

Abstract
Adrenocortical tumors are rare in children and are associated with a poor prognosis when malignant. The fund of knowledge regarding etiology, presentation and clinical outcomes remains limited. Evaluation of genetic disorders associated with the development of adrenocortical disorders has allowed researchers to identify a number of mutations that may be involved in tumorigenesis, including alterations in the GNAS1, PRKAR1A, TP53 and IGF2 genes. Clinical presentation in children is associated most commonly with young age, female gender and symptoms of virilization. Most children have localized disease at presentation which may be associated with a better prognosis when compared to adults. Surgical resection remains the only potentially curative treatment and mitotane, the most frequently used chemotherapeutic agent, has a poor response rate and is highly toxic. Broader participation in multi-center research, such as the International Pediatric Adrenocortical Tumor Registry, is needed to collect sufficient data to better guide our clinical management.
Keywords: Adrenocortical, Pediatric, AIMAH, PPNAD, Carcinoma, Adenoma, Hyperplasia, Mitotane
Adrenocortical neoplasms are rare in children and adolescents. Tumors account for less than 0.2% of all pediatric neoplasms and 1.3% of all carcinomas in patients less than 20 years old (1,2). Single tumors usually are benign unilateral adenomas and more rarely malignant carcinomas. Less commonly, patients can present with benign multinodular hyperplastic lesions including pigmented or non-pigmented micronodular adrenal disease and ACTH-independent macronodular adrenal hyperplasia (AIMAH). The latter is also known as massive macronodular adrenocortical disease (MMAD) (3-5). The purpose of this review is to highlight what is known about the etiology, clinical presentation and treatment of these disorders.
Etiology and Genetics
Little is known about the pathogenesis of these proliferative disorders. It would seem plausible that mutations in the normal signaling pathways that stimulate adrenal steroidogenesis would contribute to their development. The normal signaling pathways begin with the binding of ACTH to a G-protein coupled receptor. This leads to activation of the Gαs subunit followed by activation of adenylyl cyclase. This increases the concentration of cyclic adenosine 3′,5′-monophosphate (cAMP), thereby activating protein kinase A (PKA). PKA is a serine/threonine kinase that phosphorylates transcription factors such as cAMP response element modulator (CREM), cAMP response element binding protein (CREB), activating transcription factor-1 (ATF-1) and steroidogenic factor 1 (SF-1). These transcription factors bind to the cAMP response element (CRE) sequence or the cAMP responsive sequence (CRS) and modify the expression of steroidogenic genes (6-9). Investigations into familial syndromes that are associated with adrenocortical tumors or nodular disease have indeed identified mutations that affect this signaling pathway (see Figure).
Figure.
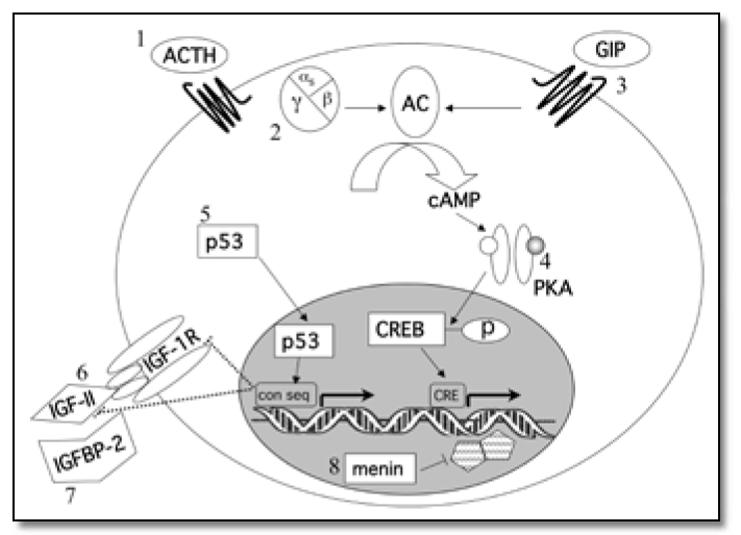
Alterations in signaling pathways thought to be involved in the development of adrenocortical tumorsor or hyperplasias. 1. Excessive ACTH secretion in Cushing’s Disease or CAH, 2. GNAS1 activating mutations in McCune Albright Syndrome, 3. Ectopic expression of G-protein associated receptors, 4. Mutations in PRKAR1A leading to increased cAMP stimulated PKA activity in PPNAD and the Carney Complex, 5. TP53 mutations associated with Li-Fraumeni Syndrome and adrenocortical carcinomas in Brazil, 6. IGF-II over-expression in Beckwith-Wiedemann Syndrome, 7. IGFBP-2 over-expression, and 8. Inactivating mutations of MEN1 in Multiple Endocrine Neoplasia type 1. Con seq = p53-binding consensus sequence; CRE = cAMP response element.
ACTH is important for normal adrenal gland development and growth. It functions in utero as the principal stimulator of adrenocortical fetal zone growth. Afterwards, ACTH stimulates the differentiation, maintenance and hormonal secretion of glucocorticoids from the zona fasciculata, the middle zone of the adrenal cortex and androgens from the zona reticularis, the inner-most zone (10). ACTH hypersecretion, which can be seen with either Cushing’s Disease or congenital adrenal hyperplasia (CAH), leads to bilateral adrenocortical hyperplasia and increased steroidogenesis (5,6,11,12). The size of the adrenal glands correlates with the plasma levels of ACTH and the duration of disease (13). Nodular transformation has been noted in a minority of patients with long standing hyperplasia (12,14).
