

Abstract
A novel platform for the electronic detection of nucleic acids on microarrays is introduced and shown to perform well as a selective detection system for applications in molecular diagnostics. A gold electrode in a printed circuit board is coated with a self-assembled monolayer (SAM) containing DNA capture probes. Unlabeled nucleic acid targets are immobilized on the surface of the SAM through sequence-specific hybridization with the DNA capture probe. A separate signaling probe, containing ferrocene-modified nucleotides and complementary to the target in the region adjoining the capture probe binding site, is held in close proximity to the SAM in a sandwich complex. The SAM allows electron transfer between the immobilized ferrocenes and the gold, while insulating the electrode from soluble redox species, including unbound signaling probes. Here, we demonstrate sequence-specific detection of amplicons after simple dilution of the reaction product into hybridization buffer. In addition, single nucleotide polymorphism discrimination is shown. A genotyping chip for the C282Y single nucleotide polymorphism associated with hereditary hemochromatosis is used to confirm the genotype of six patients’ DNA. In addition, a gene expression-monitoring chip is described that surveys five genes that are differentially regulated in the cellular apoptosis response. Finally, custom modification of individual electrodes through sequence-specific hybridization demonstrates the potential of this system for infectious disease diagnostics. The versatility of the electronic detection platform makes it suitable for multiple applications in diagnostics and pharmacogenetics.
The analysis of nucleic acids has gained broad acceptance in diagnostic testing, pharmacological research, and numerous other fields including animal husbandry and detection of transgenes. The growing number of nucleic acid-based tests has stimulated a demand for automated, inexpensive testing devices that also afford miniaturization of the test platform and, ideally, the associated instrumentation. DNA chips have proven attractive for satisfying the demands of nucleic acid-based testing. On a chip, DNA capture probes are immobilized on a solid support in a miniature array. 1 The capture probes are deposited individually at defined locations in close proximity, allowing the opportunity to test for the presence of many distinct nucleic acid targets in a single hybridization reaction. Nucleic acid targets are allowed to hybridize to the capture probes and, after hybridization, the target is detected through the presence of a reporter molecule. The reporter molecule, frequently a fluorescent species, can be incorporated into the target before hybridization or in a separate signaling probe that forms a ternary complex with the capture probe and the target nucleic acid. DNA chip systems that require an optical reader often use a high density of capture probe sites. These high-density DNA chips are most useful for gene and drug discovery efforts where the high cost of instrumentation can be justified. 2, 3 Other systems will be required for the more economically sensitive environment of the molecular diagnostics laboratory, where relatively few interrogations are relevant for clinical investigation of a patient specimen.
We report here (and in Terbrueggen RH, Vielmetter J, Chen Y-P, Millan KM, Mucic RC, Olsen GT, Swami N, Umek RM, Wang H, Welch TW, Yowanto H, Yu CJ, Blackburn GF, and Kayyem JF, submitted for publication) the development of a novel electronic detection format for nucleic acids that utilizes inexpensive instrumentation including a disposable DNA chip. 4 Signaling probes containing ferrocene moieties, 5 redox active metal centers that facilitate the detection of nucleic acid targets in homogeneous assays that eliminate the need for separate labeling and washing steps, form the basis of this electronic detection platform. We demonstrate here that the electronic detection platform facilitates sequence-specific detection of amplicons, mismatch discrimination for the characterization of single nucleotide polymorphisms (SNPs), and gene expression monitoring. The versatility of the electronic detection platform makes it suitable for multiple applications in molecular diagnostics and pharmacogenetics.
Materials and Methods
Manufacturing of Electrode Arrays Modified with DNA Capture Probes
Printed circuit board technology is used to manufacture chips with 14 exposed gold electrodes, each of which is wired to a connector at the chip edge. A solder mask defines the electrode diameter (250 or 500 μm) and covers the lead to the connector. DNA capture probes are deposited as a mixed solution with the other components of the self-assembled monolayer (SAM). After deposition, chips are rinsed, dried, and sealed in a housing for hybridization.
Synthesis of DNA Capture Probes and Signaling Probes
DNA capture probes were prepared by solid phase synthesis using an alkane chain of 16 residues terminated with a thiol (abbreviated C16S) as substrate after attachment to a controlled pore glass support. The probes were synthesized 3′ to 5′ such that the 5′ end is in solution and the 3′ end in the SAM (Figure 1A) . The capture probes synthesized are shown in Table 1 . Signaling probes (Table 2) were synthesized with modified adenine residues that have a ferrocene substitution on the ribose ring. 6 Probes are designed such that signal and capture probes are complementary to adjacent regions of the target.
Figure 1.
