

Abstract
Wolfram (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness) syndrome is a rare autosomal-recessive neurodegenerative disorder that is characterized by juvenile-onset diabetes mellitus, optic atrophy, diabetes insipidus, and sensorineural hearing impairment. A gene responsible for Wolfram syndrome (WFS1) has been identified on the short arm of chromosome 4 and subsequently mutations in WFS1 have been described. We have screened 12 patients with Wolfram syndrome from nine Dutch families for mutations in the WFS1-coding region by single-strand conformation polymorphism analysis and direct sequencing. Furthermore, we analyzed the mitochondrial genome for gross abnormalities and the A3243G point mutation in the leucyl-tRNA gene, because Wolfram syndrome shows phenotypic similarities with mitochondrial disease. Seven mutations in WFS1 were identified in six of nine families: two missense mutations, one frameshift mutation, one splice donor site mutation, and three deletions. In addition, a splice variant near the 5′UTR of WFS1 was identified, present in patient as well as control RNA samples in various percentages, alternating the translation initiation consensus sequence. Whether this WFS1 splice variant displays impaired translation efficiency remains to be determined. No MtDNA lesions were identified in any of the Wolfram patients. Our results demonstrate the usefulness of molecular analysis of WFS1 in the refinement of clinical diagnostic criteria for Wolfram syndrome that helps to dissect the clinically overlapping syndromes sharing diabetes mellitus and optic atrophy.
Wolfram syndrome (WS, MIM 222300) is an autosomal recessive disorder characterized by the association of juvenile-onset diabetes mellitus and optic atrophy. 1, 2 This disorder is also associated with diabetes insipidus and deafness, hence the acronym DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness). 2, 3, 4 WS is a progressive neurodegenerative disorder in which patients present with nonautoimmune and non-HLA-linked diabetes mellitus followed by optic atrophy in the first decade; cranial diabetes insipidus and sensorineural deafness in the second decade; renal tract abnormalities early in the third decade; and multiple neurological abnormalities, such as cerebellar ataxia, myoclonus, and psychiatric illness early in the fourth decade. WS patients usually die from central respiratory failure as a result of brainstem atrophy in their third or fourth decade. 5 WS is rare with an estimated prevalence of 1 in 770,000, and with a carrier frequency of 1 in 354. 5
The clinical phenotype of WS shows resemblance with mitochondrial disorders, such as maternally inherited diabetes and deafness, mitochondrial encephalopathy, mitochondrial myopathy, lactic acidosis and stroke-like episodes, or Leber’s hereditary optic neuropathy and much research has focused on mitochondrial pathology in WS. Mitochondrial disturbances at the biochemical, morphological, and molecular level have been described in WS patients, but this has not been a consistent finding. 6, 7, 8, 9, 10
Genetic linkage studies linked WS to the short arm of chromosome 4 11 and in 1998 the gene for WS, wolframin/WFS1 was identified. 12, 13 The WFS1 gene spans 33.4-kb of genomic DNA and is comprised of eight exons, of which the first exon is noncoding. The 3.6-kb WFS1 mRNA encodes a polypeptide of 890 amino acids predicted to have nine putative transmembrane domains, and an apparent molecular mass of 100-kd. The protein shows predominant subcellular localization to endoplasmic reticulum, 14 but no physiological function has been ascribed to the protein as yet. Since the identification of the WFS1 gene, more than 50 distinct mutations have been found in affected individuals of Wolfram families worldwide. 12, 13, 15, 16, 17, 18 These vary from nonsense, missense, to frameshift insertion/deletion lesions.
Here we have characterized the coding region of WFS1 in 12 WS patients from nine Dutch families. Also, mtDNA was examined for the presence of gross alterations and for the A3243G mutation in the leucyl tRNA gene, as has been described previously in patients with familial diabetes and deafness. 19
Materials and Methods
Patients
Twelve Wolfram patients from nine Dutch families participated in this study (the pedigrees are depicted in Figure 1 ). The patient’s major clinical characteristics are compiled in Table 1 . Consanguinity was present in two pedigrees (WF1 and WF3). Remarkable homogeneity is observed among the affected siblings reflecting minor intrafamiliar variability. The families were of Turkish (WF1), Dutch (WF2, WF4 to WF8), Dutch/former-Yugoslavian (WF9), and Moluk (WF3) descent. Patients of six families (WF1 to WF6) met the minimal diagnostic criteria for WS being juvenile-onset diabetes mellitus (younger than 30 years) and optic atrophy. 5 Patients from WF7 and WF8 showed an atypical phenotype in a way that they all developed diabetes mellitus (as well as optic atrophy) at the age of 35 years or older. A patient from family WF9 displayed an alternative phenotype in a way that diabetes insipidus as well as deafness was present, although diabetes mellitus and optic atrophy have not been diagnosed. However, a single abnormal oral glucose tolerance test at the age of 11 years and signs of myopia and enlarged papil nervi optici had been documented.
Figure 1.