AIMAH is a benign proliferative disorder of the adrenal cortex that presents with ACTH-independent Cushing ’s syndrome. Histologically it is composed of nodules with two cell types, lipid rich cells with a clear cytoplasm and lipid poor cells with a compact cytoplasm. (4) Steroid hormone secretion by these cells is ACTH independent and associated with both undetectable plasma levels of ACTH and the inability to suppress cortisol secretion with high dose dexamethasone. (15) The cells, though, express the ACTH receptor and patients will respond to exogenous ACTH. (3,16) This is in contrast to primary pigmented nodular adrenocortical disease (PPNAD), adenomas and carcinomas, which are ACTH unresponsive. (3,17) The increased steroid hormone synthesis in AIMAH is thought to be due to the overall increase in adrenocortical mass rather than augmented synthesis within each cell (16).
The majority of patients with AIMAH present in the fifth decade of life with sporadic isolated disease (3). In children, though, AIMAH can be associated with McCune-Albright Syndrome (OMIM 174800), an autosomal dominant disorder characterized by polyostotic fibrous dysplasia, café-au-lait spots, precocious puberty and hyperfunctional endocrine glands. Hypercortisolism and AIMAH is found in 5% of patients with McCune-Albright Syndrome (18,19). The disorder is caused by an activating mutation in the GNAS1 gene (chromosome 20q13.2, OMIM 139320), which encodes the Gαs subunit of the G-protein receptor. By inhibiting the protein’s intrinsic GTPase function, GNAS1 activating mutations lead to the constitutive activation of adenylyl cyclase, an increase in cAMP levels and enhanced intracellular signaling (19). AIMAH has also been found to be associated with ectopic expression of G-protein receptors for hormones other than ACTH, such as gastric-inhibitory peptide (GIP), vasopressin, catecholamines, luteinizing hormone and human chorionic gonadotropin. These receptors also activate adenylyl cyclase, and their over expression increases cAMP-mediated signaling. For example, food-dependent Cushing’s Syndrome is characterized by low fasting cortisol levels and suppressed ACTH levels yet elevated cortisol and GIP levels after enteral meals (8).
Macronodular adrenocortical hyperplasia and adrenocortical nodules have also been reported in up to 36% of patients with the Multiple Endocrine Neoplasia Syndrome type 1 (MEN 1) with bilateral cortical hyperplasia present in 6-21% of patients (20,21). MEN 1 is an autosomal dominant tumor syndrome caused by an inactivating mutation of the MEN1 gene (chromosome 11q13;OMIM 131100) which encodes the tumor suppressor protein, menin (22). Menin is found mainly in the nucleus where it interacts with DNA processing and repair proteins and transcription factors such as JunD. In binding JunD, menin suppresses its transcriptional activity and is necessary for JunD’s action as a growth suppressor (22). MEN 1 is most commonly associated with pituitary, parathyroid and pancreatic tumors (11,18,22). In the vast majority of these patients their adrenal disease is hormonally silent (20).
PPNAD is another benign bilateral proliferative disorder. Histologically, it is characterized by small nodules, usually less then 4-6 mm in diameter, with a brown or black color due to large cells with a granular, pigment-containing cytoplasm. The internodular adrenal cortex is atrophic and disorganized and the adrenal glands retain normal weight and size (18,23). PPNAD can be seen in isolation, but is usually associated with the Carney Complex (OMIM 160980). This is an autosomal dominant syndrome that includes perioral, ocular or genital spotty skin pigmentation (lentiginosis), cardiac and peripheral myxomas, melanotic schwannomas, and endocrine over-activity. Affected patients often have tumors of two endocrine glands, most commonly PPNAD, but also including prolactin or growth hormone secreting pituitary tumors, thyroid adenomas or carcinomas, testicular large-cell calcifying Sertoli cell tumors or ovarian cysts (23,24,25). Clinically evident PPNAD is seen in 25-30% of patients with the Carney Complex and usually presents in childhood, late adolescence, or early adulthood (11,18,24). Germline inactivating mutations in the gene encoding the type 1α regulatory subunit of protein kinase A (PRKARIA), located on chromosome 17q22-24, have been found in 45-65% of the familial forms and 35% of the sporadic forms of Carney Complex (7,11,18,24,25). Somatic and de novo germline mutations of PRKAR1A are also found in isolated PPNAD (7,11). These mutations lead to decreased basal but increased cAMP-stimulated PKA activity (7,18,25). A second locus for Carney Complex (CNC2) was mapped to chromosome 2p16, but a causative gene has not been identified (26). These patients have been treated successfully with bilateral adrenalectomies (23).