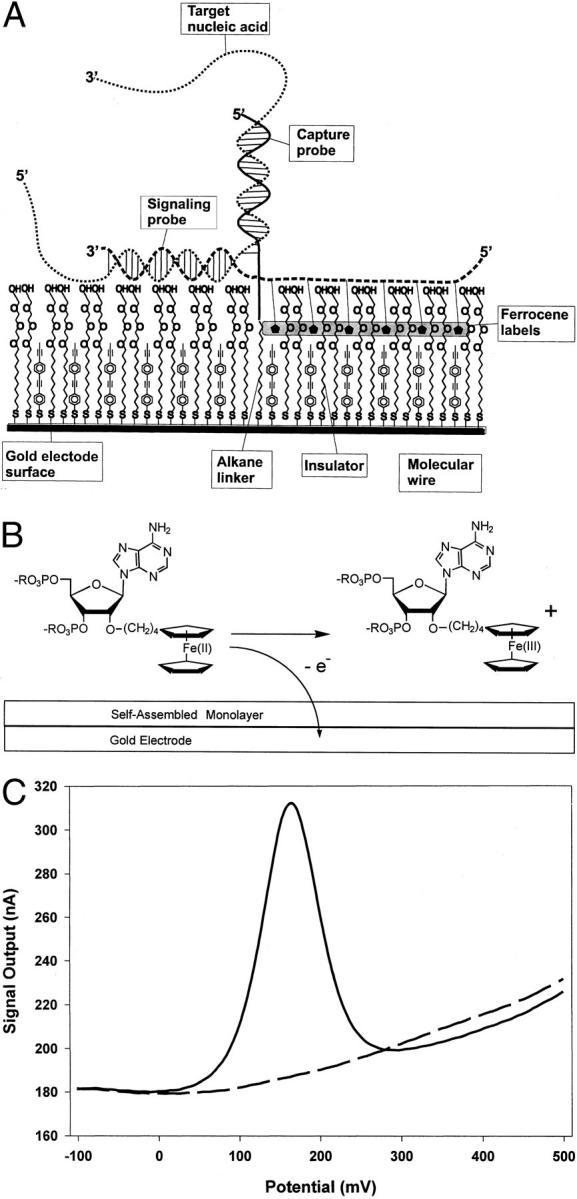
Electrochemical detection of nucleic acids on the bioelectronic sensor using a sandwich assay. A: Schematic diagram of the sandwich assay for electronic detection of nucleic acids. The gold electrode is coated with a self-assembled monolayer (SAM) that includes (1) DNA-alkanethiols containing the capture probe sequence (one shown for simplicity), (2) thiol-terminated oligophenylethynyl molecules, also called molecular wires, and (3) alkanethiols terminated in polyethylene glycol insulator. The molecular wires provide a pathway for electron transfer between the ferrocenes and the gold in response to potential changes at the electrode. The alkanethiols terminated in ethylene glycol serve as insulators to block access of redox species in solution to the electrode, including free signaling probes. A target nucleic acid is shown annealed to a capture probe and a ferrocene-labeled signaling probe. B: Scheme depicting electrochemical oxidation of a ferrocene-labeled adenosine derivative from Fe(II) to Fe(III). Interfacial electron transfer from ferrocene to the gold electrode is detected as faradaic current. C: A representative voltammogram is shown. An electrode array was prepared with capture probes complementary to HIV or HCV. A 1-μmol/L solution of HIV target mimic and 2.5 μmol/L of both the HIV and HCV signaling probes were introduced into the hybridization chamber. After 1 minute of hybridization, an electrode modified with HIV capture probes was interrogated by AC voltammetry and the output current was recorded (solid line). An electrode modified with HCV capture probes was interrogated immediately afterward, and the output current was recorded (dashed line). Automated software has been developed to measure the peak height of the faradaic current relative to the baseline capacitive current. The peak height in the example shown is approximately 120 nA (310 nA peak −190 nA capacitive current = 120 nA, at the center). The faradaic current is recorded as a peak with a center at approximately 175 mV, coincident with the redox potential of ferrocene relative to the Ag/AgCl reference electrode.
Table 1.
Sequences of Capture Probes Used
| HIV wild-type | 5′TCATTGATGGTCTCTTTTAACA(C16S) |
| HIV mutant | 5′CATTGATGGTGTCTTTTAACA(C16S) |
| HCV wild-type | 5′ATGGCTAGACGCTTTCTGCGTG(C16S) |
| Hfe wild-type | 5′AGATATACGTGCCAGGTGGAG(C16S) |
| Hfe mutant | 5′AGATATACGTACCAGGTGGAG(C16S) |
| ACE D-type | 5′AGTGACTGTATAGGCAGCAGGT(C16S) |
| fas | 5′TTATGGCAGAATTGGCCATCAT(C16S) |
| bax | 5′GTGTCCACGGCGGCAATCATCC(C16S) |
| p53 (1) | 5′GGCGCGGACGCGGGTGCCGGGC(C16S) |
| p53 (2) | 5′CAGCGCCTCACAACCTCCGTCA(C16S) |
| p21 | 5′GTGGGAAGGTAGAGCTTGGGCA(C16S) |
| bcl-2 | 5′TCCCCCAGTTCACCCCGTCCCT(C16S) |
| Actin | 5′TCGTCGCCCGCGAAGCCGGCCT(C16S) |
| GAPDH | 5′CATCGCCCCACTTGATTTTGGA(C16S) |
Table 2.
Sequences of Signaling Probes Used
| HIV | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3TTTGCATGGCTGCTTGATGTC-3′ |
| HCV | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3AAGACAGTAGTTCCTCACAGGG |
| Hfe | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3CACCCAGGCCTGGATCAGC-3′ |
| ACE | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3CTAGAGAAATGGGAGAAAGGAT3′ |
| fas | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3GATGCAGGCCTTCCAAGTTCTG3′ |
| bax | [Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3TCTGCAGCTCCATGTTACTGTC |
| p53 (1) | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3GGGGGTGTGGAATCAACCCACA3′ |
| p53 (2) | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3TGTGCTGTGACTGCTTGTAGAT3′ |
| p21 | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3GGCCAAGGCCCCGCACACGCTC3′ |
| bcl-2 | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3GAAGAGCTCCTCCACCACCGTG3′ |
| Actin | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3TGCACATGCCGGAGCCGTTGTC3′ |
| GAPDH | 5′[Fc]C[Fc]G[Fc]C[Fc]GCTTA[Fc]C[Fc]G[Fc]C[Fc]G(EO)3GGGATCTCGCTCCTGGAAGATG3′ |
The modified adenine is referred to as [Fc] in the signaling probes shown here. The portion of the signaling probe complementary to the target is at the 3′ end and is separated from the ferrocene-labeled tail by a spacer comprised of three ethylene glycol units, abbreviated (EO)3.
Synthesis of Target Mimics
For initial characterization of the system, unmodified DNA oligonucleotides were synthesized to serve as target mimics, which are co-linear with their respective targets in the region complementary to the capture and signal probes (Table 3) .
Table 3.
