chain residues in the peptoid.
Figure 2.

Peptoids interact with DNA and protect from DNase I degradation. The indicated peptoids were complexed with DNA at a 3:1 (+/−) charge ratio and divided into three aliquots: A shows the ability of peptoids to retard the migration of plasmid DNA into the gel; B shows the DNA after the complexes are disrupted with 1% SDS prior to electrophoresis; and C shows the DNA after DNase treatment and SDS disruption.
Nuclease protection. Condensation of DNA by cationic polymers, such as polylysine and histones, has been shown to protect supercoiled DNA from degradation by DNase I (4, 22). To test the ability of peptoids to protect DNA from nuclease degradation, complexes were formed as described and divided into two fractions. One fraction was treated with 1% SDS and electrophoresed directly on an agarose gel (Fig. 2B). The second fraction was treated with 1 unit of DNase I at 37°C for 30 min and then with 1% SDS to dissociate complexes and to inactivate DNase before the analysis (Fig. 2C). While DNase treatment led to complete degradation of naked DNA (compare Fig. 2B lane 19 and Fig. 2C lane 19), the majority of peptoids provided some protection to plasmid DNA. Many of the peptoids analyzed were able to completely protect DNA. Other peptoids offered partial protection; for example, (NmeNmeNae)12 and (Nag)12(NpeNpeNae)12 protect the nicked circle DNA (upper band) but not the supercoiled DNA (lower band). At the charge ratio used in this experiment, (Nae)36 and (NieNieNae)12 fail to protect DNA from nuclease treatment. This result is consistent with the inability of (NieNieNae)12 to bind to DNA. Interestingly, (Nae)36 interacts with DNA in a manner that apparently eliminates the nicked form in the untreated sample. (NagNagNae)12 also leads to an altered migration in the absence of DNase treatment. This altered migration may result from an insensitivity of the complex to SDS treatment.
Because cationic peptoids do not have peptide bonds, they should be insensitive to degradation by serum proteases (18). This stability should lead to protection of DNA from serum nucleases. We have tested the effects of serum on DNA–peptoid complexes by measuring the ability of complexed DNA to withstand nuclease degradation (Fig. 3). Plasmid DNA was incubated with cationic peptoid at a 1.5:1 (+/−) charge ratio or with polylysine at a 1:1 (wt/wt) ratio for 30 min and complexes were subsequently incubated with 50% BALB/c mouse serum for 30 min before electrophoresis (Fig. 3). This treatment led to complete degradation of naked DNA (lane 4). Although the peptoid used in this study, (Nae)36, failed to protect plasmid DNA from purified DNase (Fig. 2), it offered substantial protection of DNA from nucleolytic activity of mouse serum (lane 1). Interestingly, (Nae)36 provided substantially more protection to supercoiled DNA than did (Nae)18 (lane 2). In contrast, polylysine was capable of condensing DNA at this ratio (not shown) but was not able to protect DNA from nuclease digestion (lane 3).
Figure 3.
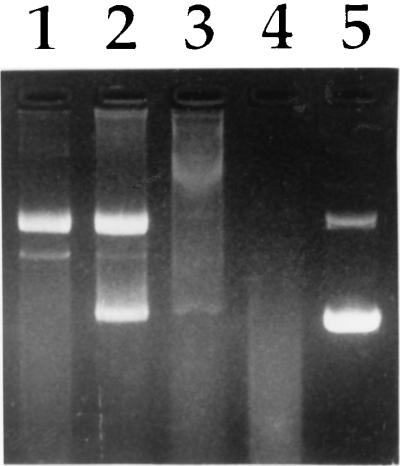
Protection from serum degradation. Lane 1, (Nae)18; lane 2, (Nae)36; lane 3, poly(l-lysine); lane 4, DNA alone; and lane 5, untreated DNA.