Malignant adrenocortical carcinomas that develop in children can be either sporadic or associated with Li-Fraumeni Syndrome (OMIM 151623), a dominantly inherited familial cancer syndrome. Patients with this syndrome may develop a number of cancers that typically include soft tissue sarcomas, osteosarcomas, breast cancer, brain tumors, leukemia and lymphoma, and adrenocortical adenomas and carcinomas (27,28). Clinical criteria for diagnosing a family with classic Li-Fraumeni Syndrome include: (1) a proband with a sarcoma diagnosed under 45 years of age; (2) a first-degree relative with one of the associated tumors diagnosed under 45 years of age and (3) a first or second degree relative with any cancer diagnosed under 45 years or with a sarcoma diagnosed at any age (29,30,31). Adrenocortical tumors usually occur during the first decade of life and second primary tumors develop in up to 15% of patients (28,32). Germline mutations in the gene encoding the tumor suppressor p53 located at chromosome 17p13.1 (TP53) are found in 70% of affected families (33,34). These germline mutations have also been found in 50-80% of children with sporadic adrenocortical carcinoma in two small studies in North America and Europe (32,35). In southern Brazil, there is a fifteen-fold greater incidence of isolated pediatric adrenocortical carcinoma compared to other populations. These children have a unique germline missense mutation of TP53 (R337H) that affects the protein’s oligomerization domain, leading to pH-dependent instability, and predisposes them to develop adrenocortical tumors (36,37,38,39). The reason for this increased frequency in southern Brazil was unclear, as a founder effect was initially thought to be unlikely given the pattern of two intragenic and two flanking polymorphic markers (36,40). However, Pinto et al. recently demonstrated an identical TP53 haplotype in 95% of 22 apparently unrelated Brazilian patients with adrenocortical tumors carrying the R337H p53 mutation, suggesting that it originated from a single common ancestor. The same haplotype was also found in all the tumor DNA, usually compounded by loss of the normal TP53 allele (41). Normally the p53 protein is activated by cellular stress like DNA damage, irradiation, hypoxia and oncogenic stress and, through transcriptional activation, repression and protein-protein interactions, stimulates cell cycle arrest, apoptosis or senescence. p53 is therefore important in protecting the body from the proliferation of damaged and dangerously aberrant cells (42,43). Loss of heterozygosity at TP53 or the loss of a normal allele within tumor DNA that unmasks an inherited mutant allele, has also been associated with an increase in the incidence of malignancy and recurrence in adults (11,44). Germline mutations in the human checkpoint kinase 2 (hCHK2) gene, at chromosome 22q12.1, have also been reported in patients with Li-Fraumeni syndrome who do not harbor TP53 mutations. hCHK2 is a serine/threonine kinase that phosphorylates and activates p53. However, there have been no reports of adrenocortical carcinoma in patients with hCHK2 mutations (29).
Adrenocortical carcinomas have also been associated with Beckwith-Wiedemann Syndrome (OMIM 130650), an overgrowth disorder characterized by macrosomia, macroglossia, organomegaly and abdominal wall defects. This syndrome can occur either sporadically or in an autosomal dominant pattern and patients are predisposed to developing embryonal tumors such as Wilms’ tumors, neuroblastomas, hepatoblastomas and adrenocortical carcinomas (11,45). Over expression of IGF-II is thought to contribute to tumorigenesis in Beckwith-Wiedemann Syndrome (46,47). The IGF2 gene, located on chromosome 11p15.5, is normally expressed from only the paternal allele due to imprinting, or the expression of only one parent’s allele (in this case, the father’s) while the other parent’s allele (the mother’s) is silenced (11,18,47). Over expression of IGF-II can be caused by paternal isodisomy, in which the maternal allele is lost and the paternal allele is duplicated, or by maternal inheritance of microdeletions of the imprinting center such that the maternal allele is no longer silenced (47). IGF-II stimulates cell survival and proliferation by binding and activating the type 1 IGF receptor (46,48) and transcription of both the IGF2 and IGF1R genes can be repressed by p53 (42). IGF-II over expression was also found in NCI-H295R cells, which are derived from a human adrenocortical tumor (49) and in approximately 90% of malignant adrenal tumors in studies reviewed by Fottner et al. (46) Loss of heterozygosity at the IGF2 locus in tumor DNA is also associated with an increased incidence of malignancy and recurrence in adults with adrenocortical tumors. (50)
Bioavailability and action of the IGFs are modulated by the six high-affinity IGF binding proteins (IGFBPs), some of which have been shown to also carry IGF-independent functions (51). In addition to IGF-II, significantly elevated levels of IGFBP-2 are seen with malignant adrenocortical carcinomas (52). The significance of this elevation in IGFBP-2 is unclear. It was initially thought that IGFBP-2 negatively regulates cell growth by sequestering IGFs, but there is growing evidence that IGFBP-2 may actually promote tumor growth through IGF-independent mechanism(s) (53,54).
Presentation of Single Adrenal Tumors
There are very few studies evaluating the clinical characteristics and treatment outcomes for adrenocortical tumors in children due to the rarity of the condition. The largest to date presents data from 254 patients with either adenomas or carcinomas enrolled in the International Pediatric Adrenocortical Tumor Registry (IPACTR). This population has a predominantly Brazilian contribution (79.5%), but it also includes patients from the United States (13%) and nine other countries (7.5%). The patients were entered into the database if they were reported by their primary physician(38). Two additional studies retrospectively evaluated a series of 54 patients from France(55). and 30 patients from Turkey (56). The median age at diagnosis for these three studies was 3-4 years, with a range of 0-19 years(38,55,56). There appears to be two peak times for presentation, during the first 2 years of life, with 60% of children in the IPACTR presenting before 4 years, and peripubertally between 12-14 years(38,55). There also appears to be a greater incidence of adrenocortical tumors in female patients, with an overall predominance of 1.6:1(38,56). In the IPACTR this difference was seen in children aged ≤3 years (1.7:1) and ≥ 13 years (6.2:1) but not between 4-12 years(38). In the Turkish study, the gender difference was greater for children with adenomas (4:1)(56).