Sequences of Target Mimics Used
| HIV wild-type | 5′GACATCAAGCAGCCATGCAAATGTTAAAAGAGACCATCAATGAGGAAGCTGCAGAATGGGATAGAGTGCATCCAGT-3′ |
| HIV mutant | 5′GACATCAAGCAGCTATGCAAATGTTAAAAGACACCATCAATGAGGAAGCTGCAGAATGGGATAGAGTGCATCCAGT-3′ |
| Hfe WT | 5′GCTGATCCAGGCCTGGGTGCTCCACCTGGCACGTATATCTCTGCTCTTCCCCAGGGGGTACAGCCAAGGTTATCCA-3′ |
| Hfe C282Y mutant | 5′GCTGATCCAGGCCTGGGTGCTCCACCTGGTACGTATATCTCTGCTCTTCCCCAGGGGGTACAGCCAAGGTTATCCA-3′ |
| ACE D-type | 5′CACTCCCATCCTTTCTCCCATTTCTCTAGACCTGCTGCCTATACAGTCACTTTTATGTGGTTTCGCCAATTTTA3′ |
| fas | 5′CAGAACTTGGAAGGCCTGCATCATGATGGCCAATTCTGCCATAAGCCCTGTCCTCCAGGTGAAAGGAAAGCTAGGGACTG3′ |
| bax | 5′GACAGTAACATGGAGCTGCAGAGGATGATTGCCGCCGTGGACACAGACTCCCCCCGAGAGGTCTTTTTCCGAGTGGCAGC3′ |
| p53 (1) | 5′TGTGGGTTGATTCCACACCCCCGCCCGGCACCCGCGTCCGCGCCATGGCCATCTACAAGCAGTCACAGCACATGACGGAG3′ |
| p53 (2) | 5′ATGGCCATCTACAAGCAGTCACAGCACATGACGGAGGTTGTGAGGCGCTGCCCCCACCATGAGCGCTGCTCAGATAGCGA3′ |
| p21 | 5′GAGCGTGTGCGGGGCCTTGGCCTGCCCAAGCTCTACCTTCCCACGGGGCCCCGGCGAGGCCGGGATGAGTTGGGAGGAGG3′ |
| bcl-2 | 5′CACGGTGGTGGAGGAGCTCTTCAGGGACGGGGTGAACTGGGGGAGGATTGTGGCCTTCTTTGAGTTCGGTGGGGTCATGT3′ |
| Actin | 5′GACAACGGCTCCGGCATGTGCAAGGCCGGCTTCGCGGGCGACGATGCCCCTCGGGCCGTCTTCCCCTCCATCGTGGGGCG3′ |
| GAPDH | 5′CCCATCACCATCTTCCAGGAGCGAGATCCCTCCAAAATCAAGTGGGGCGATGCTGGCGCTGAGTACGTCGTGGAGTCCAC3′ |
Single nucleotide differences between wild-type and mutant alleles are indicated in bold type for the appropriate pairs.
Hybridization and Electrochemical Analysis
Target mimics were mixed with signaling probes in hybridization buffer to achieve a final concentration of 50 nmol/L and 125 nmol/L, respectively, unless otherwise noted. Hybridization buffer was 1 Mol/L NaClO4 in 50% lysed blood, prepared by combining 8 equivalents of anticoagulated blood with 1 equivalent of Proteinase K solution (Qiagen, Valencia, CA) and then adding 8 equivalents of Qiagen Lysis Buffer AL. Lysed blood served to reduce nonspecific binding of signaling probes to the DNA sensor; other ingredients were later substituted for this component (data not shown). For amplicon analysis, an aliquot of the reaction products was combined with signaling probe, heated 3 minutes at 100°C, placed on ice, and added to hybridization buffer such that the final concentration of signaling probe was 200 nmol/L. Hybridization buffer was then introduced into the hybridization chamber above the electrode array.
The AC voltammetry technique used to gather the electrochemical signal at the DNA sensor is described in more detail elsewhere 4, 5 (Terbrueggen RH, Vielmetter J, Chen Y-P, Millan KM, Mucic RC, Olsen GT, Swami N, Umek RM, Wang H, Welch TW, Yowanto H, Yu CJ, Blackburn GF, and Kayyem JF, submitted for publication). A DC voltage bias is applied to the electrode and ramped, typically from −100 to +500 mV, over a range of electrical potentials that encompasses the redox potential of the ferrocene moiety. An AC voltage of 25 or 100 mV is applied in conjunction with the DC bias to increase the frequency of the redox reaction, thus amplifying the signal. At the redox potential of the ferrocene, the peak of the faradaic current is directly proportional to the number of ferrocene moieties immobilized at the electrode surface that is, in turn, proportional to the number of target nucleic acid molecules. (Faradaic current is that current specifically generated as a result of reduction or oxidation of the ferrocene label.) The total current from the cell passes through microelectronics devices used to detect and measure the electrical signal generated.
Amplification Reactions
Nucleic Acids
Anonymized human genomic DNAs characterized for ACE and Hfe genotypes were gifts of Dr. Wayne Grody (UCLA, Los Angeles, CA) and Karl Voelkerding (University of Wisconsin, Madison, WI), respectively. The template for the HIV gag DNA amplification was plasmid V103[lowhy]4, 7 an M13-based plasmid containing the entire HIV sequence. To genotype the Hfe samples, 50 to 200 ng of human genomic DNA were amplified by asymmetric polymerase chain reaction (A-PCR) with the forward primer in fivefold excess over the reverse primer and the total primer concentration adjusted to 720 nmol/L. Primers for Hfe A-PCR were: 5′TGGCAAGGGTAAACAGATCC3′ (forward) and 5′CTCAGGCACTCCTCTCAACC3′ (reverse).
PCR Reagents
Deoxynucleotide 5′-triphosphates, 10× PCR buffer, 25 mmol/L MgCl2, and Taq polymerase were purchased and used according to the supplier’s recommendation (Applied Biosystems, Foster City, CA).
PCR and A-PCR
PCR of ACE type D DNA was carried out using various amounts of human genomic DNA (12.5–200 ng), 200 μmol/L dNTP, 50 mmol/L KCl, 10 mmol/L Tris-HCl (pH 8.3), 2 mmol/L MgCl2, 0.25 U Taq polymerase, and 300 nmol/L forward and reverse primer. Cycling parameters were 94°C for 3 minutes to denature human DNA, followed by 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 45 seconds, and ending at 72°C for 5 minutes to extend all unfinished DNA ends. The ACE type D DNA amplicon (191 bp) was confirmed by gel electrophoresis alongside molecular weight standards and by ApoI restriction enzyme digestion and subsequent gel electrophoretic analysis. A-PCR 8 differed from PCR in that forward (5′CTGGAGACCACTCCCATCCTTTCT3′) to reverse primer (5′GATGTGGCCATCACATTCGTCAGA3′) concentration was 5:1 and total primer concentration was 720 nmol/L.
Reverse Transcriptase (RT)-A-PCR for Gene Expression Monitoring
RNA Preparation
Total white blood cell RNA was isolated using the Qiagen RNA Easy kit. RNA integrity was assessed by formaldehyde gel electrophoresis followed by ethidium bromide staining and rRNA visualization (not shown). Human heart RNA was purchased from Ambion, (Austin, TX).
Reverse Transcription
cDNAs were synthesized using Retroscript (Ambion) according to the supplier’s recommendation. One to 2 μg of total white blood cell RNA was mixed with 4 μl dNTP (10 mmol/L), 2 μl primers (random decamer), and H2O to a volume of 16 μl. The sample was heated for 3 minutes at 70°C and then chilled on ice. One microliter of Moloney murine leukemia virus reverse transcriptase (100 units), 2 μl reaction buffer (10×), and 1 μl RNasin (10 units) were added to the sample, followed by incubation at 42°C for 1 hour for cDNA synthesis. Reverse transcriptase was then inactivated at 92°C for 10 minutes and cDNAs were stored at −20°C.
