

Abstract
Two quantitative polymerase chain reaction (PCR) methods for HER2/neu gene quantification were evaluated for implementation into a clinical laboratory. Assays were developed using sequence-specific hybridization probes to detect a target (HER2/neu) and a reference gene (β-globin) simultaneously. One method utilizes real-time quantification while the second uses internal competitors and melting curves to quantify the unknown sample. These two methods were evaluated using three cell lines and 97 breast tumor samples. Two hundred ninety-four samples were subsequently evaluated using the real-time quantification and immunohistochemical (IHC) staining. Real-time PCR gave HER2/neu gene doses of 10 for SKBR3 and 2 for T47D while the competitive PCR gave doses of 11 for SKBR3 and 2.2 for T47D. Both methods produced coefficients of variation (CV) of less than 3% for within-run and less than 6% for between-run analysis. Examination of 97 breast tumors found a correlation of r = 0.974 between the two methods. IHC and PCR results agreed for 234 of the subsequent 294 samples analyzed (79% concordance). A subset of ten discrepant samples was microdissected. After microdissection all ten were positive by PCR, thus resolving the discrepancy. Real-time quantification and microdissection is useful clinically for HER2/neu quantification. Its ease of use and broad dynamic range allows screening for amplification of HER2/neu.
HER2/neu is a proto-oncogene located on chromosome 17 which codes for a 185-kd transmembrane protein closely related but distinguishable from the EGF receptor. 1, 2 It is amplified in 25% to 30% of primary breast tumors and is associated with poor prognosis in both node-positive and -negative patients. 3, 4, 5, 6, 7, 8 A monoclonal antibody, Herceptin, has been developed against HER2/neu. Preliminary trials in which the drug is administered in addition to chemotherapy have shown an increase in tumor response and a slowing of disease progression. 9, 10, 11 Because of the clinical implication of HER2/neu amplification, many methods exist for quantification of Her2/neu. Traditional methods for detection include immunohistochemical staining, fluorescent in situ hybridization (FISH), and more recently, molecular methods.
Recent advances in polymerase chain reaction (PCR) technology using the real-time fluorescent monitoring capabilities of the LightCycler allow mutation detection and quantification of gene amplification. 12 This technology combines real-time PCR and fluorescent detection using sequence-specific hybridization probes labeled with fluorophores. Fluorescent hybridization probes are designed to monitor product accumulation during the log phase of the PCR reaction 13 and allow end-point analysis through the use of melting curves. 14 We have developed two PCR assays with different levels of control, that quantify the HER2/neu gene amplification in DNA extracted from cell lines and from fresh-frozen and paraffin-embedded tissues. Both assays target the HER2/neu gene while using a single-copy gene, β-globin, as an internal control. β-globin was chosen as the reference gene because this area of chromosome 11p has not been shown to be commonly altered in breast cancer. 15 Use of a non-chromosome 17 reference gene would detect increased HER2/neu copies due to gene amplification as well as to aneuploidy. 16 HER2/neu and β-globin are co-amplified in both assays and the HER2/neu gene copy number is normalized for β-globin gene copy number. To control for run-to-run differences, a gene dose is obtained by adjusting the HER2/neu and β-globin copy numbers with the MRC-5 cell line that carries a single copy of each gene.
In the first method, samples are amplified and quantified in real-time using external standard curves. HER2/neu and β-globin gene copy numbers are obtained by comparing the second derivative maximum (crossing point) of sample curves with their respective standard curves. 17
The second method uses competitive PCR for an additional level of control. Samples are co-amplified with a dilutional range of competitors that differ from the HER2/neu and β-globin amplicons by one bp. Copy numbers for HER2/neu and β-globin are determined by comparing the derivative melting curve area of target gene with the area of the competitor with known copy number. 18 The target and competitor are differentiated based on melting temperature (Tm) of the probe. The Tm difference between the probe dissociating from a perfectly matched template and dissociating from a template with a single mismatch is 4 to 15°C, depending on the mismatch, the base composition of its nearest neighbors, length of the probe, and salt conditions. 12 For this system, the Tm differences are 12°C for HER2/neu and 11°C for β-globin.
The purpose of this study is to compare these two methods, examining correlation between the methods, the within- and between-run variation, the dynamic range and level of control necessary for precise analysis. On completion of the methods analysis, the real-time PCR method was chosen and accuracy of the method was determined against IHC staining.