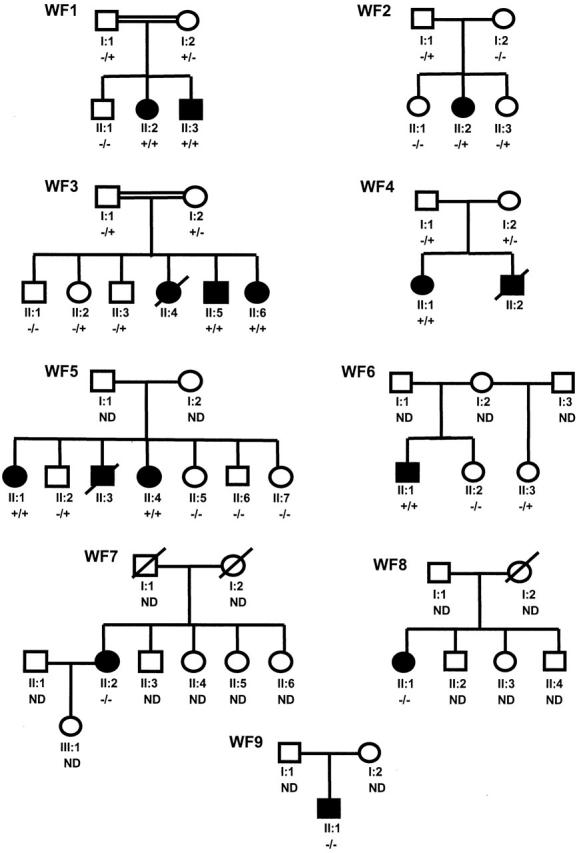
The pedigrees of Wolfram families WF1 to WF9. The first line below each symbol represents generation (roman numeral) and identification number. −, Absence of WFS1 mutation; +, presence of WFS1 mutation; ND, not determined.
Table 1.
Clinical Characteristics of Dutch Wolfram Patients
| Family and patient number | Age | Sex | Consang. | DM | OA | DI | D | Renal tract abnormalities | Neurologic abnormalities | Other complications (age at onset) |
|---|---|---|---|---|---|---|---|---|---|---|
| WF1, II-2 | 28 | F | + | 3 | 22 | 18 | 7 | + (7) | − | Paranoid depression |
| II-3 | 22 | M | + | 3 | 11 | 19* | 13 | − | Seizures | Depression |
| WF2, II-2 | 20 | F | − | 5 | 6 | 11* | 11 | + (11) | Cerebellar atrophy, Babinski reflex | |
| WF3, II-5 | 32 | M | + | 1 | 10 | 6 | 9 | + (20) | Anosmia | Pulmonal stenosis (18), primary gonadal atrophy |
| II-6 | 30 | F | + | 4 | 13 | − | 12 | + (13) | − | − |
| WF4, II-1 | 35 | F | − | 4 | 8 | 4.5 | 9 | − | Peripheral neuropathy (DM) | Irritable bowel syndrome |
| WF5, II-1 | 47 | F | − | 10 | 28 | − | 45 | − | − | − |
| II-4 | 42 | F | − | 12 | 26 | − | − | − | − | Hypothyroidism, hypertension |
| WF6, II-1 | 28 | M | − | 4 | 12 | 11 | 25 | + (17) | − | Scoliosis, primary gonadal atrophy |
| WF7, II-1 | 54 | F | − | 37 | 37 | − | 54 | − | − | Diplopia |
| WF8, II-1 | 59 | F | − | 35 | 45 | − | 6 | − | − | Hypoparathyroidism, hypercalcaemia, bipolar disorder |
| WF9, II-1 | 38 | M | − | ?* | ?* | 1.5 | 5.5 | ? | Epilepsy | Hyperkeratosis, adipositas |
Age in years; M, male; F, female; DM, diabetes mellitus; DI, diabetes insipidus; D, deafness; OA, optic atrophy; consang., consanguinity. Age of onset indicated in years. +, presence of condition;−,absence of condition; *, possible; ?,unknown.
DNA Analysis
Patient blood samples and skin biopsies were collected on informed consent. Total DNA was extracted from leukocytes or cultured fibroblasts by standard procedures.
WSF1 Analysis
Exons 2 to 8 (exon 1 is noncoding) of the WFS1 gene were amplified by polymerase chain reaction (PCR) using sets of primers as previously published by Strom and colleagues, 13 with the exception of exon 4 in which primers as described by Inoue and colleagues 12 were used.
In WF1, WF2, WF3, WF6, and WF9, mutation screening was performed by the single-strand conformation polymorphism (SSCP) technique. An equal volume of formamide-containing buffer was added to an aliquot of the amplified PCR product and the sample was denatured at 95°C for 5 minutes, quick-cooled, and electrophoresed on glycerol-free 6% polyacrylamide gels at 20°C for 2.5 hours at 30 W (DCode Universal Mutation Detection System; Biorad, Veenendaal, The Netherlands). Bands were visualized by silver staining (Amersham Pharmacia Biotech, Amersham Biosciences Europe Gmdh, Roosen daal, The Netherlands). DNA samples of Wolfram patients with well-characterized mutations in the WFS1 gene 13 and samples known to carry no WFS1 mutation were included in the SSCP analysis as positive and negative controls, respectively. Abnormal migrating bands were reamplified, purified either from solution or from agarose gel by GFX columns (Amersham Pharmacia Biotech), and directly sequenced with both forward and reverse primers using the Thermo Sequenase Cy5.5 dye terminator cycle-sequencing kit (Amersham Pharmacia Biotech). Sequencing products were run on a SEQ4x4 personal DNA sequencer (Amersham Pharmacia Biotech). In the case of WF9, the negative result from SSCP analysis prompted us to sequence the entire WFS1-coding sequence.
