

Abstract
FLT3 is a receptor tyrosine kinase that is expressed on early hematopoietic progenitor cells and plays an important role in stem cell survival and differentiation. Two different types of functionally important FLT3 mutations have been identified. Internal tandem duplication mutations arise from duplications of the juxtamembrane portion of the gene and result in constitutive activation of the FLT3 protein. This alteration has been identified in ∼20% to 30% of patients with acute myelogenous leukemia and appears to be associated with a worse prognosis. The second type of FLT3 mutation, missense mutations at aspartic acid residue 835, occurs in ∼7.0% of acute myelogenous leukemia cases. These mutations also appear to be activating and to portend a worse prognosis. Identification of FLT3 mutations is important because it provides prognostic information and may play a pivotal role in determining appropriate treatment options. We have developed an assay to identify both internal tandem duplication and D835 FLT3 mutations in a single multiplex polymerase chain reaction. After amplification, the polymerase chain reaction products are analyzed by capillary electrophoresis for length mutations and resistance to EcoRV digestion. Here we describe the performance characteristics of the assay, assay validation, and our clinical experience using this assay to analyze 147 clinical specimens.
FMS-like tyrosine kinase 3 (FLT3 also known as STK1 and flk2) is a member of the class III receptor tyrosine kinase family that also includes PDGF-R, KIT, and FMS. 1 The FLT3 protein is normally expressed on hematopoietic stem progenitor cells and appears to play an important role in stem cell survival, and the development of dendritic and natural killer cells. 2, 3 FLT3 is overexpressed in most cases of acute myeloid leukemia (AML). 4, 5 In addition, analysis of leukemic blasts from AML patients has identified two specific somatic mutations of the FLT3 gene. 6, 7 Identification of these mutations in AML patients provides independent prognostic information that may also prove important for treatment optimization.
The first and best-studied FLT3 mutation is an internal tandem duplication (ITD) mutation. ITD mutations typically result from the duplication and tandem insertion of a portion of the juxtamembrane (JM) region (exons 11 to 12) of the FLT3 wild-type gene. 6 The lengths of the duplicated segments have been reported to range in size from 6 to 180 bases and are always in frame. 8, 9 ITD mutations result in the constitutive autophosphorylation of the FLT3 receptor and are thus gain-of-function mutations of the FLT3 proto-oncogene. 10 FLT3 ITD mutations have been reported to occur in 20 to 30% of patients with AML and have been associated with an increased relapse risk, decreased disease-free survival, decreased event-free survival, and decreased overall survival. 8, 9, 11 In a multivariate analysis of FLT3 ITD mutations, cytogenetic risk group, presentation white blood cell count, percentage BM blasts at diagnosis, age, gender, and FAB type in 854 AML patients, the presence of a FLT3 ITD mutation was the most significant factor adversely affecting relapse risk (P < 0.0001) and disease-free survival (P < 0.0001). 9 FLT3 ITD mutations are amenable to polymerase chain reaction (PCR)-based molecular diagnostic DNA testing because they are limited to a small, predictable region of the FLT3 gene.
Recently, an additional type of FLT3 mutation has been described. These alterations are missense mutations that alter the wild-type aspartic acid residue at position 835 (D835) within the activation loop of the FLT3 protein. 7, 12 Alteration of D835 also appears to result in constitutive activation of the FLT3 receptor and portends a worse disease-free survival in at least some studies. 7 D835 mutations have been reported to occur in ∼7% of patients with AML, 3% of patients with myelodysplastic syndrome (MDS), and 3% of patients with acute lymphocytic leukemia. 7 D835 and ITD mutations appear to occur independently but not exclusively of one another and the presence of concurrent D835 and ITD mutations has been reported. 7 The D835 wild-type gene sequence is located within an EcoRV restriction endonuclease cut site, a feature exploited by the reported assay.
Detection of ITD and D835 FLT3 mutations is clinically important for several reasons. First, patients harboring these mutations generally have a worse prognosis and may benefit from aggressive up-front treatment interventions. Secondly, both ITD and D835 mutations may serve as markers for the detection of residual disease, which may become an important part of posttreatment disease monitoring. 13 Finally, investigators have recently developed specific tyrosine kinase inhibitors of the FLT3 receptor for use as tumor-specific chemotherapeutic agents, 14, 15, 16, 17 analogous to the use of STI-571 [imatinib mesylate (Gleevac); Novartis, Basel, Switzerland] in the treatment of chronic myelogenous leukemia. 18 Clinical trials are currently underway looking at the utility of FLT3 inhibitors in the treatment of relapsed or refractory AML with FLT3 mutations. Clinical testing for FLT3 mutations in AML may thus become critical to the determination of appropriate therapeutic interventions in AML.