Materials and Methods
Sample Preparation
The assay parameters were developed using SKBR3 (ATCC No. HTB-30), a cell line derived from an adenocarcinoma, containing approximately 11 copies of HER2/neu 19, 20 and T47D (ATCC No. HTB-133), a ductal carcinoma cell line with approximately 4 copies HER2/neu. 19, 21 The single copy HER2/neu control DNA was extracted from MRC-5 (ATCC No. CCL-171), a fetal lung cell line. DNA was prepared from cell lines using phenol/chloroform extraction and ethanol precipitation. 22
DNA concentration was determined using the Pico-Green dsDNA Quantification kit (Molecular Probes, Eugene, OR) adapted to the LightCycler. The Lambda DNA standards were diluted twofold to give a concentration range from 200 ng/ml to 6.5 ng/ml. The reaction volume was reduced from 200 μl (manufacturer’s instructions) to 20 μl (10 μl sample + 10 μl dye) and loaded into LightCycler capillaries for both standards and unknowns. The fluorescence was read using the LightCycler fluorimeter after disabling the “Use Seek Threshold” option. The cycling program was set to hold the samples at 30°C while acquiring three fluorescence readings. Fluorescence was read in the 520-nm (fluorescein) channel. Fluorescent background was subtracted and a standard curve was generated by plotting fluorescence values against known standard concentrations. Unknown concentrations were determined by using the equation of the standard curve. DNA concentration of each of the three cell lines was determined to be 4 ng/μl (approximately 1200 copies of disomic genes/μl) using PicoGreen. A 1-μl volume of each cell line was used in the PCR.
DNA samples from fresh and frozen breast tumor tissues were obtained under a research protocol from Penrose Hospital (Colorado Springs, CO) and Louisiana State University Medical Center (Shreveport, LA). DNA concentrations were adjusted to 4 to 5 ng/μl in 10 mmol/L Tris (pH 8.0), containing 0.1 mmol/L EDTA. All samples were boiled for 10 minutes before PCR. DNA samples from paraffin-embedded tissues were obtained from Sunnybrook and Women’s College, Health Sciences Center, Toronto, Canada.
Three hundred fifty-five paraffin-embedded breast tumor samples were obtained from ARUP (Salt Lake City, UT) under an Internal Review Board-approved protocol. Two 20-μm sections adjacent to the tissue used for the IHC staining were cut and one section was extracted using Puregene DNA Isolation Kit (Gentra, Minneapolis, MN). The other section was placed onto a glass slide to hold for possible microdissection. The monoclonal antibody c-erbB2/c-neu, clone 3B5, (Oncogene, CN Biosciences, Boston, MA) was used for the IHC staining. A subset of ten samples was prepared according to standard protocol (www.arctur.com) and microdissected using the Pixcell II Laser Capture Microdissection (LCM) system (Arcturus, Mountain View, CA). Approximately 100 nuclear targets were captured for each sample. DNA was isolated using the PicoPure DNA extraction kit (Arcturus).
Primers and Probes
Primer synthesis and labeling were performed as previously described. 13, 23, 24 The forward primer for HER2/neu was 5′-CCT CTG ACG TCC ATC GTC TC-3′. The reverse primer for HER2/neu was 5′-CGG ATC TTC TGC TGC CGT CG-3′. 18 The probe sequences were 5′-CTT GAT GAG GAT CCC AAA GAC CAC CCC CAA GAC CAC-3′ with the 3′ end labeled with fluorescein and 5′-ACC AGC AGA ATG CCA ACC A-3′ with the 5′ end labeled with LCRed 640 (Roche Molecular Biochemicals, Indianapolis, IN), and the 3′ end blocked with a phosphate group. 18 The primers for β-globin were 5′-ACA CAA CTG TGT TCA CTA GC-3′ (forward) and 5′-CAA CTT CAT CCA CGT TCA CC-3′ reverse. 25 The probe sequences for β-globin were 5′-CCA CAG GGC AGT AAC GG-3′ with the 3′end labeled with fluorescein and 5′-AGA CTT CTC CTC AGG AGT CAG GTG CAC CAT G-3′ with the 5′ end labeled with LCRed 705 (Roche Molecular Biochemicals, Indianapolis, IN), and the 3′ end blocked with a phosphate group.
External Standards Preparation
External standards for HER2/neu and β-globin were constructed by amplifying genomic DNA using rapid cycle PCR. 12, 26 The PCRs were performed in two separate reactions using 0.5 μmol/L of each appropriate primer, 200 μmol/L each dNTP, 3.0 mmol/L MgCl2, 50 mmol/L Tris (pH 8.3), 500 mg/L bovine serum albumin, 0.4 U Taq polymerase (Boehringer Mannheim, Mannheim, Germany) and 250 ng DNA per 50 μl total reaction volume in a thin-walled microfuge tube adapted for use in a rapid air thermocycler (Rapid Cycler, Idaho Technology, Salt Lake City, UT). PCR conditions were 94°C for 30 seconds, 50°C for 30 seconds, and 75°C for 30 seconds for 30 cycles. The transition rate between 50°C and 75°C was 1.0°C/second.