A-PCR of cDNAs
Using the cDNA as input, A-PCR was performed using primers specific for actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and the apoptosis-related genes fas, p53, p21, bax, and bcl-2. The gene-specific primers were purchased as a set (Stratagene, San Diego, CA). Reactions were conducted with 1 to 3 μl of cDNA solution, 200 μmol/LdNTP, 50 mmol/L KCl, 10 mmol/L Tris-HCl (pH 8.3), 2 mmol/L MgCl2, and 0.25 U Taq polymerase, using 600 nmol/L forward primer and 120 nmol/L reverse primer. A-PCR cycling parameters were 94°C (2 minutes), followed by 35 cycles (94°C for 45 seconds, 58°C for 1 minute, and 72°C for 1 minute), and ending with 72°C for 5 minutes to extend unfinished DNA ends. Amplicons of actin (400 bp), GAPDH (600 bp), fas (550 bp), p53 (431 bp), p21 (561 bp), bax (365 bp), and bcl-2 (293 bp) were confirmed by gel electrophoresis, as above.
Results
A novel system based on electrochemical detection of nucleic acids has been developed. Gold electrodes on printed circuit board serve as the solid support for the formation of SAMs containing DNA capture probes. The gold electrode also serves as the system’s detection electrode. A schematic of the SAM with an associated nucleic acid target and its cognate signaling probe is shown in Figure 1A . The SAM contains three species of molecules including DNA capture probes with alkane linkers, polyglycol-terminated alkane chains that serve as insulators for the sensor, and highly conjugated phenylacetylene molecular wires, that provide a pathway for electron transport between labeled probe and the gold electrode 4, 5 (Terbrueggen RH, Vielmetter J, Chen Y-P, Millan KM, Mucic RC, Olsen GT, Swami N, Umek RM, Wang H, Welch TW, Yowanto H, Yu CJ, Blackburn GF, and Kayyem JF, submitted for publication). The DNA capture probes assemble in the SAM by virtue of the alkane linker at the 3′ end of the DNA strand. All of the molecules have a thiol group that, on deposition, results in a sulfur-gold bond that immobilizes the molecules on the gold surface. The illustration is simplified inasmuch as the ethylene glycol heads are likely not ordered when present in the SAM, 9 and the SAM is likely less ordered in the presence of the molecular wires. 10
When target nucleic acid is present, sequence-specific hybridization to the capture probe occurs at the electrode surface (Figure 1A) . The target participates in a second sequence-specific hybridization reaction with a signaling oligonucleotide that is labeled with ferrocene moieties that act as reporter molecules. In most cases, the region of signaling probe hybridization is immediately proximal to the region of capture probe hybridization on the target. The signaling oligonucleotide is modified through the incorporation of multiple ferrocene-modified phosphoramidites during synthesis. Ferrocene is a redox-active metal compound. When a given potential is applied to the gold electrode, electron transfer occurs between the ferrocene and the gold electrode (Figure 1B) . Target-specific electrodes can be engineered by varying the sequence of the deposited DNA capture probe. In the chip configuration described in this report, 14 electrodes are arrayed on a chip along with reference and auxiliary electrodes to complete the electrochemical cell. A polycarbonate cartridge containing inlet and outlet ports houses the electrodes in a hybridization chamber while the connector edge remains exposed such that the chip can be analyzed in an electrochemistry workstation. Devices more amenable to use in the molecular diagnostics laboratory have been developed and will be described elsewhere.
We fabricated an electrode array containing capture probes specific for sequences in the human immunodeficiency virus (HIV) or hepatitis C virus (HCV) on separate electrodes. We synthesized an oligonucleotide co-linear with a portion of HIV to serve as a target mimic. A ferrocene-labeled signaling probe was synthesized that is complementary to the HIV target in the sequence immediately adjacent to the region of complementarity to the capture probe. A ferrocene-labeled signaling probe complementary to HCV was also synthesized. The HIV target mimic and both signaling probes were introduced into the hybridization chamber, and the electrodes were interrogated electronically and their signal outputs recorded. Representative voltammograms from HIV- and HCV-specific electrodes demonstrate fundamental properties of the system (Figure 1C) . Both electrodes exhibit a background current, arising from the capacitance of the electrode-solution interface, of approximately 180 nA at −100 mV that rises to approximately 240 nA at 500 mV. However, only the electrode containing HIV capture probes also exhibits a specific faradaic current. Thus, there is no detectable faradaic current from the HCV-specific electrodes despite the fact that the HCV signaling probe is present. We reason that sequence-specific hybridization of the HIV target mimic facilitates the formation of a structure similar to that depicted in Figure 1A only on the electrodes modified with HIV-specific capture probes. Moreover, only signaling probes immobilized in the ternary complex are electronically coupled to the electrode and thus capable of generating a signal, an observation consistent with development of one-step detection protocols that do not require steps to remove unbound signaling probes (see below).
Amplicon Detection on the Electrochemical Sensor
The utility of the electrochemical sensor for sequence-specific detection of amplicons was explored. As an example, we examined an insertion/deletion polymorphism in intron 16 of the angiotensin converting enzyme (ACE) gene that results from the presence (allele I) or absence (allele D) of an Alu sequence. The polymorphism has been studied for correlation with various aspects of cardiovascular disease. 11 A 192-bp fragment of the D allele was amplified from genomic DNA. We synthesized a 22-bp capture probe homologous to the D allele and a ferrocene-labeled signaling probe complementary to an adjacent 22 bp of the D allele. Also, a 74-mer oligonucleotide, co-linear with a portion of the D allele and encompassing the capture and signaling probe annealing regions, was synthesized to serve as a target mimic (Figure 2A) . The sister strand to the 74-mer target mimic was also synthesized. Electrode arrays were prepared that contained capture probes complementaryto the target strand of the ACE D allele. The arrays also contained negative control electrodes modified with an unrelated, heterologous DNA capture probe. ACE electrode arrays were challenged separately with ACE amplicon or single- or double-stranded target mimics. In each hybridization reaction, the same ferrocene-labeled signaling probe was also present.
Figure 2.