Characterization of peptoid–DNA complexes. Peptoids and plasmid DNA complexes at a 2:1 (+/−) charge ratio were examined by negative-stained transmission electron microscopy. As with other common polycationic polymers such as polylysine, DNA complexes with (NmeNmeNae)12 and (Nae)36 displayed heterogeneous populations of complexes including both rod-shaped and toroidal particles (Fig. 4 a and b). In contrast, (NpeNpeNae)12, the most effective transfection reagent, condensed DNA into highly homogenous spherical particles whose diameters were around 50–60 nm (Fig. 4d). These values are in good agreement with the size determined by dynamic light scattering (around 80 nm). Interestingly, (NhpeNhpeNae)12, which differs from (NpeNpeNae)12 by only one hydroxyl group per aromatic monomer, formed only extended heterogeneous aggregates (Fig. 4c). Thus, the spacing of charged residues on the peptoid chain as well as the degree of hydrophobicity of the side chains has a dramatic effect on the ability to form homogenous complexes with DNA in high yield.
Figure 4.

Electron microscopy of peptoid–DNA complexes. (a) (Nae)36. (b) (NmeNmeNae)12. (c) (NhpeNhpeNae)12. (d) (NaeNpeNpe)12. (Bars indicates 200 nm.)
In Vitro Transfection with Cationic Peptoid–DNA Complexes.
Although cationic homopolymers, such as polylysine, are capable of condensing and protecting DNA, they are not efficient transfection reagents. In contrast, amphipathic peptides have been shown to be relatively efficient at mediating DNA uptake by cells. To identify new transfection reagents, a library of peptoids, in which cationic and noncationic side chains are distributed in specific patterns, was analyzed. Peptoids were initially screened by mixing at a 2:1 (+/−) charge ratio with the plasmid pCMVkmLuc, which carries the firefly luciferase marker gene, and adding complexes to HT1080 and COS cells. All transfections were done in the presence of 10% FCS.
Fig. 5 shows the results of a single transfection experiment for a representative set of peptoids. The ability of a peptoid to mediate transfection of the indicator cell lines is highly dependent on the primary sequence of the peptoid and, to a lesser degree, on the length of the peptoid. The results are not dependent on the indicator cell line; active peptoids were active on both lines and inactive peptoids did not work on either line. As with polylysine, peptoids that were homopolymers with cationic side chains, such as (Nae)36, did not mediate transfection of marker cell lines. All of the peptoids that showed high transfection activity contained a repeating trimer motif (aromatic-aromatic-cationic) in which aromatic residues were interspersed with cationic residues. Optimal activity was seen with the peptoid (NaeNpeNpe)12, in which the aromatic side chain was analogous to that of phenylalanine. In contrast, peptoids in which cationic side chains are interspersed with nonaromatic side chains were poor transfection reagents. Examples of this group include (NmeNmeNae)12, in which cationic residues are interspersed with a short aliphatic side chain, (NagNagNae)12, which contains hydrophilic galactosyl side chains, and (NchmNchmNae)12, which contains cyclic nonaromatic side chains. Both (NmeNmeNae)12 and (NagNagNae)12 behaved aberrantly in the binding and protection assays (Fig. 2). Interestingly, some peptoids that contain the aromatic-aromatic-cationic motif perform poorly in the transfection assay. (NieNieNae)12, which contains an aromatic ring analogous to the indole side chain of tryptophan, showed no activity in the transfection assay, consistent with its failure to bind and protect DNA. The peptoid (NhpeNhpeNae), which contains side chains analogous to that of tyrosine, also showed no activity in the transfection assay. This peptoid was fully functional in the DNA binding and protection assays, suggesting that the modification of the aromatic ring with the hydroxyl group affects a function that is required for cell entry. This may be a result of the morphology of its DNA complex, which is very heterogeneous (Fig. 4c). Other modifications of the ring, such as addition of a chloro or methoxy group in the para position, have only modest effects on the transfection activity.
Figure 5.