Here we describe a molecular diagnostic approach capable of detecting both ITD and D835 mutations of the FLT3 gene in a single multiplex PCR assay. We discuss the assay design strategy, assay validation, and our experience with the clinical application of the assay.
Materials and Methods
Samples
For the validation study, viably frozen AML blast cells were obtained from leukemia patients through an IRB-approved protocol at our institution. Appropriate informed consent, approved by the Johns Hopkins School of Medicine (JHUSOM) Joint Commission on Clinical Investigation was obtained before sample procurement. For clinical testing, DNA was extracted from peripheral blood or BM samples. The diagnosis of AML includes 1° AML, 2° AML, and AML arising out of MDS. Other diagnoses consist of three cases of myeloproliferative disease, three cases of acute lymphocytic leukemia, two biphenotypic leukemias, one connective tissue disorder with aplasia, and three cases without a known diagnosis. For all samples, DNA was extracted using the QIAamp DNA kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
PCR
Multiplex PCR reactions were composed of 1× PCR Buffer (Applied Biosystems, Foster City, CA), 200 μmol/L each dNTP (Applied Biosystems), 0.2 μmol/L each of four primers (Oligos Etc., Wilsonville, OR), 0.002% gelatin and 2.5 U Taq Gold (Applied Biosystems). Primers for the ITD portion of the assay were 11F, 5′-6-FAM-GCAATTTAGGTATGAAAGCCAGC-3′ and 12R, 5′-HEX-CTTTCAGCATTTTGACGGCAACC-3′, previously described unlabeled. 19 The primers for the D835 portion of the assay were D835F: 5′-TET-GTAAAACGACGGCCAGCCGCCAGGAACGTGCTTG-3′ and D835R: 5′-CAGGAAACAGCTATGACGATATCAGCCTCACATTGCCCC-3′. Thermocycling conditions were: 95°C for 9 minutes followed by 35 cycles of 95°C for 30 seconds, 56°C for 1 minute, 72°C for 2 minutes, with a final extension at 72°C for 7 minutes.
Restriction Digestion and Capillary Electrophoresis (CE)
After amplification, 8.5 μl of multiplex PCR product was digested with 0.5 μl of EcoRV (10U/μl) and 1 μl of restriction buffer REACT 2 (Invitrogen, Inc.,Carlsbad, CA). Each digestion reaction was incubated at 37°C for 1 hour, followed by heat inactivation at 65°C for 10 minutes. One μl of digested multiplex PCR product was mixed with deionized formamide and TAMRA size standard per the manufacturer’s protocol, heated to 95°C for 5 minutes, and placed on ice for at least 1 minute before electro-kinetic injection to the ABI 310 capillary electrophoresis instrument (Applied Biosystems).
Polyacrylamide Gel Electrophoresis and Band-Stab
A small proportion of clinical cases contained low numbers of neoplastic cells resulting in ITD mutant peaks with very low CE peak intensity. To enrich the proportion of mutant PCR products so that they could be confirmed by sequencing as ITD mutants, amplification products were separated by PAGE, band-stabbed, and reamplified. PCR products were separated by 5% PAGE, stained with ethidium bromide, and visualized under UV light. A 22-gauge needle was stabbed into the band of interest and then rinsed into an Eppendorf tube with 20 μl of PCR-grade dH2O. Five μl of the band-stab product was then subjected to PCR and CE as described above.
Sequencing
Five of the ITD mutations and the single D835 mutation identified during the assay validation were cycle sequenced in the forward and reverse direction to verify the results. PCR products were purified using QIAQuick columns (Qiagen) and cycle sequenced using Big Dye, Version 2 (Applied Biosystems) according to the manufacturer’s protocol. For ITD sequencing the ITD PCR primers were used. For D835 sequencing, M13F and M13R primers were used. Sequences were aligned and examined using Sequencher software (Gene Codes Corp., Inc., Ann Arbor, MI).
Results
Assay Design
The overall design of this assay is a multiplex PCR with fluorescently labeled primers followed by EcoRV restriction endonuclease digestion and separation/sizing by CE. FLT3 ITD mutations are readily detectable by PCR amplification because the duplication occurs at a relatively small, predictable area of the FLT3 gene, the JM region (Figure 1) . The D835 mutation site is also amenable to PCR testing because the mutation is conveniently located within an EcoRV restriction endonuclease digestion site. Mutation of the wild-type sequence results in loss of this cut site, generating a larger fragment after EcoRV digestion (Figure 1) .
Figure 1.