In WF4, WF5, WF7, and WF8, the entire coding sequence of the WFS1 gene was sequenced, without previous SSCP analysis, using primers previously described 13 with an ABI3100 automated DNA sequencer (Applied Biosystems, Foster City, CA, USA). DNAs from Dutch controls were screened for the D211N and P607R mutations (WF5) using the Snapshot kit on an automated sequencer ABI3100.
MtDNA Analysis
The presence of major rearrangements in mtDNA was determined by restriction fragment length polymorphism analysis. Five μg of total DNA was digested with PvuII (or BamHI) overnight at 37°C, and fragments were resolved on a 0.8% agarose gel, followed by Southern blotting. The blot was hybridized with a 32P-labeled HeLa cell mtDNA probe. Hybridization signals were quantified with a PhosphoImager and visualized by autoradiography. A positive mtDNA deletion sample obtained from a patient with Pearson syndrome 20 was included in the procedure, as well as a negative control.
The presence of the A3243G mutation in the mitochondrial tRNALeu(UUR) gene was determined by ApaI digestion of a 427-bp PCR-amplified fragment encompassing the mutated site (forward primer, 3029 to 3048 (5′-AAGGTTCGTTTGTTCAACGA-3′); reverse primer, 3437 to 3456 (5′-AGCGAAGGGTTGTAGTAGCC-3′)). DNA fragments were separated on polyacrylamide gel and visualized by silver staining (Pharmacia Biotech). Heteroplasmy levels as low as 1% could easily be detected. Positive and negative controls for the A3243G mutation 19 were included in the procedure.
RNA Analysis
Skin fibroblasts were grown in Dulbecco’s modified Eagle’s medium containing 4.5 mg/ml of glucose and 110 μg/ml of pyruvate supplemented with 10% fetal calf serum. RNA was isolated from cultured cells [WF2 (II-2) and WF9 (II-1)] or from freshly obtained blood samples [WF2 (I-1, II-2 and II-3), WF6 (II-1) and 15 controls], using a QIAamp RNA Blood Mini Kit (Qiagen, Westburg B.V., Leusden, The Netherlands). A one-step reverse transcriptase-PCR was performed using the Titan One Tube RT-PCR System (Roche Diagnostics, Nederland B.V., Almere, The Netherlands) using an exon 1 forward primer, 5′-GCAGATCTCCCGTTTGCG-3′, and an exon 8 reverse primer (R8-7 from Strom and colleagues 13 ). The amplicons of 3 kb in length were purified from agarose gel by GFX column (Amersham Pharmacia Biotech) and sequenced using the Thermo Sequenase Cy5.5 dye terminator cycle sequencing kit (Amersham Pharmacia Biotech) using various primer sets; in particular, the exon 2 acceptor splice-site region was analyzed using an exon 1 forward primer (see above) and an exon 2 reverse primer (5′-TCTGCTCTTTCCCGGCTC-3′). Fragments were run on a SEQ4x4 personal DNA sequencer (Amersham Pharmacia Biotech). The same primer set was used to test for the presence and the amount of splice-variant WFS1 mRNA templates in 17 RNA samples by restriction fragment length polymorphism analysis. A PCR-amplified product of 333 bp was digested with both HaeII and Hinf I, resulting in the formation of four smaller fragments (146, 81, 73, and 33 bp, respectively) and run on a 12% polyacrylamide gel. In case of variant exon 2 splicing (giving rise to a 4-bp deletion), the smallest 33-bp fragment became 29 bp in length. Bands were visualized by silver staining.
Results
Mutations in WFS1
Mutation screening was initiated as described in Materials and Methods. Abnormally migrating bands by SSCP analysis were subsequently subjected to DNA sequencing. DNA-sequencing patterns were compared to the WFS1 cDNA sequence (GenBank access no. AF084481). The A of the ATG initiator codon was denoted as “nucleotide + 1.” Seven mutations in WFS1 were identified in six of nine families: two missense mutations, one frameshift mutation, one splice donor site mutation, and three deletions (Table 2) . No WFS1 mutations were detected in families WF7, WF8, and WF9.
Table 2.
Mutations in the WFS1 Gene
| Family | Exon | Nucleotide change | Amino acid change | Type of mutation | Status |
|---|---|---|---|---|---|
| WF1; II-2, II-3 | 4 | 460 + 1G->A | − | 5′Splice signal | Homozygous |
| WF2; II-2 | 8 | 1581insC; ND | Multiple* | Insertion/frameshift | Heterozygous |
| WF3; II-5, II-6 | 8 | 1522–1536del15 | Y508-L512del | In-frame deletion | Homozygous |
| WF4; II-1 | 8 | 1522–1536del15 // 1230–1234del4 | Y508-L512del // L410del4nt | In-frame deletion // Frameshift deletion | Heterozygous |
| WF5; II-1, II-4 | 5//8 | 631G->A // 1820C->G | D211N // P607R | Missense | Heterozygous |
| WF6; II-1 | 8 | 1525–1537del13 | V509del13nt | Frameshift deletion | Homozygous |
The A of the ATG of the initiator Met codon is denoted nucleotide +1.