PCR products (101 bp for HER2/neu and 110 bp for β-globin) were visualized as single bands by agarose gel electrophoresis. PCR products were purified and concentrated using Amicon Ultrafree-DA (Millipore Corporation, Bedford, MA) membranes following manufacturer’s instructions. The amplicons were sequenced by dye-terminator chemistry (ABI 377 Sequencer, Applied Biosystems, Foster City, CA) for sequence confirmation. Concentrations were determined by PicoGreen as described above and converted to copy number. External standards of HER2/neu and β-globin templates were combined in equal concentrations.
Internal Competitor Preparation
Internal competitors for HER2/neu and β-globin were constructed in the same manner as the external standards except using an extended primer with a single base alteration from the wild-type sequence. The forward competitive primer for HER2/neu was 5′-CCT CTG ACG TCC ATC GTC TCT GCG GTG GTT [A]GC ATT CTG CTG GT-3′. 14 The introduced base change is indicated in brackets. The reverse primer for β-globin was modified with the sequence 5′-CAA CTT CAT CCA CGT TCA CCT TGC CCC ACA GGG [T]AG TAA CGG-3′.
LightCycler PCR Amplification
In both assays, the HER2/neu target sequence and a single-copy gene, β-globin, were co-ampified in the same reaction. Five tenfold serially diluted external standards ranging from 1000,000 to 100 copies along with 4 ng of each unknown sample comprised the real-time PCR runs. The competitor PCR method used 4 ng of unknown sample co-amplified with five serial dilutions of competitor ranging from 25,000 to 1500 copies.
PCR for both methods was performed using 200 μmol/L each dNTP, 3.0 mmol/L MgCl2, 50 mmol/L Tris (pH 8.3), 500 mg/L bovine serum albumin, Klentaq polymerase (4 units/10 μl; AB Peptides, St. Louis, MO), and TaqStart antibody (0.32 μg/10 μl; Clontech, Palo Alto, CA). Primer concentrations were 0.5 μmol/L for the forward primer and 0.2 μmol/L for the reverse primer. Probe concentrations were 0.2 μmol/L. PCR conditions for the external standard method were 94°C for 3 seconds, 58°C for 10 seconds, and 75°C for 0 seconds for 40 cycles. The programmed transition rates were 20°C/second from denaturation to annealing, 1°C/second from annealing to extension (during which primer extension is occurring), and 20°C/second from extension to denaturation. Fluorescence was detected once per cycle at the end of the annealing stage. PCR conditions for the competitive PCR method were 94°C for 3 seconds, 53°C for 10 seconds, and 72°C for 0 seconds for 37 cycles. The programmed transition rates and detection of fluorescence were as described above. On completion of the amplification, the samples were denatured at 94°C for 3 seconds, cooled to 35°C at transition rate of 20°C/second, and held for 2 minutes. The samples were then heated to 90°C at a rate of 0.1°C/second with continuous fluorescence monitoring. The data were plotted as negative derivative fluorescent curves with respect to temperature (−dF/dT).
Analysis
Analysis was performed using two detection channels on the LightCycler. The LCRed640 channel was used to monitor the HER2/neu target and the LCRed705 channel was used to monitor the β-globin target. A color compensation file was used to correct for fluorescence bleed-through. 24
The real-time PCR method used crossing point analysis, the cycle number at which amplicon is first detectable, for quantification. The crossing points were obtained using the second derivative maximum (LightCycler 3 Data Analysis version 3.5.28 software). All calculations were made using the RealQuant software version 1.00 program (Roche Molecular Biochemicals, Indianapolis, IN).
The competitive PCR method used end-point analysis to compare competitor to target peak areas. Areas of the derivative melting curves were determined using non-linear least-squares regression analysis. 14 The log of each target area/competitor area was plotted against the log of the competitor copy number. A line was fit through the plotted points, and the x axis intercept determined the target copy number.
For both methods, HER2/neu copy number was calculated as the ratio of HER2/neu copy number to β-globin copy number. HER2/neu/β-globin copy number was normalized between runs using the MRC-5 cell line to achieve a gene dose. A gene dose greater than 2.5 was considered amplified. Gene doses smaller than 1.5 were considered non-amplified, while gene doses between 1.5 and 2.5 were indeterminate.
Results
Both assays use artificial templates for quantification. The real-time method uses wild-type amplicons while the competitive PCR method uses amplicons with a single base alteration. Preferential amplification of either the wild-type or the competitor amplicon was assessed before beginning the assay comparison. To examine this, equal concentrations of each template were amplified and quantified using both methods. When run in separate tubes, equal starting concentrations of wild-type and competitor generated equal crossing point values. When both competitor and wild-type were included in the same tube at equal concentrations, equal melting peak areas (Figure 1) resulted. These findings illustrate no preferential amplification of one amplicon over the other.
Figure 1.

Equal starting concentrations of competitor and wild-type amplicons exhibit equal crossing points and equivalent melting-peak areas. Wild-type and competitor templates were assayed separately to determine crossing points (Cp). Allele areas are shown for the wild-type and competitor assayed in the same reaction. A: HER2/neu B: β-globin.