Adrenocortical tumors can be either functional or non-functional. In children most tumors are functional, with 80-90% having endocrine manifestations at diagnosis and up to 94% secreting excess hormones on further evaluation(38,55,56). Most children (50-84%) present with virilization (pubic hair, accelerated growth and skeletal maturation, an enlarged penis or clitoris, hirsutism and acne) due to excess androgen secretion(38,55,56). Less frequently, children present with Cushing’s Syndrome (15-40%) with hypertension, obesity and decreased linear growth due to excess glucocorticoids, feminization (7%) or gynecomastia due to excess estrogens, signs of hyperaldosteronism (1-4%) including hypertension and hypokalemia, or a mixture of symptoms (38,55,56). Cushing’s Syndrome appears to occur more frequently in adrenocortical carcinomas, larger tumors (>10 cm), and older children (38,55,56). Non-functional tumors also tend to occur more frequently in older children (38). An abdominal mass could be palpated in approximately half of the patients in the two retrospective studies (55,56). There often is a delay between the onset of symptoms and diagnosis with a median time of 5-8 months (38,55,56) Children, though, seem to be diagnosed earlier than adults, possibly due to the relative ease with which virilization can be recognized before puberty, when androgen levels are normally very low; during puberty, sex steroid production normally increases and results in phenotypic changes, such that any additional androgen production by a tumor is often masked (55).
Disease stage at diagnosis is important because smaller tumors are associated with better surgical outcome and complete surgical resection is the only known curative treatment for adrenocortical carcinoma (55). The staging system that is employed may vary slightly between studies, but it usually divides tumors into local, regional or metastatic disease. Most children (∼75%) present with local disease, either stage I (<5 cm or ≤200g with complete resection) or stage II (>5cm or >200g with complete resection). A smaller percentage (∼10%) present with regional invasion to adjacent areas such as lymph nodes, the kidney and the inferior vena cava or have residual tumor after resection (stage III). Another small percentage (∼15%) present with distant hematogenous metastasis to the lungs, liver or both (stage IV) (38,55). In one study, all of the children with adrenocortical adenomas had localized disease and were cured by total resection (56). For adrenocortical carcinoma, local disease with a smaller tumor burden (<10cm) appears to be associated with an improved prognosis (55).
In adult adrenocortical tumors, the Weiss score, a microscopic diagnostic score, and other immunohistochemical properties are used to determine malignant potential and prognosis. The Weiss score looks at nuclear grade, mitotic rate and atypia, vascular and capsular invasion and necroses to predict malignancy. An increased mitotic rate has the greatest predictive value (57). In addition, antibody staining for increased expression of the proliferation antigen Ki67 (58) and defining altered gene expression by cDNA micorarrays, including increased expression of IGF-II, appear to be promising techniques in differentiating carcinomas from adenomas (59,60). Unfortunately, histological classification in pediatric adrenocortical tumors has been unreliable and classification based on existing systems does not predict prognosis. In a retrospective study of 33 pediatric adrenocortical tumors, clinical and surgical factors proved prognostic, while none of the immunohistochemical markers or histopathologic criteria were significantly associated with outcome (61)
Therapy and Prognosis of Single Adrenal Tumors
The prognosis for children with malignant adrenocortical tumors remains poor. The 5-year overall survival rate is 49-55% with an event- or disease-free survival rate of 46-54% (38,55). Complete surgical resection is the only effective and potentially curative treatment for adrenocortical carcinoma (38,55,56). Surgical resection can include an adrenalectomy alone or be more extensive involving a nephrectomy, partial hepatectomy, splenectomy, or the removal of an intracaval thrombus depending on the disease stage (62). In one study, complete macroscopic resection was initially achieved in 45 of 54 children, but 40% of those with apparently complete resection still had disease recurrence after a median disease-free survival time of 7 months (55). Patients with microscopically complete resection were found to fare better with a 5-year overall survival rate of 70% compared to 7% in those children in whom it was not achieved (55).
Chemotherapy is often used before or after tumor resection to either reduce the tumor burden to enable a more complete resection, prevent recurrence or to treat recurrence if it occurs. The most common agent is mitotane (O,P’-DDD), an insecticide derivative that inhibits the conversion of cholesterol to pregnenolone and 11-deoxycortisol to cortisol and induces necrosis in adrenal tumors and metastases (38,55,56,63-65). Mitotane use in children continues to be controversial due to modestly low response rates (25-30%) and well documented toxicity which includes gastrointestinal complaints, hepatotoxicity, fatal adrenal insufficiency making glucocorticoid replacement mandatory, growth failure and neuropsychiatric symptoms such as weakness, confusion, lethargy and ataxia (55,63,66-68). Damage to the developing central nervous system leading to permanent developmental delays is one of the greatest concerns regarding the use of mitotane in young pediatric patients (63). Studies in adults suggest that achieving plasma levels greater than 14mg/L are necessary for a therapeutic response (69,70), but that levels less than 20mg/L limit toxicity (70,71). Additional chemotherapeutic regimens include different combinations of fluorouracil, doxorubicin, cisplatin and etoposide (38,55,56). Little is known about the efficacy of these medications for adrenocortical carcinoma due to their limited use. In one study, children who achieved complete remission with these chemotherapeutic agents relapsed in only two to five months after stopping the medications (55). A more recently published study using etoposide, doxorubicin and cisplatin plus mitotane in adult patients with advanced disease shows promising results with improved response rates and overall survival (72). Radiation therapy for the treatment of metastases, incomplete local resection or local recurrences is now being studied as well (38,55,62,67).