This mutation leads to a frameshift at amino acid residue 527 resulting in a 14-amino acid extension before a stop codon is encountered.
In WF1, a homozygous intronic G to A transition in the splice donor site of exon 4 (460 + 1G->A) was detected in both affected siblings. Both parents are heterozygous for this splice mutation, confirming consanguinity. The unaffected sibling has recently requested genetic testing for potential carrier status. He was found to be wild type with respect to the splice donor-site mutation. Assuming that this mutation abolishes correct splicing, a frameshift is created that on translation results in a truncated protein of 157 amino acids.
In WF2, a heterozygous C insertion at nucleotide position (np) 1581 was found in the patient, as well as in her father and her younger sister. This mutation leads to a frameshift at amino acid residue 527 resulting in a 14-amino acid extension before a stop codon is encountered. As a result, 39% of the wolframin protein becomes lacking. At the genomic level, no second mutation could be detected in the WFS1-coding region in this patient.
In WF3, a homozygous 15-bp deletion (1515-1530del15nt) in exon 8 was present in both affected siblings. The parents are cousins and are heterozygous for this deletion. According to the predicted structure of the wolframin protein, residues 508 to 512 are located in the fifth transmembrane domain.
In WF4, a heterozygous 4-bp deletion (L410del4nt) in exon 8 was detected in both the patient II-1 and her mother. This deletion leads to a frameshift at amino acid residue 410 resulting in a 30-amino acid extension of mainly serines and prolines before a stop codon is encountered. As a result, half of the wolframin becomes missing. From her father, she has inherited an allele harboring the identical 15-bp deletion (1515-1530del15nt) in exon 8 that is also present in both affected siblings from WF3.
In WF5, both affected siblings are compound heterozygous for two missense mutations; 631G->A (D211N) and 1820C->G (P607R). Two of their sisters were homozygous wild type, whereas one of their brothers is heterozygous for P607R. The missense mutations D211N and P607R were not present in 92 and 88 control samples, respectively.
In WF6, a homozygous 13-bp deletion was identified, which leads to a frameshift at amino acid residue 509 resulting in a 7-amino acid extension before a stop codon is encountered. As a result, 42% of the wolframin protein becomes lacking, including half of the predicted transmembrane region and the complete C-terminal part of the protein.
Sequence analysis also revealed a number of polymorphic variants in the coding sequence (R228R, I333V, V395V, N500N, H611R, K811K, S855) as well as intronic variants (IVS4–16 A->G, IVS4–9 A->G, IVS6 + 73 G->A). 15, 17, 21
Variant Splice-Site in WFS1
During mutation analysis on cDNA of WF9 (II-1) we came across a mixed sequencing pattern just before the ATG translation start region that could be explained by the coexistence of a wild-type sequence with a sequence displaying a 4-bp deletion (GCAG) just before the ATG located in exon 2 (Figure 2A) . The 4-bp deletion was not detected at the genomic level using exon 2 primers, 13 suggesting that the deletion has arisen at the transcriptional level. Inspection of the exon 2 acceptor splice region revealed the presence of a second CAG motif adjacent to the first acceptor splice motif (Figure 2 B and C) , which might be used as an alternative splice site. We tested the incidence of alternative splicing in 17 control RNA samples by restriction fragment length polymorphism analysis (see Materials and Methods). Mixed populations were observed present in 11 of 17 control RNA samples, with ratios varying from 3 to 60% (result not shown).
Figure 2.
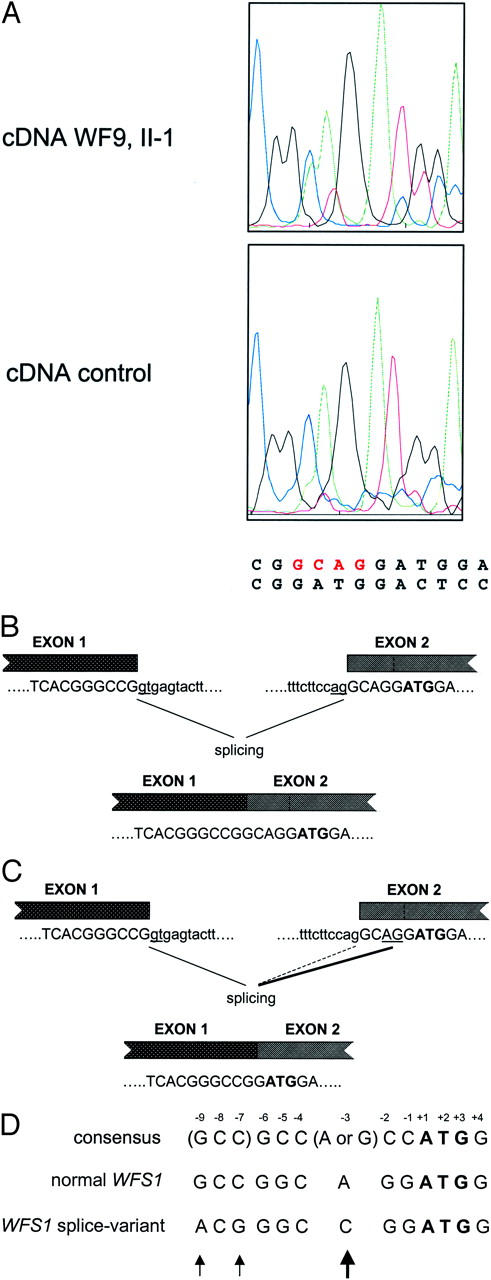
Alternative splicing site in WFS1 resulting in the deletion of four nucleotides (GCAG). A: Sequence chromatograms of the reverse transcriptase-PCR fragment of a control and of patient II-1 of WF9. Besides the normal sequence, a second sequence with the GCAG deletion is present accounting for ∼30 to 40% in patient II-1 of WF9 and to a lesser extent in a control (<10%). B and C: Schematic diagram of the normal (B) and alternative (C) splicing mechanisms in WFS1. Boxes denote exons. Normal (B) and alternative (C) acceptor splice sites are underlined. Alternative splicing in C is shown by the thickened line. The initiator codon is in bold. Intron and exon sequences are shown in lower and upper case letters, respectively. D: Effect of the alternative splicing on the sequences flanking the initiation codon. Translation initiation signal sequences are shown of the normal WFS1 transcript, 2 the variant WFS1 transcript, 3 and of the consensus 1 as proposed by Kozak. 26 Numbers denote position from the A(+1)TG initiator sequence. Arrows mark difference between the normal WFS1 sequence and the sequence that results from alternative splicing.