Comparison of peptoid transfection efficiency. Indicated peptoids were formulated with DNA at a 2:1 charge ratio and added to either HT1080 (solid bars) or COS (open bars) in the presence of 10% serum. Luciferase activity was analyzed 48 hr after transfection. Each data point represents the average of two experiments.
Derivatization of the amino terminus of the active peptoid was also well tolerated. Peptoid AMCA-(NpeNpeNae)12 has a fluorescent group appended to the amino terminus by a 6-carbon linker. Peptoid DMPE-(NpeNpeNae)12 is derivatized with the lipid dimyristoyl phosphatidylethanolamine (DMPE). We have taken advantage of the amino group of phosphatidylethanolamine to create a series of lipid-peptoid hybrid molecules, which are highly efficient transfection reagents (C. Huang, T.U., J.E.M., S. Lee, J.D.H., J. Escobedo, F.E.C., R. Radhakrishnan, V.D., and R.N.Z., unpublished data). The ability to derivatize a defined site in the peptoid side chain will allow for the controlled synthesis of delivery vehicles modified with targeting ligands.
To study the effect of peptoid chain length on transfection efficiency, peptoids consisting of various lengths of the same motif (NspeNspeNae)n were synthesized (Fig. 5). In general, the longer peptoids appear to have higher activity, with a maximum reached at about 36 residues. In the results shown here and in other experiments, the 36-mer performs about 2-fold better than the 24-mer and the 48-mer on both cell lines. What is most striking is the complete inability of the 18-mer peptoid to mediate transfection. At the tested ratio, this peptoid is capable of condensing DNA and protecting it from serum nucleases (data not shown), therefore the failure does not result from an inability to form complexes and withstand serum treatment. The 18-mer peptoid is also inactive as a transfection reagent in serum-free medium. It is possible that the presence of physiologic levels of NaCl in the transfection medium disrupt the interaction of this peptoid with DNA.
Comparison to Cationic Lipid-Mediated Transfection.
The most advanced systems for both in vivo and in vitro nonviral gene delivery are based on cationic lipids. To determine the efficiency of peptoid-mediated delivery, (NaeNpeNpe)12, the most effective structure, was compared directly with DMRIE-C, Lipofectin, and Lipofectamine on three indicator cell lines. In these experiments (NaeNpeNpe)12 was complexed with DNA at a 2:1 (+/−) charge ratio. Preliminary studies indicated that this was the optimal ratio; toxicity became a problem at higher ratios and efficiency decreased at lower ratios. The cationic lipids were also used at 2:1 charge ratios. In all cases this ratio fell within the manufacturer’s suggested range. In all of the tested cell lines, the efficiency of peptoid-mediated transfection is similar to transfection by cationic lipids in serum-free medium (Fig. 6). In contrast to Lipofectin and Lipofectamine, which are respectively 10- and 100-fold less efficient in the presence of serum, gene transfer by the cationic peptoid is insensitive to the presence of serum in the transfection medium. Among the tested reagents, the absolute level of gene transfer is greatest on 293 cells (note that the scale is one order of magnitude greater than for other cell lines). Similarly, NIH 3T3 cells are the most refractory to transfection with all of the reagents tested. Peptoid-mediated transfection and lipid-mediated transfection likely share a common cellular pathway that is limiting in NIH 3T3 and HT1080 cells. The toxicity of the transfection reagent was determined by visual examination of cells and by measuring total protein recovered from cells 48 hr after transfection. Toxicity of (NaeNpeNpe)12 was cell line dependent: under the conditions tested, no toxicity was seen in NIH 3T3 cells, whereas there was a 20–30% reduction in viability (relative to untransfected cells) in 293 and HT1080 cells.
Figure 6.

Comparison of (NaeNpeNpe)12 peptoid with commercially available cationic lipids. Transfections were carried out in the presence (solid bar) or absence (open bar) of 10% serum and analyzed 48 hr after transfection. Compound 1 is Lipofectin, 2 is Lipofectamine, 3 is DMRIE-C, and 4 is (NaeNpeNpe)12. Each data point represents the mean ± SEM of three transfections.