Assays were tested on DNA extracted from three cell lines: MRC-5 (a single HER2/neu gene copy), T47D (approximately 4 HER2/neu copies), and SKBR3 (approximately 11 HER2/neu copies). Figure 2A shows HER2/neu and β-globin amplification in the three cell lines. Figure 2B shows the external standard curves generated by plotting the log concentration of the standards versus the crossing point. The slopes are not identical, indicating a small efficiency difference between the two reactions. Because each target is compared with its own standard curve in every reaction, this efficiency difference has no effect on copy number. The three cell lines contained a single copy of β-globin as illustrated by similar crossing points (LCred 705 channel). Varying copy numbers of HER2/neu were found in the three cell lines (LCred 640 channel). The calculated HER2/neu gene doses when compared with the single copy MRC-5 cell line were 10 copies for SKBR3 and 2 for T47D. FISH analysis was performed which confirmed copy numbers in these cell lines. 14 The differece in copy number from the published values possibly could be due to cell lines undergoing multiple passages.
Figure 2.

Crossing points in cell lines as determined by real-time PCR. HER2/neu and β-globin were assayed in the same reaction and detected in different fluorescent channels on the LightCycler. A. β-globin Cps (----) were similar in the three cell lines, HER2/neu Cps (—–) differed depending on the amount of HER2/neu amplification. (……) No template; (•) MRC-5; (▴) T47D; (▪) SKBR3. B. Standard curves for HER2/neu (□) and β-globin (∗). Twofold dilutions of standards were used ranging from 106 to 2 × 103 copies/μL.
In the competitive PCR method, the three cell lines displayed varying amounts of HER2/neu amplification (Figure 3A) , as expected due to their varying Her2/neu copy number, while the β-globin product remained nearly constant (Figure 3B) when compared with the appropriate competitor. Likewise, the linear-fit lines in the analysis show different x-intercepts for HER2/neu (Figure 4A) and similar intercepts for β-globin (Figure 4B) . When normalized to the MRC-5 cell line, a HER2/neu gene dose of 11 was detected in the SKBR3 cell line and a gene dose of 2.2 in the T47D cell line.
Figure 3.
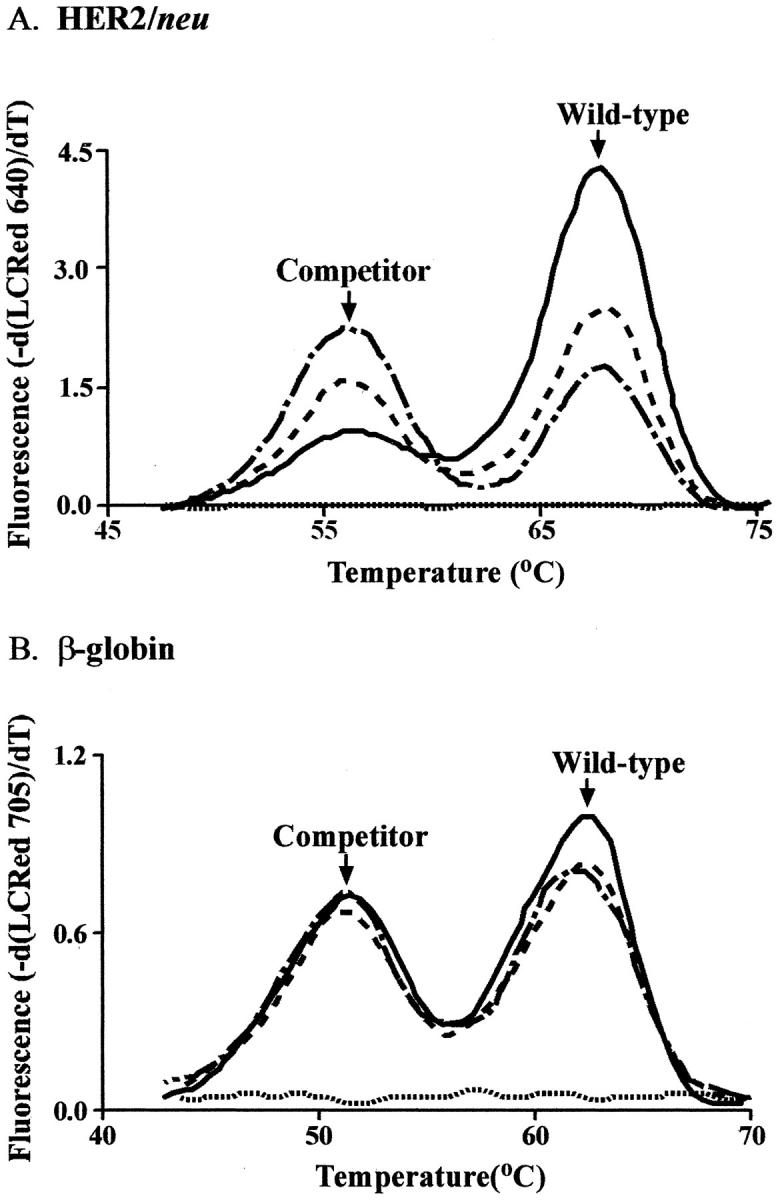
Derivative melting curves in cell lines as determined by competitive PCR. HER2/neu and β-globin were assayed with competitors in the same reaction. A: Ratios of HER2/neu melting-peak areas differed depending on the amount of HER2/neu amplification. B: Ratios of melting-peak areas were similar in the three cell lines for β-globin. ( - . - ), MRC-5; (- - -), T47D; (—), SKBR3; (….), no template.