MtDNA Analysis
The presence of the A3243G point mutation in the leucyl-transfer RNA gene, known to be associated with familial diabetes and deafness, was examined in patients in whom no mutations in WFS1 were detected (WF7, WF8, and WF9), as well as in patients from WF2 and WF3. No mutations were detected in this cohort. In addition, the presence of major rearrangements in mtDNA was determined in patients from families WF2, WF3, and WF9. No gross rearrangements were detected.
Discussion
WFS1 Mutation Analysis
In this study we have identified seven different WFS1 mutations in six of nine Dutch kindreds with WS: one splice-site mutation, two missense mutations, a 1-bp insertion leading to a frameshift, a 15-bp in-frame deletion, a 4-bp deletion, and a 13-bp deletion. The splice-site mutation at 460 + 1G->A has been reported previously in a Turkish family from Germany. 13 It is not known whether this mutation has arisen twice independently, or whether both Turkish families share a common ancestor. Also, the in-frame deletion (1515-1530del15nt), as present in homozygous form in a Moluk family (WF3) and in heterozygous form in a Dutch family (WF4), has been identified earlier in a Japanese family with WS. 12 In this case, it is plausible to assume that this deletion has arisen at least twice independently.
An evaluation of the location of WFS1 mutations in 33 families with WS showed that the majority of frameshifts and nonsense mutations resided in the predicted transmembrane domains of wolframin. 15 In our study, we observed a similar clustering of mutations within the transmembrane domains (four of seven). Three of these mutations cause a premature translation stop because of frame-shift resulting in a complete absence of the carboxy tail of the wolframin protein. No function has been ascribed to wolframin as yet, but a role in membrane trafficking, protein processing, or calcium homeostasis in the endoplasmic reticulum has been postulated. It is speculated that the carboxy tail is interacting with other, yet unknown proteins. Expression studies of mutant proteins are awaited to determine which parts of the protein are essential for biological function.
In three Dutch Wolfram kindreds no WFS1 mutations were detected at the genomic DNA level. In these cases, we have not examined the promoter or intronic sequences for mutations, therefore this possibility cannot be ruled out. In WF2, only one affected allele could be identified, the other allele might harbor a mutation in the regulatory sequences WFS1 affecting proper transcription. Genetic heterogeneity of WS has recently been demonstrated by the identification of an additional locus (WFS2) on chromosome 4q. 22 Supporting a possible WFS2 linkage in WF7 and WF8 is the fact that in WFS2-linked patients as well as in our two affected individuals lacking WFS1 mutations diabetes insipidus is absent. However, WFS2 linkage was not addressed here. An alternative explanation could be that WF6, WF7, as well as WF8 clinically resemble WS of unknown etiological cause. A number of other disorders feature the presence of diabetes mellitus and one or more of the conditions commonly found in WS such as optic atrophy, sensorineural hearing loss. 23 One of these disorders is the maternally inherited diabetes mellitus and deafness that is associated with the A3243G mutation in the mitochondrial tRNALeu(UUR) gene. 19 However, no A3243G mutation was detected in WF7, WF8, and WF9. Furthermore, in agreement with other findings 10, 15 we were unable to detect large-scale deletions in mtDNA in Wolfram kindreds WF2, WF3, and WF9. Several other reports, however, have shown that in patients with WS, of which some of them showed linkage to chromosome 4p16, single 9, 24 or multiple deletions 25 in mtDNA were present. Recently, mutations in WFS1 were discovered co-existing with mtDNA deletions in Wolfram kindreds. 16 It is hard to believe that the co-existence of WFS1 mutations and mtDNA deletions in several patients has occurred accidentally. Therefore, a role for WFS1/wolframin in mitochondrial function is suggested.