Electronic detection of PCR products. A: Line diagrams showing the sequence relationships among angiotensin-converting enzyme (ACE) alleles D and I as well as the D allele oligonucleotide target mimic, the signaling probe (SP), and the capture probes (CPs). The nucleotide coordinates are from GenBank (accession no. AF118569). The target mimic is 74 nucleotides and co-linear with the D allele. The SP is co-linear with both alleles. Two CPs were synthesized, one co-linear with the D allele through the sequence interrupted by the Alu insertion, the other co-linear with the sequence of the junction in the I allele. B: Fifty nanograms of genomic DNA (gDNA) were used to amplify a 192-bp amplicon from the ACE gene (allele D) which was heat denatured in the presence of a ferrocene-labeled signaling probe and applied as a 125 nmol/L solution of amplicon to an electrode array containing the D allele capture probe. For comparison, a 50 nmol/L solution of a 74-mer double-stranded target mimic and a 50 nmol/L solution of the single-stranded target mimic were used to challenge separate ACE chips. The products of a PCR that did not contain gDNA was similarly analyzed for comparison (neg). Hybridization proceeded for two hours before AC voltammetry was performed on individual electrodes of each chip. The mean and SD are reported from 7 electrodes/array and 2 arrays/target. No faradaic signal was detected on electrodes in the array that are modified with capture probes non-complementary to the ACE amplicon (not shown). C: A-PCR was performed and the reaction products were hybridized in the electrode array chambers for 20 minutes before analysis, as in B. The mean and SD of peak current are reported for two chips (1 and 2) from electrodes modified with the ACE D allele- or ACE I allele-specific capture probes.
The ACE amplicon is specifically detected on electrodes containing ACE capture probes after 2 hours of hybridization (Figure 2B) at room temperature. Only hybridization solutions containing the ACE amplicon or a target mimic generate a signal above the background. Notably, the single-stranded oligonucleotide target mimic generates considerably more electrochemical signal than the amplicon or the double-stranded model target. This observation suggests that, despite initial heat denaturation, amplicon sister strand re-annealing in solution competes with hybridization to the capture probes at the DNA sensor.
To test the hypothesis that sister-strand re-annealing inhibits amplicon detection, we introduced strand bias into the PCR product by performing a modified A-PCR. A-PCR D allele products were used to challenge arrays containing electrodes with capture probes homologous to the D and I alleles. After 20 minutes of hybridization at room temperature, the D allele amplicon generated signals specifically on electrodes modified with D capture probes, whereas the electrodes modified with I allele-specific capture probes did not generate detectable signals (Figure 2C) . In this amplicon detection protocol, an aliquot of the amplification reaction is simply diluted into hybridization/electrochemical detection buffer containing signaling probes and introduced into the hybridization chamber. Thus, the electronic detection platform facilitates sequence-specific detection of amplicons in a single-step, homogeneous assay. Amplicons generated with the strand displacement amplification technique 12 are also readily detected with the electronic detection platform (not shown).
Single-Base Mismatch Discrimination on the Electrochemical Sensor
Next, we investigated whether discrimination of single-base mismatches among amplicons is feasible with the system. We synthesized a 76-mer oligonucleotide co-linear with a portion of the HIV genome to serve as a target mimic. We synthesized a second 76-mer containing a single G to C transition at position 32 from the 5′ end of the oligonucleotide, centered in the region of the target complementary to the capture probe. For ease of reference, we have designated the original 76-mer as wild-type (WT) and the oligonucleotide containing the substitution as mutant. Both 76-mer oligonucleotides are perfectly complementary to the signaling probe synthesized and are anticipated to form similar sandwich structures (Figure 1A) under non-stringent conditions.
We prepared electrode arrays with capture probes perfectly complementary to the WT HIV oligonucleotide target mimic. The WT and mutant targets were both introduced into these chips and, after an initial hybridization at ambient temperature, the electrochemical signal on the electrodes was characterized at various temperatures. As the temperature in the hybridization chamber is increased, the faradaic peak current is retained preferentially on electrodes occupied by targets that are perfectly matched (Figure 3A) . Thus, SNPs are discriminated on the electronic detection platform by virtue of their differential stability at elevated temperature. We prepared a genotyping chip containing subsets of electrodes that are perfectly complementary to the WT or mutant HIV oligonucleotide target mimics. The arrays also contain capture probes that are not complementary to either target to serve as controls for nonspecific binding. At the elevated temperature, samples containing only one type of target exhibit a preferential reduction in the electrochemical signal from the electrodes that have capture probes mismatched with the target (Figure 3B) . In contrast, arrays challenged with a mixture of the two targets exhibit a similar amount of signal from the two types of electrodes at elevated temperature (Figure 3B) . In an effort to normalize the signal output with respect to variation on the electrodes, we defined the signal output at 25°C for each electrode in the array as 100%. Using the normalized data, a simple pattern emerges for the characterization of the SNP. The normalized signal output is always more than twofold greater (usually 3- to 15-fold) on the electrodes containing capture probes that are perfectly matched compared to electrodes containing mismatched capture probes for amplicons derived from homozygous samples. In contrast, the normalized signal output is less than 1.5-fold different between the two kinds of electrodes for heterozygous samples.
Figure 3.