Figure 4.
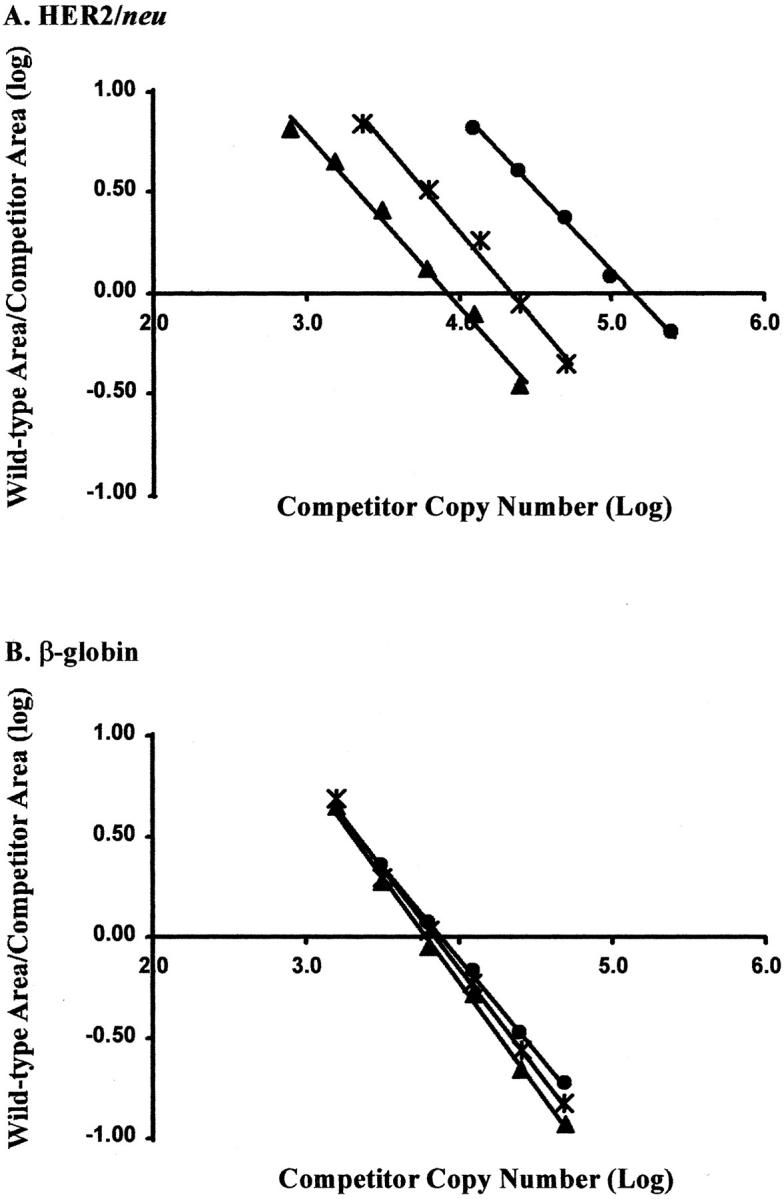
Wild-type:competitor area ratios. The wild-type and competitor area ratios were plotted against varying amounts of competitor copy number. The x-intercept is the calculated copy number in the sample. (▴) MRC-5; (∗) T47D; (▪) SKBR3. A: HER2/neu copy number differed depending on the amount of HER2/neu gene amplification. B: β-globin copy numbers were similar in the three cell lines.
To determine the within-run precision of the assay, the three cell lines, MRC-5, T47D, and SKBR3, were analyzed in triplicate using both assays. The between-run variation was calculated from three separate runs in both assays. Both methods produced coefficients of variation of less than 3% for within-run and less than 6% for between-run analysis. The mean and SD for HER2/neu and β-globin are shown in Table 1 .
Table 1.