Little is known about prognostic factors in children with adrenocortical tumors. Factors significantly associated with survival in patients with localized disease (Stage I or II) in the IPACTR include a smaller tumor burden or stage I disease (complete resection and ≤200 g), presentation with virilization alone, or age less than 4 years even when children with adrenocortical adenomas were excluded. Children less than 4 years of age had a 5-year survival rate of 85.6% compared to 59.9% for children aged 4-12 years and 38.1% for children aged 13-20 years. The researchers did not evaluate prognostic factors for those with regional or metastatic disease given the small numbers and extremely poor 5-year survival (38). These findings agree with an earlier study in which survival rates were significantly improved when tumors were revealed by endocrine symptoms (60%) or if the tumor was less than 10 cm at presentation (70% vs. 32%). A significant age effect was not found in this study, but the sample size was much smaller (55).
Management Issues for Endocrinologists
The clinical management of children with adrenocortical tumors is best handled by a multi-disciplinary team including an endocrinologist, oncologist and surgeon. Below you will find a number of issues that should be addressed by the patient’s primary endocrinologist after the diagnosis of an adrenal tumor is confirmed:
Pre-operatively, it is imperative that a full panel of adrenocortical hormones and their precursors be measured to identify any potential tumor markers (67,68).
Patients should receive empiric stress dose glucocorticoid treatment perioperatively for tumor resection. Glucocorticoid coverage for stressful situations should be continued throughout treatment until normal adrenal function is confirmed. Mitotane use requires glucocorticoid replacement therapy due to its inhibition of cortisol synthesis.
Within a week post-operatively, a repeat hormone panel should be obtained to confirm that any tumor markers, or levels that were abnormally high pre-operatively, normalize or dramatically decrease after tumor resection.
Monitor for evidence of recurrence with hormonal testing every 2 months during the first year followed by every 4 months during the second year. Afterwards, this can then be spaced to every 6 months (62).
Obtain imaging studies to evaluate for recurrent disease. An international consensus conference of physicians and researchers held in September of 2003 recommended imaging studies only if hormonal abnormalities exist (62). However, other groups have advocated routine imaging for at least five years even with a normal hormone profile (73). CT scans or MRI can be used to assess for adrenal masses, but MRI is superior to document vascular invasion. CT scans of the abdomen and thorax should be used to detect metastases in the liver or lungs (67,68).
Follow clinically for potential secondary effects of excess hormone secretion. For example, if a patient presents with signs of virilization and an advanced bone age, she/he may subsequently develop hypothalamic-pituitary activation and central precocious puberty. Such patients may benefit from treatment with a gonadotropin-releasing hormone (GnRH) agonist to delay further pubertal progression.
A careful family history should be elicited. If suspicious for a familial tumor syndrome, genetic testing may be offered, but only in the context of thorough genetic counseling. Family discussion with the oncologist is strongly recommended.
- Due to the rarity of pediatric adrenocortical tumors, collecting sufficient patients to enable rigorous study that will advance our understanding will require collaboration by as many clinicians as possible. Try to contribute to on-going research, such as referring all patients to the IPACTR:Raul C. Ribeiro, MD Director, International Outreach Member, Hematology/Oncology Department St. Jude Children s Research Hospital 332 North Lauderdale Street Memphis, TN 38105 Phone: 901-495-3694 or 901-495-5318; Fax: 901-495-3122 E-mail: raul.ribeiro@stjude.org
Summary and Future Directions
In summary, adrenocortical tumors are rare in children and are associated with a poor prognosis when malignant. Little is known about their etiology and the clinical outcomes of their treatment. Evaluation of genetic and familial disorders associated with the development of adrenocortical proliferative disorders has allowed researchers to identify a number of possible mutations that may be involved in tumorigenesis. These include mutations in the GNAS1, PRKAR1A and TP53 genes, as well as IGF2 over-expression from loss of its normal imprinting controls. The clinical presentation of adrenocortical tumors in children is associated most commonly with young age, female gender and symptoms of virilization. Most children have localized disease at presentation. This is associated with a better prognosis when compared to adults because microscopically complete resection is more easily achieved. Surgical resection remains the only potentially curative treatment and mitotane, the most commonly used chemotherapeutic agent, has a poor response rate and is highly toxic.
Physicians treating children with adrenocortical tumors are handicapped by a paucity of data when making difficult clinical decisions. Given the overall poor prognosis of adrenal carcinomas and the known toxicities of treatment, the ability to distinguish adenomas from carcinomas would be a crucial piece of information when considering the risk-benefit analysis for an individual patient. Unfortunately, markers for risk stratification, such as histologic and molecular markers that identify adenoma versus carcinoma, are not yet well defined. Thus, the IPACTR has evolved from just collecting data on the clinical course of patients to also collecting tumor samples for a tumor bank. They aim to use these samples to study gene expression and the p53 pathway to correlate molecular data with clinical and outcome data to hopefully identify useful prognostic markers that will help guide treatment decisions. Broader participation in collaborative research, like the IPACTR, is needed to increase our understanding of this disease process in order to develop more targeted therapies and improve the survival rates for children who develop adrenocortical tumors.
Acknowledgements
We would like to thank the National Institute of Diabetes, Digestive and Kidney Disease (grant 5K08 DK64352) and the Lester and Liesel Baker Foundation for financial support (A.G).