Single-base mismatch discrimination on the bioelectronic sensor. A: Electrode arrays were prepared with capture probes complementary to a region of the HIV genome and a region of the HCV genome. The arrays were challenged with a 76-mer oligonucleotide containing a region of perfect complementarity to the HIV capture probe (designated wild-type) or a second 76-mer oligonucleotide containing a single base substitution (designated mutant), centered in the region complementary to the capture probe. Five hundred nmol/L of the target oligonucleotides and 2.5 μmol/L of a corresponding signaling probe were hybridized for 20 minutes at 25°C (two arrays were studied for each target) and the electrodes were interrogated by AC voltammetry (ACV). The mean of the peak current detected from 4 electrodes at 25°C was defined as 100% for each array. Subsequently, the temperature of the hybridization solution was elevated in 5°C increments. At each elevated temperature, the solution was held for 1 minute before interrogation of the electrodes by ACV. The mean of the peak faradaic current detected on four electrodes at each temperature is reported for each of the four arrays studied. Electrodes containing capture probes complementary to HCV gave no detectable faradaic current at any temperature (not shown). Note that the same electrodes are being scanned repeatedly at the various temperatures. B: Electrode arrays were prepared containing capture probes that are perfectly complementary to the wild-type or mutant HIV targets. The arrays also contained capture probes complementary to a portion of the HCV genome. Three separate arrays were challenged with a 500 nmol/L solution of the wild-type HIV target mimic (WT Target), the mutant target mimic (mut Target), or an equimolar mixture of the two oligonucleotides (Mix). After 20 minutes at 25°C, the electrodes were interrogated and the resulting peak current measured (not shown). Subsequently, the temperature of the solutions was elevated to 52°C. The reaction volume was held at the elevated temperature for 1 minute before ACV interrogation of the electrodes. The mean and SD of the peak current detected from 4 electrodes of each type, wild-type or mutant, are reported for the three arrays. Above the signal outputs recorded at 52°C, the ratio of the means is reported as wild-type/mutant. No signal was detected at any temperature from the electrodes containing capture probes complementary to HCV (not shown). On the left-hand side, the results are reported after normalizing the signal output for each electrode type to 100% at 25°C. Above the normalized signal outputs at 52°C, the ratio of the normalized means is reported as wild-type/mutant. The targets are given the suffix n in the normalized data. C: Line diagrams showing the position of the C282Y SNP (a G-to-A substitution at nucleotide position 845) in the Hfe gene and the relationships among the amplicon (top line), the target mimics, the CPs, and the SP. The target mimics contain a G (wild-type) or an A (mutant) at position 845. The CPs have a complementary C (wild-type) or T (mutant), respectively. A single signaling probe is complementary both targets. D: A genotyping chip was developed that contains capture probes perfectly matched to the wild-type or mutant allele with respect to the C282Y SNP in the Hfe gene. Amplicons were prepared from previously characterized genomic DNAs using A-PCR. Six electrode arrays were separately challenged with A-PCR products generated from homozygous wild-type genomic DNAs (WT-1 and WT-2), homozygous mutant genomic DNAs (Homo-1 and Homo-2), and heterozygous genomic DNAs (Hetero-1 and Hetero-2). The electrodes were interrogated at 25°C and again 1 minute after the hybridization chamber reached 50°C. The mean and SD of the peak faradaic current are reported after normalizing to the mean determined for each electrode type in each array at 25°C (defined as 100%). The ratio of the normalized means at the restrictive temperature is reported above the bars reporting the values at 50°C as wild-type/mutant. The values are derived from two electrodes of each type in each array. Electrodes modified with an unrelated DNA capture probe did not yield detectable faradaic current at any temperature.
Our ability to achieve sequence-specific detection of amplicons combined with our success at discriminating single-base mismatches with the electronic detection system prompted us to study whether we could discriminate genetic variation associated with a disease state using amplicons generated from human genomic DNA. Hereditary hemochromatosis is the most common inherited genetic disorder known to date. 13 Mutations in the Hfe gene are associated with iron overload and numerous resulting pathologies. 13 The most common mutation in the population is the C282Y mutation resulting from a C-to-T transition in codon 282. We prepared capture probes that are perfect complements to the WT or mutant Hfe alleles (Figure 3C) . The probes were designed such that mismatches between the WT capture probe and the mutant sequence (and vice versa) occur in the middle of the capture probe. We also synthesized a common signaling probe and a pair of target mimics, co-linear with the WT and mutant amplicons, respectively. The optimal temperature at which to discriminate between the two targets was defined using target mimics (not shown). Next, amplicons harboring the SNP were prepared, using the A-PCR technique from patient DNAs previously characterized as homozygous WT, homozygous mutant, or heterozygous. Two patient samples of each genotype were studied. The amplicons were analyzed independently using electrode arrays containing WT and mutant C282Y capture probes. The A-PCR products can be readily genotyped using the normalized signal output at the stringent temperature (Figure 3D) . Specifically, the A-PCR products from the three different genotypes result in three distinct ratios of WT to mutant electrode signals. Thus, the electronic detection platform described here can be readily used to genotype DNA samples.
Gene Expression Monitoring through Electronic Detection
To explore the utility of a gene expression monitoring chip we chose the cellular apoptotic response as a model. Several gene products are known to be up- or down-regulated during apoptosis. 14 We chose to characterize 5 apoptosis-regulated genes fas, p53, bax, p21, and bcl-2, and actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as control genes. In the electrode array, each gene has two corresponding electrodes with cognate capture probes, an additional two electrodes are dedicated to the detection of the control gene, and two electrodes are modified with an unrelated DNA capture probe to monitor nonspecific signaling.
We designed capture and signaling probes in such a way as to minimize the chance of direct interaction between signaling probes and capture probes and cross-hybridization between amplicons and unrelated capture probes for the genes listed. No direct interaction between the signaling probes and capture probes was detected (not shown). Oligonucleotides were synthesized to serve as target mimics during the characterization and optimization of the apoptosis gene expression monitoring chip. When electrode arrays are challenged with the bcl-2 target mimic and its cognate signaling probe, the electrodes modified with the bcl-2 capture probe generate a peak faradaic current that is more than 100-fold greater than that detected on any other electrode type (Figure 4A) . When the same DNA chip is subsequently challenged with a pool of the remaining target mimics and their cognate signaling probes, all of the electrodes generate faradaic current (Figure 4A) . Similar results were obtained when bax was used as the target of interest (Figure 4B) . The amount of peak faradaic current observed varies among the electrodes with respect to capture probe type (Figure 4A and 4B) . Although the source of this variation is unknown and is under investigation, we have been able to design alternative capture and signaling probe pairs for poorly performing sets, and we have realized nearly equivalent performance among various probe sets, eg, ) (Figure 4A versus Figure 4D . Results similar to those shown in Figure 4 , A and B, were obtained for all of the target mimics studied including p53, p21, fas, actin, and GAPDH (not shown).
Figure 4.

An apoptosis gene-expression monitoring chip. A: An array was prepared containing electrodes modified with capture probes that are complementary to the human genes fas, p53, bax, p21, and bcl-2, as well as actin. Synthetic oligonucleotides were synthesized to serve as target mimics to validate and optimize the apoptosis gene-expression monitoring chip. Ferrocene-labeled signaling probes were also synthesized for each target. Fifty and 250 nm of the synthetic bcl-2 target mimic and its cognate signaling probe, respectively, were hybridized with the array for 1 hour at 25°C (t = 1 hour). All of the electrodes were interrogated, and the resulting current was recorded. Subsequently, a pool of the other target mimics and their signaling probes was applied, hybridization was continued for another hour, and the electrodes were once again interrogated by AC voltammetry and the resulting current recorded (t = 2 hours). Each bar represents the mean and SD observed for two electrodes in the array. B: The electrode array described in A was challenged first with the bax synthetic target mimic and signaling probe and subsequently the entire pool of the target mimics and their cognate signaling oligonucleotides. The array was analyzed as in A. C: A fas amplicon was obtained through RT-A-PCR with fas-specific primers using total human lymphocyte RNA. The amplicon was placed in hybridization solution along with its cognate signaling probe and incubated overnight in the hybridization chamber of an electrode array identical to the one described in A. D: A p53 amplicon was obtained through RT-A-PCR using p53-specific primers. The apoptosis gene-expression electrode array was challenged with the amplicon and its cognate signaling probe and analyzed as in A. E:fas and GAPDH amplicons were generated through RT-A-PCR and analyzed in the presence of both cognate signaling probes simultaneously, as in A.