Diagram of the assay design. The FLT3 gene consists of five extracellular immunoglobulin-like domains, a transmembrane domain (TM), a JM domain, and an interrupted kinase domain (TK1 and TK2). PCR primers flanking the JM domain [forward primer labeled with FAM (blue), reverse primer labeled with NED (yellow)] and primers specific for the TK2 domain [forward labeled with TET (green), reverse unlabeled] are multiplexed into a single PCR reaction. After amplification, the PCR products are digested with EcoRV. The dotted lines in the TK2 PCR product represent the EcoRV cut sites, with the recognition sequence (GATATC). The JM portion of the PCR yields a wild-type PCR product of 330 bases labeled with both FAM and NED. FLT3 ITD mutations result in PCR products that are longer than wild type (>330 bp), also labeled with both FAM and NED. After digestion, the D835 portion of the assay yields wild-type products sizing at 80 bases that are TET labeled. D835 mutant TET-labeled products size at 129 bases, and undigested TET-labeled products size at 150 bases.
For PCR amplification of the ITD mutation, primers were selected that amplify exons 11 and 12 of FLT3. Both the forward and reverse ITD PCR primers were fluorescently labeled with FAM and NED, respectively. A wild-type result consists of FAM- and NED-labeled PCR products at 330 bases (Figure 2A) . ITD mutants yield FAM- and NED-labeled PCR products that are greater in size than wild type (Figure 2B) . We chose to double-label the ITD PCR product to increase both the sensitivity and specificity of the assay. Because ITD mutations are of variable size, nonspecific peaks in a single color resulting from nonspecific PCR products or from pull-up peaks (capillary baseline noise), could be interpreted as a positive result if only a single fluor is used in the assay. In the assay reported here, wild-type and ITD peaks must report in both colors for the result to be valid, thus decreasing the likelihood of a false-positive result. In addition, although unlikely, it is possible that an ITD mutation could create an EcoRV restriction digestion site. In a worst-case scenario, although highly unlikely, digestion of products labeled with a single fluor at the newly formed cut site could yield products exactly the same size as wild type, generating a false-negative result. In the reported assay if an EcoRV digestion site is generated by an ITD mutation, it would be virtually impossible for digestion to yield both FAM- and NED-labeled fragments exactly the same size as wild-type PCR product. Digestion would lead to an errant blue or black peak that should raise suspicion and indicate to the interpreter that further analysis may be necessary. Thus the use of two fluors for the ITD portion of the assay should decrease the likelihood of both false-positive and false-negative results.
Figure 2.
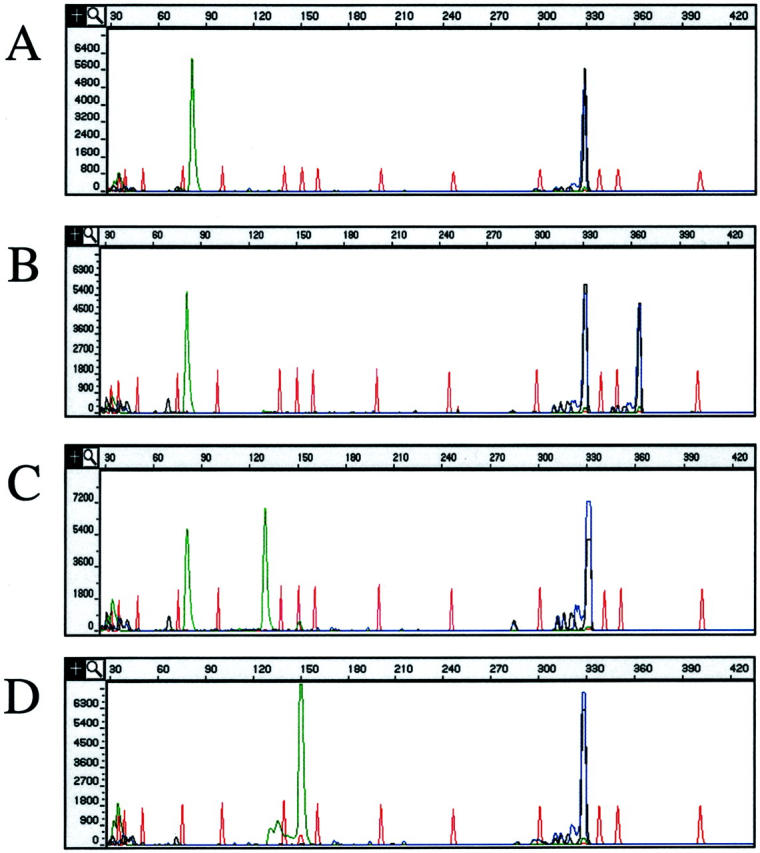
Examples of results of the FLT3 assay. A–D: CE pherograms; x axis represents size of the PCR products in bases, y axis represents relative fluorescence intensity. Red peaks represent internal size standard. Green (TET) PCR product peaks result from the D835 portion of the assay. Blue (FAM) and black (NED) peaks result form the ITD portion of the assay. A: Example of a FLT3 D835 wild-type/ITD wild-type result. B: Example of a FLT3 D835 wild-type/ITD mutant result. C: Example of a D835 mutant/ITD wild-type result. D: Example of an incomplete digest for the D835 assay and a wild-type ITD.