Precision Analysis of Competitive PCR and Real-Time Quantitation with External Standards
| Cell lines | Competitive PCR | Real-time PCR | |||
|---|---|---|---|---|---|
| Absolute | MRC-adjusted | Absolute | MRC-adjusted | ||
| Within-run | MRC-5 | 1.4 ± 0.01 | (1.0) | 0.5 ± 0.01 | (1.0) |
| T47D | 3.1 ± 0.07 | 2.3 ± 0.05 | 1.1 ± 0.03 | 2.1 ± 0.05 | |
| SKBR-3 | 15.3 ± 0.02 | 11.0 ± 0.15 | 5.6 ± 0.12 | 10.6 ± 0.23 | |
| Between-run | MRC-5 | 1.4 ± 0.02 | (1.0) | 0.5 ± 0.02 | (1.0) |
| T47D | 3.1 ± 0.08 | 2.2 ± 0.07 | 1.1 ± 0.04 | 2.0 ± 0.04 | |
| SKBR-3 | 15.9 ± 0.88 | 11.4 ± 0.40 | 5.6 ± 0.15 | 10.5 ± 0.38 | |
The mean ± standard deviation are given. The absolute HER2/neu gene dose was obtained by normalizing the HER2/neu copy number by the β-globin copy number. The MRC-5-adjusted HER2/neu gene dose, adjusting for run-to-run variance, was obtained by adjusting the absolute gene dose by the gene dose of the single copy cell line, MRC-5.
To compare assays on clinical samples, 97 DNA extractions from fresh-frozen and paraffin-embedded tissues were tested in duplicate with both methods (Figure 5) . In both assays, the gene dose of HER2/neu relative to β-globin should be 1.0 in normal tissues. Using a gene dose cutoff of 2.0 to divide “amplified ” from “non-amplified” tissue, discrepant results were found on five samples extracted from paraffin-embedded tissues. If a gene dose between 1.5 to 2.5 is considered indeterminate, the positive/negative/indeterminate results on the 97 samples correlate completely. The correlation between calculated gene doses was r = 0.974.
Figure 5.

Comparison of relative HER2/neu gene dose by real-time and competitive PCR methods. The two methods showed a high degree of correlation (r = 0.974).
Three hundred fifty-five paraffin-embedded breast tumor samples were evaluated with IHC staining. IHC scores of 0 and 1+ were considered to be negative for HER2/neu, while scores of 2+ to 3+ were considered positive. Amplifiable DNA was obtained from 294 samples and evaluated using real-time PCR (Table 2) . IHC and PCR results agreed on 79% of the samples. One-percent were negative by IHC and positive by PCR. Nineteen percent were positive by IHC and negative by PCR. Ten of the 57 discrepant samples (positive IHC and negative PCR) were microdissected by LCM. All 10 previously negative samples were positive by PCR after microdissection, indicating that the discrepancy was due to normal cell contamination.
Table 2.
Correlation of Quantitative PCR Using External Standards with IHC
| IHC/PCR correlation n = 294 | IHC | ||
|---|---|---|---|
| Positive | Negative | ||
| PCR | Positive | 32 | 3 |
| Negative | 57* | 202 | |
| (2 + IHC n = 30) | |||
| (3 + IHC n = 27) | |||
10 of 10 (2 + IHC n = 5; 3 + IHC n = 5) were positive by PCR after microdissection.
Discussion
New developments in real-time PCR technology allow detection of gene amplification using external standards and internal competitors. Questions arise about the level of control necessary for accurate quantitative PCR, specifically the advantage, if any, of an extra level of competitive control. To address this, we developed two assays using these techniques to assess HER2/neu gene quantification, and evaluated them by comparing the following parameters: assay precision, throughput capacity, dynamic range, analysis complexity, and reproducibility. On determination of a preferred method, real-time PCR, the method was evaluated against IHC results from 294 paraffin samples.
Quantitative PCR must be accurate and precise to be implemented into a clinical laboratory. The added level of control using an internally amplified competitor may improve the precision of the assay. However, data acquisition by real-time analysis may eliminate the need for this extra level of control. We found that the within-run and between-run coefficients of variation (CVs) were comparable by both methods, indicating that both methods yield precise results.
The real-time assay has a higher throughput capacity. Results can be obtained on 24 samples per run, with four standards, two Her2/neu-positive controls (a low and a high level of amplification), a HER2/neu-negative control and a no template control included in each run. Competitive PCR is limited to five samples tested with four competitor dilutions per run. Positive and negative controls require three competitor dilutions flanking the equivalence point for each control. A no template control brings the number of capillaries necessary for controls to 10 per run.
The use of external standards provides a greater dynamic range than competitive PCR. The range covered by the real-time assay is over four logs. Any patient sample should fall within this range. With competitive PCR, samples are first run with up to four competitor concentrations. Depending on the sample:competitor ratio, the sample is re-run with adjusted competitor concentrations so that a linear curve fit can be obtained for analysis. The real-time assay requires less time for analysis, taking only 5 minutes when analyzing the samples using the RealQuant software. Analysis for the competitive PCR assay requires about 30 minutes.