After characterizing the apoptosis gene expression monitoring chip with target mimics, we analyzed RT-PCR amplicons for fas expression on the chips using human RNA. An A-PCR protocol was used that limits one primer, thereby creating strand bias in the amplicon pool in favor of the target strand. A second reaction was conducted in parallel without reverse transcriptase to assure that the amplicons arise from RNA. After RT-A-PCR and without further processing, the products were placed in the hybridization chamber of apoptosis gene expression monitoring chips. After hybridization at room temperature followed by AC voltammetry, a faradaic peak was detected from electrodes modified with the fas capture probe that is approximately 36-fold greater than that observed on any other type of electrode (Figure 4C) . Moreover, the signal detected on electrodes other than fas is likely a consequence of non-specific binding of signaling probes, since similar signals are observed from the sample that lacked reverse transcriptase (Figure 4C) . The fas-specific signal detected is, in general, lower than that observed from single-strand oligonucleotide target mimics (Figure 4C versus Figure 4 , A and B), an observation consistent with the notion that sister-strand re-annealing reduces the amount of target strand that can be captured on the electrode surface (Figure 2B) . Similar to the results obtained with fas, the p53-specific reaction results in electrochemical signaling almost exclusively from electrodes modified with p53 capture probes in arrays exposed to products from a reaction that contained reverse transcriptase (Figure 4D) . Notably, the signal output for the p53 amplicon in greater than that observed for the fas amplicon (Figure 4D versus Figure 4C ). Agarose gel electrophoresis of the two amplicons followed by ethidium bromide staining confirmed that the p53 amplicon was more abundant in the samples (not shown). Thus, the gene expression monitoring chip can be a quantitative indicator of transcript abundance.
We next explored the possibility that RT-A-PCR reactions could be pooled to facilitate multiplex gene expression monitoring using the electronic detection platform. We performed RT-A-PCR using fas- and GAPDH-specific primers in separate reactions. The reaction products were combined, diluted with hybridization/electrochemical detection solution, and introduced into the apoptosis gene expression monitoring chip. The electrodes containing fas and GAPDH capture probes were the only electrodes to generate detectable faradaic current (Figure 4E) . The signal generated by the fas amplicon in the multiplex analysis is lower than that seen when fas was analyzed individually (Figure 4E versus Figure 4C ). However, less RT-A-PCR product was used and the hybridization time was shorter in the multiplex analysis. Our findings demonstrate that multiple gene expression products can be analyzed simultaneously with the electronic detection platform described here.
Discussion
We have described here a novel electronic detection platform for nucleic acid-based assays. Gold electrodes covered with a SAM containing DNA capture probes constitute the sensors arrayed on printed circuit board. The properties of the SAM result in a DNA sensor with demonstrable signal specificity. In the SAM, the capture probes are DNA-alkanethiols that self-assemble with polyglycol-terminated alkanethiols that serve as insulator molecules and phenylacetylene molecules that act as molecular wires. Signaling probe must be in close proximity to the SAM to generate a faradaic current in response to a change in the potential applied to the electrode. Although the system was designed to facilitate electron transfer through the molecular wires, some evidence suggests that alternative pathways may be active (data not shown). Our findings suggest that, regardless of the mechanism, signaling probes immobilized near the surface by virtue of their inclusion in a sandwich structure with the target are electronically coupled to the gold electrode, whereas those in solution above the surface are not. The crucial distance dependence of signaling probe function eliminates the need to remove excess signaling probe before interrogating the electrode to detect the target nucleic acid specifically. We reason that the hybridization-dependent sandwich structure (Figure 1A) electronically couples ferrocenes in the signaling probe to the gold electrode. The exquisite sensitivity of the sensor to the position of the reporter molecule eliminates the need for washing steps. For example, the PCR amplicons studied here were simply diluted into hybridization buffer and introduced into the hybridization chamber of the arrays with excess signaling probe (Figures 2 3 4) . No additional steps were required to clean up the reaction products or eliminate reaction constituents or excess signaling probe before electronic detection was conducted.
Using the sensor, we achieve single-base mismatch discrimination (Figure 3) by empirically determining a temperature at which formation of a sandwich complex between a mismatched target and capture probe is inhibited significantly compared to a perfectly matched target. We assume that destabilization of the signaling complex at the electrode containing the mismatch is a result of melting of the target and the capture probe. Such an interpretation is consistent with the observed destabilizing effect of mismatches on DNA hybrids studied in solution 15 and on solid supports. 16 However, actual melting of the target strand and the capture probe may not be a prerequisite for signal diminution, since the DNA sensor exhibits critical distance dependence for electronic coupling of the ferrocenes in the signaling probe and the electrode.
Our ability to detect different targets specifically at individual electrodes makes the electronic detection platform readily compatible with multiplex analyses. Careful selection of capture probes and signaling probes facilitates detection of multiple nucleic acid targets at electrodes modified with their respective capture probes even at ambient temperature (Figure 4E) . Multiplexing is accomplished without the need to incorporate a label into the target, eg, fluorescent or isotopic, by virtue of the sandwich assay. Thus, amplification products containing naturally occurring nucleotides (Figures 2 3 4) as well as unmodified DNA and RNA (unpublished observations) serve as targets for multiplex analysis in the electronic detection platform. The multiplex detection capability of the electronic platform combined with one-step characterization of amplicons makes the system suitable for gene expression monitoring (Figure 4) .