D835 mutation detection was accomplished using a forward PCR primer to which M13 forward (M13F) sequence and the fluorescent molecule TET were added at the 5′ end. The reverse PCR primer was unlabeled and tailed with M13 reverse (M13R) sequence. In addition, an EcoRV cut site was engineered into the reverse D835 PCR primer between the coding and M13R sequence of the primer. PCR amplification using these primers yields a 150-bp product. EcoRV digestion of wild-type D835 PCR product results in an 80-base TET-labeled fragment that can be detected by CE (Figure 2 A and B) . D835 mutations eliminate the EcoRV wild-type digestion site resulting in a longer, TET-labeled fragment of 129 bases from the mutant allele (Figure 2C) . The EcoRV cut site engineered into the reverse primer serves as an internal control for completeness of restriction enzyme cutting, such that the TET-labeled 150-base products that result from incomplete digestion (Figure 2D) can easily be distinguished from D835 mutant products (TET-labeled 129 bases) (Figure 2C) .
Assay Validation
Twenty-six samples from 23 AML patients previously characterized for FLT3 mutations were analyzed during assay validation. In addition, 25 samples from healthy donors were tested. Fifteen AML samples (15 of 26, 58%) yielded a wild-type result for both the ITD and D835 regions of the FLT3 gene. ITD mutations were detected in 10 AML samples (10 of 26, 38%). One of the 26 AML validation samples (1 of 26, 4%) revealed a D835 mutation. There were no cases in the validation group with evidence of concurrent ITD and D835 mutations. No FLT3 ITD or D835 mutations were identified in any of the samples from healthy donors (0 of 25).
The FLT3 ITD mutations identified in the validation study ranged in size from 18 to 183 nucleotides and always resulted in in-frame alterations. Five of the samples with ITD mutations were sequenced, confirming and characterizing the ITDs in these samples (data not shown). The proportion of ITD to wild-type products was heterogeneous, ranging from mutant products of low intensity, comprising only a small percentage of the total FLT3 alleles, to samples comprised virtually entirely of ITD alleles. One of the samples (1of 26, 4%) harbored two different ITD mutations as indicated by PCR products that were 39 and 90 bases greater in length than the wild-type PCR product.
The D835 mutation identified in the validation set was confirmed by sequence analysis of the uncut PCR product in both the forward and reverse direction using M13 F and R primers. Sequencing revealed an A to T transversion resulting in the replacement of the wild-type aspartic acid residue at position 835 with valine (D835V), a mutation that has previously been reported (data not shown). 7 The validation study showed 100% concordance between the results of this multiplex PCR assay and results obtained on the same samples using two separate reverse transcriptase-PCR assays.
Dilution experiments were performed to determine the lower limit of detection for the assay. Samples with essentially equal amounts of wild-type and either ITD or D835 mutant alleles (an ∼100% malignant cell population, heterozygous for either mutation) were diluted into wild-type DNA. Using this approach we were able to reliably detect a 1:12 dilution of heterozygous mutant DNA (either ITD or D835) in wild type, and frequently could detect a 1:24 dilution. For clinical diagnostic use the lower limit of detection of this assay is quoted to be 10% malignant cells heterozygous for a mutation (5% of the total alleles). To ensure the 10% limit of detection, we require a wild-type peak height of >3000 RFU to interpret a valid negative result. This cutoff ensures that we could detect a peak that represents 5% of the total alleles (which should yield a mutant allele peak height of ≥150 RFU).
Summary of Clinical Experience
At the time of submission of this article, 147 clinical samples from 135 patients have been analyzed for FLT3 mutations using the reported approach. The majority of the samples we have tested using this assay have been peripheral blood or bone marrow samples from patients with AML although we have also tested samples from patients with MDS, acute lymphocytic leukemia, myeloproliferative disorders, and biphenotypic leukemia. Many of these samples were from outside institutions and their diagnosis could not be confirmed. Table 1 summarizes the patients tested. Of 110 patients reported to have AML, 22 (22 of 110, 20%) were positive for an ITD mutation. The ITD mutations identified in our clinical experience have ranged in size from 18 bp to 186 bp. Several of the mutant samples have been sequenced to confirm and characterize the ITD (data not shown). Three of the ITD-positive samples revealed the presence of two different ITD mutations (3 of 110, 2.7% of total AML, or 3 of 22, 13.6% of ITD-positive). The assay cannot distinguish whether these two mutations occurred in the same or different FLT3 alleles. No FLT3 ITD mutations were identified in the samples from patients with MDS (0 of 13) or other diagnoses (0 of 13).
Table 1.