Effect of Chloroquine on Peptoid-Mediated Transfection.
Transfection mediated by cationic polymers, such as polylysine and histones, is greatly enhanced by addition of chloroquine to the transfection medium. To determine whether chloroquine affected peptoid-mediated transfection, HT1080 and 293 cells were transfected in the presence of chloroquine (Fig. 7). As a control, the same cell lines were transfected with polylysine in the presence and absence of chloroquine. On both 293 and HT1080 cell lines, chloroquine leads to a 100-fold increase in polylysine-mediated transfection. In contrast, the effect of chloroquine on peptoid-mediated transfection is negligible. The requirement for chloroquine reflects the inability of polylysine to escape the endosome. It can be inferred that transfection with the (NaeNpeNpe)12 peptoid is not limited by an inability to escape the endosome.
Figure 7.

Effect of chloroquine on transfection with (NaeNpeNpe)12. Either 293 (A) or HT1080 (B) cells were transfected in the presence (black bar) or absence (shaded bar) of 100 μM chloroquine. Luciferase activity and total protein content were measured 48 hr after transfection.
CONCLUSION
We have synthesized a novel class of transfection reagents based on polymers of N-substituted glycine (peptoids). These polymers have a number of desirable properties, including protease resistance and serum insensitivity. Furthermore, the peptoid chemistry lends itself to a modular approach to the design of gene delivery vehicles; side chains with different functional groups can be readily incorporated into the peptoid, and ligands for targeting specific cell types or tissues can be appended to specific sites on the peptoid.
Our data highlight the value of being able to synthesize and test a large number of polymers for gene delivery. Simple analogies to known active peptides (e.g., polylysine) did not directly lead to active peptoids. The diverse screening set used here revealed that an unexpected specific triplet motif was the most active transfection reagent. Whereas some minor changes lead to improvement in transfection [compare (NbnNbnNae)12 with (NaeNpeNpe)12], other minor changes abolished the capability of the peptoid to mediate transfection [compare (NaeNpeNpe)12 with (NhpeNhpeNae)12].
At this point the difference between the active and inactive peptoids is not clear. As seen in the electron microscopy studies, the most active peptoid forms a regular structure with DNA, whereas the inactive peptoids seem to form a less ordered structure. This regular structure suggests an underlying specific mode of interaction with DNA. The threefold periodicity corresponds to the periodicity expected for helical peptoids (24), although the inclusion of helix-inducing monomers did not increase the activity (Fig. 5). In this context, we speculate that whereas the positively charged side chains interact with the phosphate backbone of the DNA, the aromatic residues facilitate the packing interactions between peptoid monomers. In addition, the aromatic monomers are likely to be involved in critical interactions with the cell membrane during transfection.
Acknowledgments
We thank Drs. Chin-Yi Huang, Mike Innis, and Jaime Escobedo for helpful discussions, Ivy Hirsch, Dr. T. S. Ben Yen of the Veterans Affairs Medical Center and University of California San Francisco for the electron micrographs, and Dr. Ayub Khan for help with plasmid construction.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: DMF, N,N-dimethylformamide; DIC, N,N′-diisopropylcarbodiimide; FCS, fetal calf serum.