Another feature of the platform described here is the ability to interrogate the biosensor repeatedly without signal decay (Figure 4) . Several advantages emerge from this feature. First, mismatch detection is facilitated. Samples are annealed to electrodes containing capture probes at ambient temperature. Subsequently, the hybridization solution is heated and the electrodes interrogated as often as necessary to survey all of the array’s electrodes. In fact, we have surveyed up to four SNPs using three temperatures and repeated electrode scanning (unpublished results). The ability to interrogate electrodes repeatedly during hybridization facilitates kinetic analysis of target accumulation at the biosensor surface (Terbrueggen RH, Vielmetter J, Chen Y-P, Millan KM, Mucic RC, Olsen GT, Swami N, Umek RM, Wang H, Welch TW, Yowanto H, Yu CJ, Blackburn GF, and Kayyem JF, submitted for publication). Second, the electronic detection platform is inexpensive to manufacture. 4, 5 The use of exposed gold electrodes on a printed circuit board exploits existing industry protocols to prepare the solid support. In addition, the nucleic acid constituents of the monolayer as well as the signaling probes are readily prepared by conventional solid-phase synthesis. 6 The biosensor is prepared through the self-assembly properties of the DNA capture probe and the other constituents of the SAM. The relatively low manufacturing costs associated with the platform encourage the development of applications that will benefit from a disposable nucleic acid chip.
The bioelectronic detection platform described here, based on disposable DNA chips and electronic readers, will facilitate the development of relatively low-cost, arrayed nucleic acid-based tests. The arrays will have clinically appropriate densities compatible with the needs of molecular diagnostics laboratories. Moreover, labor costs for molecular diagnostics tests may be decreased by multiplex detection and the elimination of steps after amplification.
In its current design, this bioelectronic detection format remains dependent, in most but not all cases, on some form of in vitro nucleic acid amplification. It is, therefore, currently a system best suited for use in the hospital, university-based, or reference molecular diagnostics laboratory. The system is also being developed for non-human applications such as animal husbandry, transgene detection in commercially important crops, and veterinary infectious disease diagnostics. In both human and non-human applications one can imagine the need for both benchtop, laboratory-based instruments (electronic readers) and more portable, potentially field-deployable units that accommodate the kind of DNA chips described here. More portable systems would require integration of specimen preparation, amplification and electronic detection, areas of research intensively being studied here. The potential also exists for enhancements in the system that may eventually obviate the need for nucleic acid amplification.
Acknowledgments
We thank Drs. Wayne Grody and Karl Voelkerding for supplying anonymous patient DNAs that had been characterized for their ACE and C282Y genotypes, respectively. We thank Dr. Tom Welch for his help with the manuscript and acknowledge the Chemistry and Instrumentation groups at Clinical Micro Sensors Division for their vital contributions.
Address reprint requests to Daniel H. Farkas, Clinical Micro Sensors Division of Motorola, Inc., 757 South Raymond Avenue, Pasadena, CA 91105. E-mail: dan.farkas@motorola.com.
References
- 1.Abramowitz S: DNA Analysis in microfabricated formats. J Biomed Dev 1999, 1:107-112 [DOI] [PubMed] [Google Scholar]
- 2.Zhang MQ: Large-scale gene expression data analysis: a new challenge to computational biologists. Genome Res 1999, 9:681-688 [PubMed] [Google Scholar]
- 3.Hacia JG, Collins FS: Mutational analysis using oligonucleotide microarrays. J Med Genet 1999, 36:730-736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farkas DH: Bioelectronic detection of DNA and the automation of mol diagnostics. J Assoc Lab Automat 1999, 4:20-24 [Google Scholar]
- 5.Creager S, Yu CJ, Bamdad C, O’Connor S, MacLean T, Lam E, Chong Y, Olsen GT, Luo J, Gozin M, Kayyem JF: Electron transfer at electrodes through conjugated “molecular wire” bridges. J Am Chem Soc, 1999, 121:1059–1064
- 6.Yu CJ, Yowanto H, Wan Y, Meade TJ, Chong Y, Strong M, Donilon LH, Kayyem JF, Gozin M, Blackburn GF: Uridine-conjugated ferrocene DNA oligonucleotides: unexpected cyclization reaction of the uridine base. J Am Chem Soc 2000, 122:6767-6768 [Google Scholar]
- 7.Sarver N, Cantin EM, Chang PS, Zaia JA, Ladne PA, Stephens DA, Rossi JJ: Ribozymes as potential anti-HIV-1 therapeutic agents. Science 1990, 247:1222-1225 [DOI] [PubMed] [Google Scholar]
- 8.McCabe P: Production of single-stranded DNA by asymmetric PCR. Innis MA Gelfand DH Sninsky JJ White TJ eds. PCR Protocols A Guide to Methods and Applications. 1990, :pp 76-83 CA, Academic Press, San Diego [Google Scholar]
- 9.Palegrosdemange C, Simon ES, Prime KL, Whitesides GM: Formation of self-assembled monolayers by chemisorption of derivatives of oligo(ethylene glycol) of structure HS(CH2)11(OCh2CH2)Meta-OH on gold. J Am Chem Soc 1991, 113:12-20 [Google Scholar]
- 10.Cygan MT, Dunbar TD, Arnold JJ, Bumm LA, Shedlock NF, Burgin TP, Jones L, II, Allara DL, Tour JM, Weiss PS: Insertion, conductivity, and structures of conjugated organic oligomers in self-assembled alkanethiol monolayers on Au{111}. J Am Chem Soc 1998, 120:2721-2732 [Google Scholar]
- 11.O’Malley JP, Maslen CL, Illingworth DR: Angiotensin-converting enzyme and cardiovascular disease risk. Curr Opin Lipidol 1999, 10:407-415 [DOI] [PubMed] [Google Scholar]
- 12.Walker GT, Fraiser MS, Schram JL, Little MC, Nadeau JG, Malinowski DP: Strand displacement amplification-an isothermal in vitro DNA amplification technique. Nuc Acids Res 1992, 2:1691-1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Press RD: Hereditary hemochromatosis: impact of molecular and iron-based testing on the diagnosis, treatment, and prevention of a common, chronic disease. Arch Pathol Lab Med 1999, 123:1053-1059 [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 15.Guo Z, Liu Q, Smith L: Enhanced discrimination of single nucleotide polymorphisms by artificial mismatch hybridization. Nature Biotechnol 1997, 15:331-335 [DOI] [PubMed] [Google Scholar]
- 16.Wang DG, Fan J-B, Siao J-C, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, Kruglyak L, Stein L, Hsie L, Topaloglou T, Hubbell E, Robinson E, Mittmann M, Morris MS, Shen N, Kilburn D, Rioux J, Nusbaum C, Rozen S, Hudson TJ, Lipshutz R, Chee M, Lander ES: Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 1998, 280:1077-1082 [DOI] [PubMed] [Google Scholar]