Summary of Clinical Experience Testing for FLT3 Mutations
| Diagnosis | Number of patients | ITD positive D835 negative n (%) | ITD negative D835 positive n (%) | ITD positive D835 positive n (%) | Total FLT3 mutations |
|---|---|---|---|---|---|
| AML | 110 | 21 (19.1%) | 2 (1.8%) | 1 (0.9%) | 24 (21.8%) |
| MDS | 13 | 0 | 0 | 0 | 0 |
| Other* | 12 | 0 | 0 | 0 | 0 |
See Materials and Methods.
Three of the 110 patients with AML tested positive for a D835 mutation (3 of 110, 2.7%). D835 mutant samples were subjected to sequence analysis to confirm and characterize the mutation. One of the samples positive for a D835 mutation also contained an ITD mutation. This assay does not distinguish whether the two mutations are in the same or different FLT3 alleles. No FLT3 D835 mutations were identified in any of the samples from patients with MDS (0 of 13) or other diagnoses (0 of 12). Thus a total of 24 patients had identifiable FLT3 mutations (21 ITD, 2 D835, 1 both ITD and D835), yielding an overall FLT3 mutation rate of 24 of 110, 21.8%. No mutations have been identified in any cases with diagnoses other than AML. The results of our clinical experience are summarized in Table 1 .
Mutant Allele Enrichment/Advantages of CE
Two of the clinical samples with ITD mutations revealed evidence of very small amounts of ITD mutant alleles relative to wild type, an example of which is shown in Figure 3A . The blast counts of both of these patient samples were ≤5%, correlating with the findings of low-intensity mutant alleles. To confirm that the small peaks identified in these cases represented true ITD mutations, we sought to enhance the mutant band using a band-stab technique and repeat amplification. PCR products generated from the clinical assay were subjected to PAGE (Figure 3B) . ITD bands were very faint and would have been difficult to interpret clinically, possibly resulting in a false-negative (Figure 3B) . PCR products greater in size than wild type were band-stabbed and reamplified. CE analysis of the products demonstrated a nearly homogenous population of double-labeled PCR product that was exactly the same size (in bases) as the original, low-intensity ITD peak (Figure 3 compare A and C) . These products were confirmed to be true ITD mutants by sequence analysis (data not shown). This example demonstrates that the use of two fluors to double-label the PCR product, and CE detection allows for small amounts of ITD mutant products to be sensitively and specifically identified. For samples with low percentage neoplastic cells (at or below the stated limit of detection of this assay, 10%), band-stab and reamplification allows for a uniform population of mutant products to be generated that can be subjected to cycle sequencing to confirm that the products are the result of ITD mutations. In this situation, our clinical reports note that a low-intensity mutant peak was detected, correlating with the patient’s low blast count, and that the presence of a mutation was confirmed by sequence analysis.
Figure 3.

Example of enrichment of a sample containing a small number of FLT3 ITD mutant allele PCR products because of a low blast count in the sample tested. A: CE pherogram; x axis represents size of the PCR product in bases, y axis represents relative fluorescence intensity. Red peaks represent internal size standard. Blue/black peaks are the FAM/NED-labeled products of the ITD portion of the multiplexed assay. The large wild-type peak at 330 bases and small ITD mutant peak at 354 bases are indicated by black and red arrows, respectively. B: PAGE of the same PCR product as in A. Lane 1 is a 50-bp marker. Lane 2 is the same sample analyzed by CE in A. The black arrow indicates the 330-bp wild-type product, and the red arrow indicates the 354-bp ITD mutant PCR product. C: CE electropherogram after band-stab of the ITD mutant band and reamplification. Note that after band-stab and reamplification, the FLT3 ITD mutant PCR products are dominant compared to wild type.
Another advantage of CE is its superior resolution compared to PAGE. An example of this is shown in Figure 4 . In this case, the sample contains a mixture of a small amount of wild-type PCR product, and PCR product from two ITD mutations resulting from 18- and 21-bp insertions. The two ITD mutant products can readily be identified by CE (Figure 4A) . PAGE of the same PCR product revealed a small amount of wild-type PCR product, and what appears to be ITDs at 350 bp, 380 bp, 420 bp, and 500 bp. All of these bands were band-stabbed, reamplified, and subjected to CE (data not shown). As expected, the band at 350 yielded a mixture of the 348- and 351-base ITD products that could not be resolved by PAGE. Surprisingly, the 380- and 420-bp products also yielded a mixture of the 348- and 351-bp products. Also surprisingly, the 500-bp band-stab yielded a mixture of wild-type and the 348- and 351-bp ITD products. From this data it appears that the 380- and 420-bp bands on PAGE may be because of heteroduplex formation between the two different ITDs and the 500-bp band because of heteroduplex formation between the wild-type and ITD PCR products in the nondenaturing PAGE.