Effect of WFS1 Splice-Variant on Translation Efficiency
We have observed the phenomenon of alternative splicing at the acceptor site of exon 2 resulting in a 4-bp deletion in the 5′UTR of WFS1 (Figure 2) . Alternative splicing was identified by restriction fragment length polymorphism analysis in 11 of 17 (65%) control RNA samples, although with various intensities. This 4-bp deletion modifies the translation initiation consensus sequence consisting of GCC(A/G)CCAUGG) by creating a C at position −3 with respect to the AUG of the WFS1 gene (Figure 2D) . The purine (A or G) 3 bases before the AUG codon and the G immediately following it appear to be the most important that influence efficiency of translation. 26 To our knowledge, there are only two documented cases in which a similar purine to pyrimidine switch has occurred at position −3 with respect to the AUG translation start site; one involves a globin mRNA in a patient with α-thalassemia 27 and the other involves a BRCA1 mRNA in sporadic breast cancer. 28 In both cases, in vitro and in vivo expression studies have shown a marked reduction in translation efficiency up to 70%. 27, 28 At present, it is not known whether translation of the variant WFS1 transcripts will be affected, and if so, by which mechanism. The 4-bp deletion in the AUG consensus sequence of WFS1 may promote context-dependent leaky scanning of ribosomes leading to a strong reduction in translation efficiency as predicted by Iida and Masuda. 29 Otherwise, initiation from a downstream AUG codon may occur leading to the formation of truncated protein if the start codon is not in frame with the normal translation initiation site. To definitively establish the impact of the 4-bp deletion on WFS1 function, expression studies need to be performed.
Diagnostic Criteria for WS
WFS1 mutations were identified in all Wolfram kindreds (WF1 to WF6) that met the minimum ascertainment criteria for the diagnosis of WS, being the occurrence together of early-onset (<30 years) diabetes mellitus and optic atrophy. 5 Khanim and colleagues 17 has recently described their refined diagnostic criteria by adding the requisite that patients have both diabetes mellitus and optic atrophy occurring before 15 years of age. Using these refined criteria, WF5, in which we have identified compound heterozygous WFS1 missense mutations, would have been excluded from further analysis because in both patients optic atrophy occurred at relatively high age (26 and 28 years, respectively). In the families harboring mutations in WFS1 (WF1 to WF6), diabetes mellitus presented at a median age of 4 years (range, 1 to 12 years) and optic atrophy appeared at a median age of 12 (range, 5 to 28 years). Diabetes insipidus appeared in six of nine (67%) patients.
Genotype and Phenotype Correlations
We have noticed a relatively mild phenotype in affected individuals of WF5 by the relatively late onset of optic atrophy and the absence of renal tract or neurological abnormalities. Both patients were compound heterozygous for two missense mutations. Two cases have been described in the literature in which missense mutations were associated with a mild clinical presentation. In one report an individual homozygous for a missense mutation in WFS1 (A716T) was described displaying features of WS, specifically juvenile diabetes mellitus and cataracts, but lacking optic atrophy that is part of the definition of WS. 30 In the other report a patient, homozygous for a missense mutation (P885L), had a mild phenotype and has not developed diabetes insipidus, renal involvement, or neurological abnormalities. 15 These observations suggest that a correlation between the type of mutation and disease severity exists. Patients with missense mutations seem to have an attenuated disease phenotype whereas patients with protein-truncating mutations suffer from the complete, severe WS. Further support for potential genotype-phenotype relationships in WFS1 comes from the description of two nonrelated patients who were both homozygous for a 4-bp deletion (del883fs/ter949). 15, 31 This deletion results in a frameshift and readthrough, predicting an elongated protein of 949 amino acids. Both patients display a strikingly similar, severe phenotype of brain-stem atrophy and central respiratory failure with diabetes mellitus and optic atrophy but without diabetes insipidus and deafness. No central respiratory failure was observed in patients compound heterozygous for this 4-bp deletion. 15 This suggests that the extended carboxy tail may affect proper interaction with other, as yet unidentified, proteins.
Recently, it was shown that dominantly inherited nonsyndromic low-frequency sensorineural hearing loss is caused by heterozygous missense mutations in WFS1. 21, 30 Hearing loss can be progressive and mainly affects 250 to 2000 Hz, without associated features segregating with it. 32 In seven families with low-frequency sensorineural hearing loss six different missense mutations were identified, all located in the carboxy-terminal part of the wolframin protein. A relative, homozygous for a mutation, which in heterozygous form is associated with low-frequency sensorineural hearing loss, displayed features of WS. 30 Heterozygous carriers of WS have an increased incidence of psychiatric illness, including depression, suicide attempts, and anxiety with or without panic disorder. 33 Several missense mutations in WFS1 have been identified in patients with isolated psychiatric disorders that are absent in normal controls. 34 It has also been suggested that heterozygous carriers in a WS family have an increased risk of hearing loss as well as of diabetes mellitus. 35 As far as currently known, in WF6, the mother of the patient experiences recurrent depression with suicide attempts and in WF8, the mother of the patient has psychiatric illness, although here no WFS1 mutation has been detected.
In conclusion, we have confirmed the homogeneity of WS by identifying WFS1 mutations in Dutch patients with WS. Our findings expand the spectrum of mutations in WFS1 and represent the first molecular characterization of Dutch patients with WS. Molecular analysis of WFS1 allows refinement of clinical diagnostic criteria for WS, which helps to dissect the clinically overlapping syndromes sharing diabetes mellitus and optic atrophy.