Both assays were reproducible as demonstrated by low coefficients of variation in cell lines. The results from 97 DNAs extracted from fresh-frozen and paraffin samples correlated very well with a correlation coefficient of 0.974. Results correlated 100% when gene doses were scored as amplified (>2.5), negative (<1.5), or indeterminate (1.5 to 2.5). Samples in this indeterminate zone should undergo further testing by another method such as FISH or immunohistochemical staining.
Multiple methods are currently used to detect HER2/neu gene amplification. Tissue-based detection methods include FISH and immunohistochemical staining. These methods have the advantage of assessing the HER2/neu amplification in intact cells. However, both of these methods can be laborious for screening large numbers of samples. Real-time PCR has the advantage of automation and also allows levels of HER2/neu amplification to be easily evaluated. However, macro- or microdissection of tumor cells is necessary to eliminate dilution of apparent copy number by the non-tumor cells in the samples. Microdissection requires more technical time, about 10 minutes per section. A small sample size, about 100 cells, is adequate for amplification.
β-globin was chosen as the reference gene because this area of chromosome 11 is under-represented is chromosomal abnormalities in breast cancer. 15 Use of this reference gene would detect increased HER2/neu copies due to gene amplification as well as due to aneuploidy. Although chromosome 17 aneuploidy is not considered a major contributor to HER2/neu overexpression, recent studies have shown that increased HER2/neu protein expression has a higher correlation with absolute copies (signals/cell) of the Her2/neu gene in comparison to the HER2/neu gene to chromosome 17 ratio. 16 A second reference gene on chromosome 17 could be added to our assay to address aneuploidy, and also make the PCR analysis more consistent with FISH methodologies that use chromosome 17 probes.
The focus of this investigation was to identify the level of control necessary in PCR analysis for potential use in the clinical laboratory. The comparison of real-time PCR with competitive PCR allowed us to demonstrate that the use of added controls for competitive PCR are not necessary. Using real-time PCR we can now continue to answer important questions about clinical sensitivity and specificity. These clinical studies, beyond the scope of this study, can be designed to address several issues. A study should be designed to compare PCR to FISH, using a chromosome 17 reference gene. Further validation should use micro- or macrodissected cells. In retrospect, we microdissected a small subset (10 of 57) of samples, demonstrating the necessity of microdissection. A larger study could answer the question to what extent will micro- or macrodissection hinder the rapid throughput potential of PCR. Evaluation of aneuploidy versus gene amplification as a mechanism of HER2/neu protein expression could be designed using a combination of a chromosome 17 and a non-chromosome 17 reference gene. Finally, a study could use real-time PCR to resolve discordance between FISH and IHC.
In view of all these observations, real-time PCR has potential for clinical use in HER2/neu quantification. The ease of use and broad dynamic range make it a viable screening tool for detection of HER2/neu amplification in fresh-frozen, paraffin-embedded, and microdissected samples.
Acknowledgments
We thank Jennifer MacNair, for doing much of the microdissection work.
Address reprint requests to Elaine Lyon, ARUP Laboratories, 500 Chipeta Way, Salt Lake City, UT 84108. E-mail: lyone@aruplab.com.
Footnotes
Supported by the ARUP Institute for Clinical and Experimental Pathology and grant 99-3243 from the Susan G. Komen Breast Cancer Foundation.
References
- 1.Coussens L, Yang-Feng TL, Liao YC, Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J, Francke U, Levinson A, Ullrich A: Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230:1132-1139 [DOI] [PubMed] [Google Scholar]
- 2.Schechter AL, Hung MC, Vaidyanathan L, Wienberg RA, Yang-Feng TL, Francke U, Ullrich A, Coussens L: The neu gene: an erbB-homologous gene distinct from and unlinked to the gene encoding the EGF receptor. Science 1985, 229:976-978 [DOI] [PubMed] [Google Scholar]
- 3.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, Press MF: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244:707-712 [DOI] [PubMed] [Google Scholar]
- 4.Borg A, Tandon AK, Sigurdsson H, Clark GM, Ferno M, Fuqua SA, Killander D, McGuire WL: HER-2/neu amplification predicts poor survival in node-positive breast cancer. Cancer Res 1990, 50:4332-4337 [PubMed] [Google Scholar]