Figure 4.
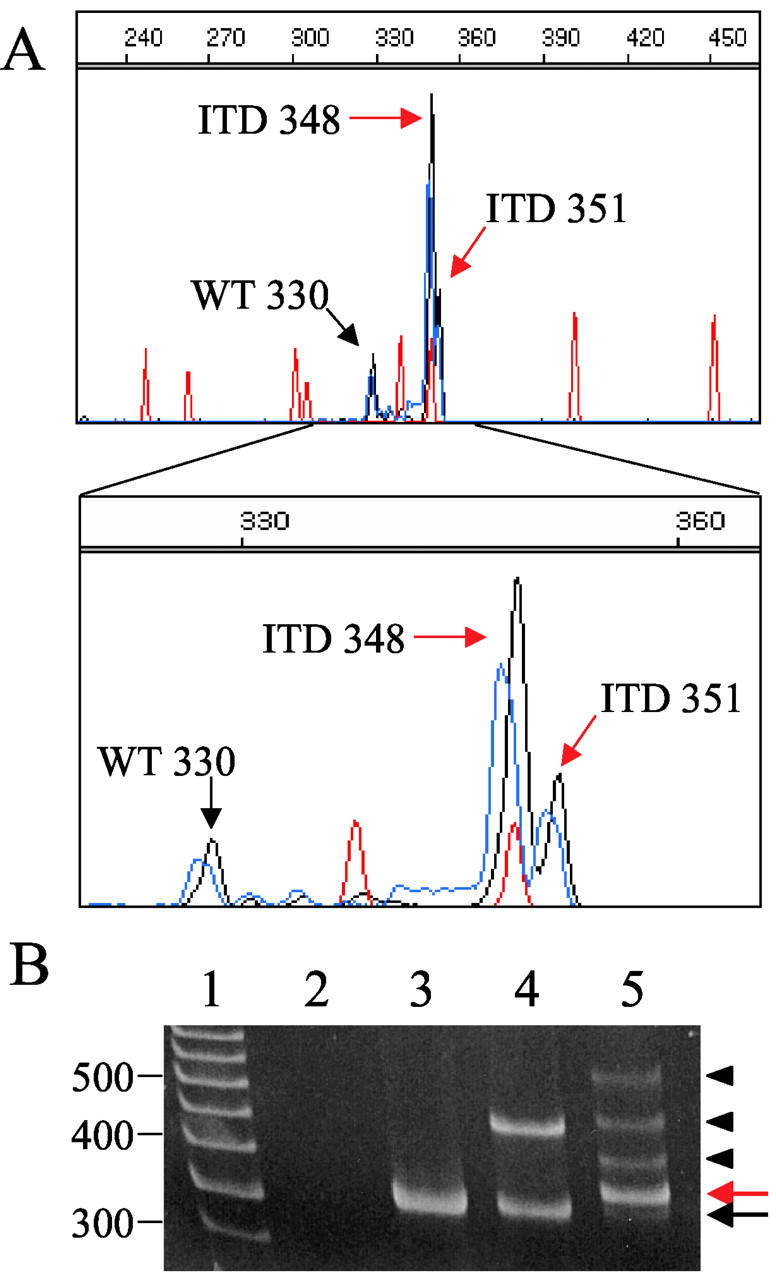
Example of improved resolution of CE compared to PAGE. A: CE pherogram of a sample positive for two different ITD mutations; x axis represents size of the PCR product in bases, y axis represents relative fluorescence intensity. Red peaks represent the internal size standard. The wild-type peak at 330 bases (black arrow) and two ITD mutant peaks at 348 and 351 bases (red arrows) are indicated. Below, the electropherogram is magnified to highlight the two different ITD mutations. B: PAGE of the same PCR product as in A. Lane 1 is a 50-bp marker, lane 2 is a no template control, lane 3 is a FLT3 wild-type control, lane 4 is a FLT3 ITD control, and lane 5 is the same sample as run by CE in A. The black arrow indicates the small amount of wild-type PCR product. The red arrow indicates the two ITD bands at 348 and 351 identified by CE. The arrowheads identify bands that are not detected by CE. These bands appear to be the result of heteroduplex formation between the two ITD products and between wild-type and ITD PCR products (see text).
Discussion
The reported FLT3 molecular diagnostic assay provides a reliable and robust method to simultaneously detect the two different types of FLT3 mutations currently known to be of importance in the natural history of AML. Experience reveals the assay to function reliably for both routine clinical testing and to be efficient enough to allow stat clinical testing. The reported assay has allowed us to achieve a 36-hour turnaround time from receipt of the clinical blood or bone marrow sample to written report of the diagnostic result. This format has facilitated rapid eligibility determination and enrollment into a FLT3 inhibitor trial currently underway at our institution.