References
- 1.Chundler RM, Kay R. Adrenocortical carcinoma in children. Urol Clin North Am. 1989;16:469–479. [PubMed] [Google Scholar]
- 2.Bernstein L, Gurney JG. Carcinomas and other malignant epithelial neoplasms. In: Ries LAG, Smith MA, Gurney JG, et al., editors. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. National Cancer Institute SEER Program; Bethesda, MD: 1999. pp. 139–147. (fill the names???) [Google Scholar]
- 3.Bourdeau I, Stratakis C. Cyclic AMP-dependent signaling aberrations in macronodular adrenal disease. Ann NY Acad Sci. 2002;968:240–255. doi: 10.1111/j.1749-6632.2002.tb04339.x. [DOI] [PubMed] [Google Scholar]
- 4.Aiba M, Hirayama A, Iri H, Ito Y, Fujimoto Y, Mabuchi M, Tazaki H, Maruyama H, Saruta T. Adrenocorticotropic hormone-independent bilateral adrenocortical macronodular hyperplasia as a distinct subtype of Cushing’s syndrome. Enzyme histochemical and ultrastructural study of four cases with a review of the literature. Am J Clin Pathol. 1991;96:334–340. doi: 10.1093/ajcp/96.3.334. [DOI] [PubMed] [Google Scholar]
- 5.Orth DN. Cushing’s Syndrome. N Engl J Med. 1995;332:791–803. doi: 10.1056/NEJM199503233321207. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg D, Groussin L, Jullian E, Perlemoine K, Bertagna X, Bertherat J. Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann NY Acad Sci. 2002;968:65–74. doi: 10.1111/j.1749-6632.2002.tb04327.x. [DOI] [PubMed] [Google Scholar]
- 7.Bossis I, Stratakis CA. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology. 2004;145:5452–5458. doi: 10.1210/en.2004-0900. [DOI] [PubMed] [Google Scholar]
- 8.Lacroix A, N’Diaye N, Tremblay J, Hamet P. Ectopic and abnormal hormone receptors in adrenal Cushing’s syndrome. Endocr Rev. 2001;22:75–110. doi: 10.1210/edrv.22.1.0420. [DOI] [PubMed] [Google Scholar]
- 9.Johannessen M, Delghandi MP, Moens U. What turns CREB on. Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Hammer GD, Parker KL, Schimmer BP. Minireview: Transciptional regulation of adrenocortical development. Endocrinology. 2005;146:1018–1024. doi: 10.1210/en.2004-1385. [DOI] [PubMed] [Google Scholar]
- 11.Libe R, Bertherat J. Molecular genetics of adrenocortical tumours, from familial to sporadic disease. Eur J Endocrinol. 2005;153:477–487. doi: 10.1530/eje.1.02004. [DOI] [PubMed] [Google Scholar]
- 12.Doppman JL, Miller DL, Dwyer AJ, Loughlin T, Nieman L, Cutler GB, Chrousos GP, Oldfield E, Loriaux Macronodular adrenal hyperplasia in Cushing disease. Radiology. 1988;166:347–352. doi: 10.1148/radiology.166.2.2827231. [DOI] [PubMed] [Google Scholar]
- 13.Imaki T, Naruse M, Takano K. Adrenocortical hyperplasia associated with ACTH-dependent Cushing’s syndrome: Comparison of the size of adrenal glands with clinical and endocrinological data. Endocr J. 2004;51:89–95. doi: 10.1507/endocrj.51.89. [DOI] [PubMed] [Google Scholar]
- 14.Smals AG, Pieters GF, van Haelst UJ, Kloppenborg PW. Macronodular adrenocortical hyperplasia in long-standing Cushing’s disease. J Clin Endocrinol Metab. 1984;58:25–31. doi: 10.1210/jcem-58-1-25. [DOI] [PubMed] [Google Scholar]
- 15.Cheitlin RA, Westphal M, Cabrera CM, Fujii DK, Snyder J, Fitzgerald PA. Cushing’s syndrome due to bilateral adrenal macronodular hyperplasia with undetectable ACTH: cell culture of adenoma cells on extracellular matrix. Horm Res. 1988:162–167. doi: 10.1159/000180995. [DOI] [PubMed] [Google Scholar]
- 16.Morioka M, Ohashi Y, Watanabe H, Komatsu F, Jin TX, Suyama B, Tanaka H. ACTH-independent macronodular adrenocortical hyperplasia (AIMAH): report of two cases and the analysis of steroidogenic activity in adrenal nodules. Endocr J. 1997;44:65–72. doi: 10.1507/endocrj.44.65. [DOI] [PubMed] [Google Scholar]
- 17.Lamberts SW, Zuiderwijk J, Uitterlinden P, Blijd JJ, Bruining HA, de Jong FH. Characterization of adrenal autonomy in Cushing’s syndrome: a comparison between in vivo and in vitro responsiveness of the adrenal gland. J Clin Endocrinol Metab. 1990;70:192–199. doi: 10.1210/jcem-70-1-192. [DOI] [PubMed] [Google Scholar]
- 18.Koch CA, Pacak K, Chrousos GP. Genetics of endocrine disease: The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab. 2002;87:5367–5384. doi: 10.1210/jc.2002-021069. [DOI] [PubMed] [Google Scholar]
- 19.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Speigel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 20.Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Theodorsson E, Wide L, Wilander E, Oberg K. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1992;75:76–81. doi: 10.1210/jcem.75.1.1352309. [DOI] [PubMed] [Google Scholar]
- 21.Burgess JR, Harle RA, Tucker P, Parameswaran V, Davies P, Greenaway TM, Shepherd JJ. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. Arch Surg. 1996;131:699–702. doi: 10.1001/archsurg.1996.01430190021006. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal SK, Kennedy PA, Scacheri PC, Chandrasekharappa SC, Burns AL, Collins FS, Spiegel AM, Marx SJ. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res. 2005;37:369–374. doi: 10.1055/s-2005-870139. [DOI] [PubMed] [Google Scholar]