- 5.Parkes HC, Lillycrop K, Howell A, Craig RK: C-erbB2 mRNA expression in human breast tumours: comparison with c-erbB2 DNA amplification and correlation with prognosis. Br J Cancer 1990, 61:39-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL: Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 253:177-182 [DOI] [PubMed] [Google Scholar]
- 7.Press MF, Pike MC, Chazin VR, Hung G, Udove JA, Marckowicz M, Danyluk J, Godolphin W, Sliwkowski M, Akita R, Paterson MC, Slamon DJ: Her-2/neu expression in node-negative breast cancer: direct tissue quantitation by computerized image analysis and association of overexpression with increased risk of recurrent disease. Cancer Res 1993, 53:4960-4970 [PubMed] [Google Scholar]
- 8.Giai M, Roagna R, Ponzone R, De Bortoli M, Dati C, Sismondi P: Prognostic and predictive relevance of c-erbB-2 and ras expression in node-positive and -negative breast cancer. Anticancer Res 1994, 14:1441-1450 [PubMed] [Google Scholar]
- 9.Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J: Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 1998, 58:2825-283 [PubMed] [Google Scholar]
- 10.Shak S: Overview of the trastuzumab (Herceptin) anti-HER2 monoclonal antibody clinical program in HER2-overexpressing metastatic breast cancer: Herceptin Multinational Investigator Group. Semin Oncol 1999, 26:71-77 [PubMed] [Google Scholar]
- 11.Burris HA, III: Docetaxl (Taxotere) in HER-2-positive patients and in combination with trastuzumab (Herceptin). Semin Oncol 2000, 27:19-23 [PubMed] [Google Scholar]
- 12.Lyon E: Mutation detection using fluorescent hybridization probes and melting curve analysis. Exp Rev Mol Diagn 2001, 1:17-26 [DOI] [PubMed] [Google Scholar]
- 13.Wittwer CT, Herrmann MG, Moss AA, Rasmussen RP: Continuous fluorescent monitoring of rapid cycle DNA amplification. Biotechniques 1997, 22:130-138 [DOI] [PubMed] [Google Scholar]
- 14.Lyon E, Millson A, Lowery MC, Woods R, Wittwer CT: Quantification of HER2/neu gene amplification by competitive PCR using fluorescent melting curve analysis. Clin Chem 2001, 47:844-851 [PubMed] [Google Scholar]
- 15.Persson K, Pandis N, Mertens F, Borg A, Baldetorp B, Killander D, Isola J: Chromosomal aberrations in breast cancer: a comparison between cytogenetics and comparative genomic hybridization. Genes Chromosomes Cancer 1999, 25:115-122 [PubMed] [Google Scholar]
- 16.Wang S, Saboorian MH, Frenkel EP, Haley BB, Siddiqui MT, Gokaslan S, Hynan L, Ashfaq R: Aneusomy 17 in breast cancer: its role in HER-2/neu protein expression and implication for clinical assessment of HER-2/neu status. Mod Pathol 2002, 15:137-145 [DOI] [PubMed] [Google Scholar]
- 17.Bièche I, Olivi M, Champème M-H, Vidaud D, Lidereau R, Vidaud M: Novel approach to quantitative polymerase chain reaction using real-time detection: application to the detection of gene amplification in breast cancer. Int J Cancer 1998, 78:661-666 [DOI] [PubMed] [Google Scholar]
- 18.Lyon E, Millson A, Suli A: HER2/neu gene amplification quantified by PCR and melting peak analysis using a single base alteration competitor as an internal standard. Meuer S Wittwer C Nakagawara K eds. Rapid Cycle Real Time PCR: Methods and Application. 2001:pp 207-217 Springer-Verlag Heidelberg, Germany
- 19.Sestini R, Orlando C, Zentilin L, Donetella L, Gelmini S, Pinzani P: Gene amplification for cer-bB-2, c-myc, epidermal growth factor receptor, int-2, and N-myc measured by quantitative PCR with a multiple competitor template. Clin Chem 1995, 41:826-832 [PubMed] [Google Scholar]
- 20.Hynes NE, Geber HA, Saurer S, Groner B: Overexpression of the c-erbB-2 protein in human breast tumor cell lines. J Cell Biochem 1989, 39:167-173 [DOI] [PubMed] [Google Scholar]
- 21.Dati C, Antoniotti S, Traverna D, Perroteau I, De Bortoli M: Inhibition of c-erbB-2 oncogene expression by estrogens in human breast cancer cells. Oncogene 1990, 5:1001-1006 [PubMed] [Google Scholar]
- 22.Thomas SM, Moreno RF, Tilzer LL: DNA: extraction with organic solvents in gel barrier tubes. Nucleic Acids Res 1989, 17:5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lay MJ, Wittwer CT: Real-time fluorescence genotyping of factor V Leiden during rapid-cycle PCR. Clin Chem 1997, 43:2262-2267 [PubMed] [Google Scholar]
- 24.Bernard PS, Pritham GH, Wittwer CT: Color multiplexing hybridization probes using the apolipoprotein E locus as a model system for genotyping. Anal Biochem 1999, 273:221-228 [DOI] [PubMed] [Google Scholar]
- 25.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N: Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230:1350-1354 [DOI] [PubMed] [Google Scholar]
- 26.Wittwer CT, Reed GB, Ririe KM: Rapid cycle DNA amplification. Mullis KB Ferré F Gibbs RA eds. Polymerase Chain Reaction. 1994:pp 174-181 Birkhauser Boston