Figure 3.

Hypothetical structure of wolframin (WFS1), and positions of mutations detected in Dutch families. The amino acid sequence of wolframin (GenBank accession number Y18064) was analyzed for hydrophobicity with the transmembrane prediction program, TMpred. Gray circles denote hydrophilic domains, and the white circles denote the best predicted transmembrane domains. Black circles denote amino acid residues that were deleted or mutated to stop codons, or sites of missense mutations. The black triangle denotes the putative stop codon resulting from the splice-donor site mutation in family WF1. The figure has been modified from Hardy and colleagues. 15
Acknowledgments
We thank the family members who participated in this study and Dr. S. Hoffmann for providing DNA samples of Wolfram patients with mutations in the WFS1 gene for use as positive controls in the SSCP analysis. Dr. J.C. van Swieten (Department of Neurology, Erasmus University, Rotterdam, The Netherlands) is acknowledged for his cooperation in this study.
Address reprint requests to J. M. W. van den Ouweland, Ph.D., Department of Pathology and Laboratory Medicine, CMC-V, 1st Floor, Room Y1.173, University Hospital Groningen, Hanzeplein 1, PO Box 30.001, NL-9700 RB Groningen, The Netherlands. E-mail: j.m.w.van.den.ouweland@path.azg.nl.
Footnotes
K. C. holds a predoctoral position with the Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen (IWT); and G. V. C. holds a research position with the Flemish fonds voor Wetenschappelijk Onderzoek (FWO).
References
- 1.Wolfram DJ, Wagener HP: Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc 1938, 13:715-718 [Google Scholar]
- 2.Cremers CWRJ, Wijdeveld PGAB, Pinckers AJLG: Juvenile diabetes melitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). Acta Paediatr Scand 1977, 264:S1-S16 [DOI] [PubMed] [Google Scholar]
- 3.Page MMCB, Asmal AC, Edwards CRW: Recessive inheritance of diabetes: the syndrome of diabetes insipidus, diabetes mellitus, optic atrophy and deafness. Q J Med 1976, 179:505-520 [PubMed] [Google Scholar]
- 4.Gunn T, Bortolussi R, Little JM, Andermann F, Fraser FC, Belmonte MM: Juvenile diabetes mellitus, optic atrophy, sensory nerve deafness, and diabetes insipidus—a syndrome. J Pediatr 1976, 89:565-570 [DOI] [PubMed] [Google Scholar]
- 5.Barrett TG, Bundey SE, Macleod AF: Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995, 346:1458-1463 [DOI] [PubMed] [Google Scholar]
- 6.Van den Ouweland JMW, Bruining GJ, Lindhout D, Wit JM, Veldhuyzen BF, Maassen JA: Mutations in mitochondrial tRNA genes: non-linkage with syndromes of Wolfram and chronic progressive external ophthalmoplegia. Nucleic Acids Res 1992, 20:679-682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bu X, Rotter JI: Wolfram syndrome: a mitochondrial-mediated disorder? Lancet 1993, 342:598-600 [DOI] [PubMed] [Google Scholar]
- 8.Jackson MJ, Bindoff LA, Weber K, Wilson JN, Ince P, Alberti KG, Turnbull DM: Biochemical and molecular studies of mitochondrial function in diabetes insipidus, diabetes mellitus, optic atrophy, and deafness. Diabetes Care 1994, 17:728-733 [DOI] [PubMed] [Google Scholar]
- 9.Barrientos A, Casademont J, Saiz A, Cardellach F, Volpini V, Solans A, Tolosa E, Urbano-Márquez A, Estivill X, Nunes V: Autosomal recessive Wolfram syndrome associated with an 8.5-kb mtDNA single deletion. Am J Hum Genet 1996, 58:963-970 [PMC free article] [PubMed] [Google Scholar]
- 10.Barrett TG, Scott-Brown M, Seller A, Bednarz A, Poulton K, Poulton J: The mitochondrial genome in Wolfram syndrome. J Med Genet 2000, 37:463-466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polymeropoulos MH, Swift RG, Swift M: Linkage of the gene for Wolfram syndrome to markers on the short arm of chromosome 4. Nat Genet 1994, 8:95-97 [DOI] [PubMed] [Google Scholar]
- 12.Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, Mueckler M, Marshall H, Donis-Keller H, Crock P, Rogers D, Mikuni M, Kumashiro H, Higashi K, Sobue G, Oka Y, Permutt MA: A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998, 20:143-148 [DOI] [PubMed] [Google Scholar]
- 13.Strom TM, Hortnagel K, Hofmann S, Gekeler F, Scharfe C, Rabl W, Gerbitz KD, Meitinger T: Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998, 7:2021-2028 [DOI] [PubMed] [Google Scholar]
- 14.Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabe Y, Shinoda K, Oka Y: WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001, 10:477-484 [DOI] [PubMed] [Google Scholar]