- 23.Storr HL, Mitchell H, Swords FM, Main KM, Hindmarsh PC, Betts PR, Shaw NJ, Johnston Di, Clark AJL, Reznek RH, Grossman AB, Savage MO. Clinic features, diagnosis, treatment and molecular studies in paediatric Cushing’s syndrome due to primary nodular adrenocortical hyperplasia. Clin Endocrinol. 2004;61:553–559. doi: 10.1111/j.1365-2265.2004.02124.x. [DOI] [PubMed] [Google Scholar]
- 24.Stratakis CA, Kirschner LS, Carney JA. Genetics of Endocrine Disease: Clinical and molecular features of the Carney Complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endorinol Metab. 2001;86:4041–4046. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 25.Kirschner LS, Carney JA, Pack SD, Tayman SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-a regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 26.Stratakis CA, Carney JA, Lin JP, Papanicolaou DA, Karl M, Kastner DL, Pras E, Chrousos GP. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest. 1996;97:699–705. doi: 10.1172/JCI118467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li FP, Fraumeni JF. Soft tissue sarcomas, breast cancer, and other neoplasm. A familial syndrome. Ann Inter Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 28.Hisada M, Garber JE, Fung CY, Fraumeni JF, Jr., Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–611. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 29.Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 30.Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, Harris M, Jones PH, Binchy A, Crowther D. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- 31.Eng C, Schneider K, Fraumeni JF, Jr, Li FP. Third international workshop on collaborative interdisciplinary studies of p53 and other predisposing genes in Li-Fraumeni syndrome. Cancer Epidemiol Biomarkers Prev. 1997;6:379–383. [PubMed] [Google Scholar]
- 32.Wagner J, Portwine C, Rabin K, Leclerc JM, Narod SA, Malkin D. High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst. 1994;86:1707–1710. doi: 10.1093/jnci/86.22.1707. [DOI] [PubMed] [Google Scholar]
- 33.Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, Varley JM. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene. 1998;17:1061–1068. doi: 10.1038/sj.onc.1202033. [DOI] [PubMed] [Google Scholar]
- 34.Varley JM, Thorncroft M, McGown G, Appleby J, Kelsey AM, Tricker KJ, Evans DG, Birch JM. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene. 1997;14:865–871. doi: 10.1038/sj.onc.1201041. [DOI] [PubMed] [Google Scholar]
- 35.Varley JM, McGown G, Thorncroft M, James LA, Margison GP, Forster G, Evan DGR, Harris M, Kelsey AM, Birch JM. Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65:995–1006. doi: 10.1086/302575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, DeLacerda L, Rabin M, Cadwell C, Sampaio G, Cat I, Stratakis CA, Sandrini R. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci USA. 2001;98:9330–9335. doi: 10.1073/pnas.161479898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latronico AC, Pinto EM, Domenice S, Fragoso MC, Martin RM, Zerbini MC, Lucon AM, Mendanca BB. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J Clin Endocrinol Metab. 2001;86:4970–4973. doi: 10.1210/jcem.86.10.7957. [DOI] [PubMed] [Google Scholar]
- 38.Michalkiewcz E, Sandrini B, Figueiredo B, Miranda ECM, Caran E, Oliveira-Filho AG, Marques R, Pianovski MAD, Lacerda L, Cristofani LM, Jenkins J, Rodriguez-Galindo C, Ribeiro RC. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol. 2004;22:838–845. doi: 10.1200/JCO.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 39.Figueiredo BC, Sandrini R, Zambetti GP, Pereira RM, Cheng C, Liu W, Lacerda L, Pianovski MA, Michalkiewicz E, Jenkins J, Rodriguez-Galindo C, Mastellaro MJ, Vianna S, Watanabe F, Sandrini F, Arram SBI, Boffetta P, Ribeiro RC. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet. 2006;43:91–96. doi: 10.1136/jmg.2004.030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Moura Gallo CV, Azevedo ES, Mendonca G, de Moraes E, Olivier M, Hainaut P. TP53 mutations as biomarkers for cancer epidemiology in Latin America: current knowledge and perspectives. Mutat Res. 2005;589:192–207. doi: 10.1016/j.mrrev.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 41.Pinto EM, Billerbeck AE, Villares MC, Domenice S, Mendonca BB, Latronico AC. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol. 2004;48:647–650. doi: 10.1590/s0004-27302004000500009. [DOI] [PubMed] [Google Scholar]
- 42.Grimberg A. p53 and IGFBP-3: apoptosis and cancer protection. Mol Genet Metab. 2000;70:85–98. doi: 10.1006/mgme.2000.3008. [DOI] [PubMed] [Google Scholar]
- 43.Yee KS, Vousden KH. Complicating the complexity of p53. Carcinogenesis. 2005;26:1317–1322. doi: 10.1093/carcin/bgi122. [DOI] [PubMed] [Google Scholar]
- 44.Jorde LB, Carey JC, Bamshad MJ, White RL. Medical Genetics. Mosby; St. Louis, MO: 1999. p. 231. [Google Scholar]