Several features of this assay make it convenient for clinical diagnostic use. The multiplex PCR format of the assay provides for the simultaneous detection of both types of clinically relevant FLT3 mutations. We chose to design the assay as a DNA-based PCR reaction rather than a RNA-based assay to avoid assay variability often associated with RNA instability. The D835 portion of the assay is optimized for clinical use by the addition of an EcoRV cut site engineered into the D835 reverse primer, providing an internal quality control measure of the success of the EcoRV restriction enzyme digestion. The addition of M13 sequence to both primers facilitates sequencing of D835-positive samples for mutation confirmation and characterization. Our clinical reports reflect the fact that any alteration of D835 or I836 yields a positive result in this assay. One insertion mutation has previously been reported that alters I836. 7 Limited data suggest this mutation also results in constitutive activation of the FLT3 receptor and may be of biological and clinical importance. 7
The ITD portion of this assay is clinically appealing because of the use of fluorescently labeled forward and reverse primers and the use of CE for separation of the PCR products. Double-labeling the PCR products decreases the likelihood of false-positives, increasing the specificity of the assay. Detection of PCR products by CE allows for more accurate sizing of ITD mutations and the ability to resolve multiple ITD bands of similar size that cannot be resolved by PAGE. In addition, it appears that heteroduplex formation between wild-type and ITD PCR products can result in multiple bands on PAGE that are not independent ITD mutations. Because the CE format applied to this assay is denaturing, it detects only single-stranded PCR products, and thus does not suffer from this artifact. The significance of multiple ITD products is unclear at this time, however accumulation of this data may help establish the biological and clinical significance of this finding.
The assay we describe has been optimized for use as a qualitative diagnostic tool and is not optimal for quantitative assessment of minimum residual disease. Compared to assays that measure translocations (Bcr-Abl, PML-RARα), the FLT3 assay has decreased sensitivity because of competition between wild-type and mutant FLT3 alleles. Without the use of mutation (patient)-specific primers, an adequately high level of assay sensitivity is difficult to achieve in this testing situation. It is currently unclear what role FLT3 mutation analysis will have as a tool for minimum residual disease detection in AML. Three groups have studied FLT3 mutations in paired diagnostic and relapse samples. 20, 21, 22 From these studies, it appears that a significant percentage of relapsed patients will have acquired an ITD mutation that was not detectable at diagnosis, or will have lost an ITD mutation that was detected at diagnosis. Although the exact frequency at which FLT3 mutation status is discrepant at diagnosis and relapse is still unclear, it is clear that molecular evolution may make minimum residual disease detection difficult in a subset of AML patients.
Although it has been reported that the ratio of ITD mutant peak height to the wild-type peak height provides additional prognostic information, 8 we currently do not report this data. Clearly this ratio is highly dependent on the percentage of neoplastic cells in the population tested. In many clinical testing situations, this ratio may simply reflect the ratio of the normal to neoplastic cells in the mixture and therefore would have to be interpreted in that context. We do however report the absence or near absence of wild-type PCR products, indicating a FLT3 loss of heterozygosity event. It has been reported that disease-free survival and overall survival are significantly inferior for patients with FLT3 ITD mutations and loss of heterozygosity of the wild-type allele (FLT3 −/−) compared to patients with FLT3 wild-type (FLT3 +/+) or FLT3 ITD mutations without loss of heterozygosity (FLT3 +/−). 23
Our experience has revealed a slightly higher rate of ITD mutations in our validation study set (38%) than expected (20 to 30%) that is also higher than that seen in our clinical experience with the assay (20%). This probably results from the use of banked samples in the validation study that are likely to have a collection bias toward patients with high peripheral white blood cell counts. FLT3 ITD mutations correlate with elevated white blood cell counts, likely explaining this result and highlighting the need to accumulate FLT3 mutation data prospectively. The FLT3 mutation rate in our clinical experience may be slightly lower than expected because of testing samples with blast counts below the limit of detection for this assay (10%).
The clinical identification of FLT3 mutations in a prospective manner will yield important information about the incidence and natural history of FLT3 mutations in AML. In addition, identification of FLT3 mutations is likely to become important for optimization of patient care. Because FLT3 ITD mutations portend a worse prognosis, it has been proposed that patients testing positive for a FLT3 mutation may benefit from aggressive up-front treatment regimens such as an allogeneic bone marrow transplantation. On-going clinical trials will determine whether AML patients with FLT3 mutations will also benefit from novel therapeutic strategies that target and inhibit FLT3 tyrosine kinase activity.
Acknowledgments
We thank Dr. James Eshleman for helpful technical discussions and Patrick Pearson for technical assistance.
Address reprint requests to Kathleen M. Murphy, Ph.D., Carnegie Bldg., Room 367, 600 North Wolfe St., Baltimore, MD 21287. E-mail: kmurphy4@jhmi.edu.