References
- 1.Mulligan R C. Science. 1993;260:926–932. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- 2.Ledley F D. Hum Gene Ther. 1995;6:1129–1144. doi: 10.1089/hum.1995.6.9-1129. [DOI] [PubMed] [Google Scholar]
- 3.Wu G Y, Wu C H. J Biol Chem. 1987;262:4429–4432. [PubMed] [Google Scholar]
- 4.Fritz J D, Herweijer H, Zhang G, Wolff J A. Hum Gene Ther. 1996;7:1395–1404. doi: 10.1089/hum.1996.7.12-1395. [DOI] [PubMed] [Google Scholar]
- 5.Mistry A R, Falciola L, Monaco L, Tagliabue R, Acerbis G, Knight A, Harbottle R P, Soria M, Bianchi M E, Coutelle C, Hart S L. BioTechniques. 1997;22:718–729. doi: 10.2144/97224rr01. [DOI] [PubMed] [Google Scholar]
- 6.Wagner E, Cotten M, Mechtler K, Kirlappos H, Birnstiel M L. Bioconjugate Chem. 1991;2:226–231. doi: 10.1021/bc00010a006. [DOI] [PubMed] [Google Scholar]
- 7.Gottschalk S, Sparrow J T, Hauer J, Mims M P, Leland F E, Woo S L C, Smith L C. Gene Ther. 1996;3:448–457. [PubMed] [Google Scholar]
- 8.Wadhwa M S, Collard W T, Adami R C, McKenzie D L, Rice K G. Bioconjugate Chem. 1997;8:81–88. doi: 10.1021/bc960079q. [DOI] [PubMed] [Google Scholar]
- 9.Legendre J Y, Trzeciak A, Bohrmann B, Deuschle U, Kitas E, Supersaxo A. Bioconjugate Chem. 1997;8:57–63. doi: 10.1021/bc960076d. [DOI] [PubMed] [Google Scholar]
- 10.Wyman T B, Nicol F, Zelphati O, Scaria P V, Plank C, Szoka F C., Jr Biochemistry. 1997;36:3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 11.Boussif O, Zanta M A, Behr J-P. Gene Ther. 1996;3:1074–1080. [PubMed] [Google Scholar]
- 12.Tang M X, Redemann C T, Szoka F C., Jr Bioconjugate Chem. 1996;7:703–714. doi: 10.1021/bc9600630. [DOI] [PubMed] [Google Scholar]
- 13.Haensler J, Szoka F C., Jr Bioconjugate Chem. 1993;4:372–379. doi: 10.1021/bc00023a012. [DOI] [PubMed] [Google Scholar]
- 14.Goldman C K, Soroceanu L, Smith N, Gillespie G Y, Shaw W, Burgess S, Bilbao G, Curiel D T. Nat Biotech. 1997;15:462–466. doi: 10.1038/nbt0597-462. [DOI] [PubMed] [Google Scholar]
- 15.Zuckermann R N, Martin E J, Spellmeyer D C, Stauber G B, Shoemaker K R, Kerr J M, Figliozzi G M, Goff D A, Siani M A, Simon R J, Banville S C, Brown E G, Wang L, Richter L S, Moos W H. J Med Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
- 16.Zuckermann R N, Kerr J M, Kent S B H, Moos W H. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 17.Figliozzi G M, Goldsmith R, Ng S C, Banville S C, Zuckermann R N. Methods Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- 18.Miller S M, Simon R J, Ng S, Zuckermann R N, Kerr J M, Moos W H. Drug Dev Res. 1995;35:20–32. [Google Scholar]
- 19.Krapcho A P, Kuell C S. Synth Commun. 1990;20:2559–2564. [Google Scholar]
- 20.Chapman B S, Thayer R M, Vincent K A, Haigwood N L. Nucleic Acids Res. 1991;19:3979–3986. doi: 10.1093/nar/19.14.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seifert H S, Chen E Y, So M, Heffron F. Proc Natl Acad Sci USA. 1986;83:735–739. doi: 10.1073/pnas.83.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X, Huang L. Biochemistry. 1996;35:1027–1036. doi: 10.1021/bi952436a. [DOI] [PubMed] [Google Scholar]
- 23.Zuckermann R N, Kerr J M, Siani M A, Banville S C. Int J Peptide Protein Res. 1992;40:497–506. doi: 10.1111/j.1399-3011.1992.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 24.Armand P, Kirshenbaum K, Falicov A, Dunbrack R L, Jr, Dill K, Zuckermann R N, Cohen F E. Folding Design. 1997;2:369–375. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]