- 15.Hardy C, Khanim F, Torres R, Scott-Brown M, Seller A, Poulton J, Collier D, Kirk J, Polymeropoulos M, Latif F, Barrett T: Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 1999, 65:1279-1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomez-Zaera M, Strom TM, Rodriguez B, Estivill X, Meitinger T, Nunes V: Presence of a major WFS1 mutation in Spanish Wolfram syndrome pedigrees. Mol Genet Metab 2001, 72:72-81 [DOI] [PubMed] [Google Scholar]
- 17.Khanim F, Kirk J, Latif F, Barrett TG: WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat 2001, 17:357-367 [DOI] [PubMed] [Google Scholar]
- 18.Tessa A, Carbone I, Matteoli MC, Bruno C, Patrono C, Patera IP, De Luca F, Lorini R, Santorelli FM: Identification of novel WFS1 mutations in Italian children with Wolfram syndrome. Hum Mutat 2001, 17:348-349 [DOI] [PubMed] [Google Scholar]
- 19.Van den Ouweland JMW, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, Van de Kamp JJP, Maassen JA: Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet 1992, 1:368-371 [DOI] [PubMed] [Google Scholar]
- 20.Van den Ouweland JMW, de Klerk JB, van de Corput MP, Dirks RW, Raap AK, Scholte HR, Huijmans JG, Hart LM, Bruining GJ, Maassen JA: Characterization of a novel mitochondrial DNA deletion in a patient with a variant of the Pearson marrow-pancreas syndrome. Eur J Hum Genet 2000, 8:195-203 [DOI] [PubMed] [Google Scholar]
- 21.Bespalova IN, Van Camp G, Bom SJH, Brown DJ, Cryns K, DeWan AT, Erson AE, Flothmann K, Kunst HPM, Kurnool P, Sivakumaran TA, Cremers CWRJ, Leal SM, Burmeister M, Lesperance MM: Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 2001, 10:2501-2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Shanti H, Lidral AC, Jarrah N, Druhan L, Ajlouni K: Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am J Hum Genet 2000, 66:1229-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuqua JS: Wolfram syndrome: clinical and genetic aspects. The Endocrinologist 2000, 10:51-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rötig A, Cormier V, Chatelain P, Francois R, Saudubray JM, Rustin P, Munnich A: Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest 1993, 91:1095-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrientos A, Volpini V, Casademont J, Genis D, Manzanares J-M, Ferrer I, Corral J, Cardellach F, Urbano-Márquez A, Estivill X, Nunes V: A nuclear defect in the 4p16 region predisposes to multiple DNA deletions in families with Wolfram syndrome. J Clin Invest 1996, 97:1570-1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozak M: Initiation of translation in prokaryotes and eukaryotes. Gene 1999, 234:187-208 [DOI] [PubMed] [Google Scholar]
- 27.Morlé F, Starck J, Godet J: Alpha-thalassemia due to the deletion of nucleotides-2 and -3 preceding the AUG initiation codon affects translation efficiency both in vitro and in vivo. Nucleic Acids Res 1986, 14:3279-3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Signori E, Bagni C, Papa S, Primerano B, Rinaldi M, Amaldi F, Fazio VM: A somatic mutation in the 5′UTR of BRCA1 gene in sporadic breast cancer causes down-modulation of translation efficiency. Oncogene 2001, 20:4596-4600 [DOI] [PubMed] [Google Scholar]
- 29.Iida Y, Masuda T: Strength of translation initiation signal sequence of mRNA as studied by quantification method: effect of nucleotide substitutions upon translation efficiency in rat preproinsulin mRNA. Nucleic Acids Res 1996, 24:3313-3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young T-L, Ives E, Lynch E, Person R, Snook S, MacLaren L, Cator T, Griffin A, Fernandez B, Lee MK, King M-C: Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001, 10:2509-2514 [DOI] [PubMed] [Google Scholar]
- 31.Sam W, Qin H, Crawford B, Yue D, Yu S: Homozygosity for a 4-bp deletion in a patient with Wolfram syndrome suggesting possible phenotype and genotype correlation. Clin Genet 2001, 59:136-138 [DOI] [PubMed] [Google Scholar]
- 32.Pennings RJE, Bom SJH, Cryns K, Flothmann K, Huygen PLM, Kremer H, Van Camp G, Cremers CWRJ: Progression of low-frequency sensorineural hearing loss (DFNA6/14-WFS1). Arch Otolaryngol Head Neck Surg (in press) [DOI] [PubMed]
- 33.Swift RG, Polymeropoulos MH, Torres R, Swift M: Predisposition of Wolfram syndrome heterozygotes to psychiatric illness. Mol Psychiatry 1998, 3:86-91 [DOI] [PubMed] [Google Scholar]
- 34.Torres R, Leroy E, Hu X, Katrivanou A, Gourzis P, Papachatzopoulou A, Athanassiadou A, Beratis S, Collier D, Polymeropoulos MH: Mutation screening of the Wolfram syndrome gene in psychiatric patients. Mol Psychiatry 2001, 6:39-43 [DOI] [PubMed] [Google Scholar]
- 35.Ohata T, Koizumi A, Kayo T, Shoji Y, Watanabe A, Monoh K, Higashi K, Ito S, Ogawa O, Wada Y, Takada G: Evidence of an increased risk of hearing loss in heterozygous carriers in a Wolfram syndrome family. Hum Genet 1998, 103:470-474 [DOI] [PubMed] [Google Scholar]