References
- 1.Blume-Jensen P, Hunter T: Oncogenic kinase signalling. Nature 2001, 411:355-365 [DOI] [PubMed] [Google Scholar]
- 2.Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC: FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood 1996, 88:3383-3390 [PubMed] [Google Scholar]
- 3.McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, Maliszewski CR, Lynch DH, Smith J, Pulendran B, Roux ER, Teepe M, Lyman SD, Peschon JJ: Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95:3489-3497 [PubMed] [Google Scholar]
- 4.Birg F, Courcoul M, Rosnet O, Bardin F, Pebusque MJ, Marchetto S, Tabilio A, Mannoni P, Birnbaum D: Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood 1992, 80:2584-2593 [PubMed] [Google Scholar]
- 5.Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, Witte L, Borowitz MJ, Civin CI, Small D: Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996, 87:1089-1096 [PubMed] [Google Scholar]
- 6.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S: Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10:1911-1918 [PubMed] [Google Scholar]
- 7.Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Saito H, Ueda R, Ohno R, Naoe T: Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97:2434-2439 [DOI] [PubMed] [Google Scholar]
- 8.Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, Wermke M, Bornhauser M, Ritter M, Neubauer A, Ehninger G, Illmer T: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99:4326-4335 [DOI] [PubMed] [Google Scholar]
- 9.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, Goldstone AH, Linch DC: The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001, 98:1752-1759 [DOI] [PubMed] [Google Scholar]
- 10.Kiyoi H, Towatari M, Yokota S, Hamaguchi M, Ohno R, Saito H, Naoe T: Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12:1333-1337 [DOI] [PubMed] [Google Scholar]
- 11.Yokota S, Kiyoi H, Nakao M, Iwai T, Misawa S, Okuda T, Sonoda Y, Abe T, Kahsima K, Matsuo Y, Naoe T: Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies: a study on a large series of patients and cell lines. Leukemia 1997, 11:1605-1609 [DOI] [PubMed] [Google Scholar]
- 12.Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT: Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol 2001, 113:983-988 [DOI] [PubMed] [Google Scholar]
- 13.Stirewalt DL, Willman CL, Radich JP: Quantitative, real-time polymerase chain reactions for FLT3 internal tandem duplications are highly sensitive and specific. Leuk Res 2001, 25:1085-1088 [DOI] [PubMed] [Google Scholar]
- 14.Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D: A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99:3885-3891 [DOI] [PubMed] [Google Scholar]
- 15.Levis M, Tse KF, Smith BD, Garrett E, Small D: A FLT3 tyrosine kinase inhibitor is selectively cytotoxic to acute myeloid leukemia blasts harboring FLT3 internal tandem duplication mutations. Blood 2001, 98:885-887 [DOI] [PubMed] [Google Scholar]
- 16.Kelly LM, Yu JC, Boulton CL, Apatira M, Li J, Sullivan CM, Williams I, Amaral SM, Curley DP, Duclos N, Neuberg D, Scarborough RM, Pandey A, Hollenbach S, Abe K, Lokker NA, Gilliland DG, Giese NA: CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell 2002, 1:421-432 [DOI] [PubMed] [Google Scholar]
- 17.Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, Gilliland DG, Griffin JD: Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1:433-443 [DOI] [PubMed] [Google Scholar]
- 18.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL: Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001, 344:1031-1037 [DOI] [PubMed] [Google Scholar]
- 19.Kiyoi H, Naoe T, Yokota S, Nakao M, Minami S, Kuriyama K, Takeshita A, Saito K, Hasegawa S, Shimodaira S, Tamura J, Shimazaki C, Matsue K, Kobayashi H, Arima N, Suzuki R, Morishita H, Saito H, Ueda R, Ohno R: Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia 1997, 11:1447-1452 [DOI] [PubMed] [Google Scholar]
- 20.Nakano Y, Kiyoi H, Miyawaki S, Asou N, Ohno R, Saito H, Naoe T: Molecular evolution of acute myeloid leukaemia in relapse: unstable N-ras and FLT3 genes compared with p53 gene. Br J Haematol 1999, 104:659-664 [DOI] [PubMed] [Google Scholar]
- 21.Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC: Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood 2002, 100:2393-2398 [DOI] [PubMed] [Google Scholar]
- 22.Shih LY, Huang CF, Wu JH, Lin TL, Dunn P, Wang PN, Kuo MC, Lai CL, Hsu HC: Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood 2002, 100:2387-2392 [DOI] [PubMed] [Google Scholar]
- 23.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, Carroll AJ, Mrozek K, Vardiman JW, George SL, Kolitz JE, Larson RA, Bloomfield CD, Caligiuri MA: Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res 2001, 61:7233-7239 [PubMed] [Google Scholar]