- 45.Beckwith JB. Macroglossia, omphalocele, adrenal-cytomegaly, gigantism, and hyperplastic viceromeglie. Birth Defects. 1969;5:188–196. [Google Scholar]
- 46.Fottner C, Hoeflich A, Wolf E, Weber MM. Role of the insulin-like growth factor system in adrenocortical growth control and carcinogenesis. Horm Metab Res. 2004;36:397–405. doi: 10.1055/s-2004-814563. [DOI] [PubMed] [Google Scholar]
- 47.Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36:958–960. doi: 10.1038/ng1410. [DOI] [PubMed] [Google Scholar]
- 48.Grimberg A. Mechanisms by which IGF-I may promote cancer. Cancer Biol Ther. 2003;6:630–635. [PMC free article] [PubMed] [Google Scholar]
- 49.Logie A, Boulle N, Gaston V, Perin L, Boudou P, Le Bouc Y, Gicquel C. Autocrine role of IGF-II in proliferation of human adrenocortical carcinoma NCI H295R cell line. J Mol Endocrinol. 1999;23:23–32. doi: 10.1677/jme.0.0230023. [DOI] [PubMed] [Google Scholar]
- 50.Gicquel C, Bertagna X, Gaston V, Coste J, Louvel A, Baudin E, Bertherat J, Chapuis Y, Duclos JM, Schlumberger M, Plouin PF, Luton JP, Le Bouc Y. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res. 2001;61:6762–6767. [PubMed] [Google Scholar]
- 51.Grimberg A, Cohen P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. J Cell Phys. 2000;183:1–9. doi: 10.1002/(SICI)1097-4652(200004)183:1<1::AID-JCP1>3.0.CO;2-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boulle N, Logie A, Gicquel C, Perin L, Le Bouc Y. Increased levels of insulin-like growth factor II (IGF-II) and IGF-binding protein-2 are associated with malignancy in sporadic adrenocortical tumors. J Clin Endocrinol Metab. 1998;83:1713–1720. doi: 10.1210/jcem.83.5.4816. [DOI] [PubMed] [Google Scholar]
- 53.Hoeflich A, Reisinger R, Lahm H, Kiess W, Blum WF, Kolb HJ, Weber MM, Wolf E. Insulin-like growth factor-binding protein 2 in tumorigenesis: protector or promoter. Cancer Res. 2001;61:8601–8610. [PubMed] [Google Scholar]
- 54.Hoeflich A, Fettscher O, Lahm H, Blum WF, Kolb HJ, Engelhardt D, Wolf E, Weber MM. Overexpression of insulin-like growth factor-binding protein-2 results in increased tumorigenic potential in Y-1 adrenocortical tumor cells. Cancer Res. 2000;60:834–838. [PubMed] [Google Scholar]
- 55.Teinturier C, Pauchard MS, Brugieres L, Landai P, Chaussain JL, Bougnere PF. Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol. 1999;32:106–111. doi: 10.1002/(sici)1096-911x(199902)32:2<106::aid-mpo7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 56.Ciftci AO, Senocak ME, Tanyel C, Buyukpamukcu N. Adrenocortical tumors in children. J Pediatr Surg. 2001;36:549–554. doi: 10.1053/jpsu.2001.22280. [DOI] [PubMed] [Google Scholar]
- 57.Weiss LM, Medeiros LJ, Vickery AL., Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol. 1989;13:202–206. doi: 10.1097/00000478-198903000-00004. [DOI] [PubMed] [Google Scholar]
- 58.Wachenfeld C, Beuschlein F, Zwermann O, Mora P, Fassnacht M, Allolio B, Reincke M. Discerning malignancy in adrenocortical tumors: are molecular markers useful. Eur J Endocrinol. 2001;145:335–341. doi: 10.1530/eje.0.1450335. [DOI] [PubMed] [Google Scholar]
- 59.Slater EP, Diehl SM, Langer P, Samans B, Ramaswamy A, Zielke A, Bartsch DK. Analysis by cDNA microarrays of gene expression patterns of human adrenocortical tumors. Eur J Endocrinol. 2006;154:587–98. doi: 10.1530/eje.1.02116. [DOI] [PubMed] [Google Scholar]
- 60.Velazquez-Fernandez D, Laurell C, Geli J, Hoog A, Odeberg J, Kjellman M, Lundeberg J, Hamberger B, Nilsson P, Backdahl M. Expression profiling of adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery. 2005;138:1087–1094. doi: 10.1016/j.surg.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 61.Sbragia L, Oliveira-Filho AG, Vassallo J, Pinto GA, Guerra-Junior G, Bustorff-Silva J. Adrenocortical tumors in Brazilian children: immunohistochemical markers and prognostic factors. Arch Pathol Lab Med. 2005;129:1127–1131. doi: 10.5858/2005-129-1127-ATIBCI. [DOI] [PubMed] [Google Scholar]
- 62.Schteingart DE, Doherty GM, Gauger PG, Giiordano TJ, Hammer GD, Korobkin M, Worden FP. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer. 2005;12:667–680. doi: 10.1677/erc.1.01029. [DOI] [PubMed] [Google Scholar]
- 63.De Leon D, Lange BJ, Walterhouse D, Moshang T. Long-term (15 years) outcome in an infant with metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2002;87:4452–4456. doi: 10.1210/jc.2001-011978. [DOI] [PubMed] [Google Scholar]
- 64.Schteingart DE, Sinsheimer JE, Counsell RE, Abrams GD, McClellan N, Djanegara T, Hines J, Ruangwises N, Benitez R, Wotring LL. Comparison on adrenalytic activity of mitotane and a methylated homolog on normal adrenal cortex and adrenal cortical carcinoma. Cancer Chemother Pharmacol. 1993;31:459–466. doi: 10.1007/BF00685036. [DOI] [PubMed] [Google Scholar]
- 65.Martz F, Straw JA. The in vitro metabolism of 1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloroethane (o,p-DDD) by dog adrenal mitochondria and metabolite covalent binding to mitochondrial macromolecules: a possible mechanism for the adrenocorticolytic affect. Drug Metab Dispos. 1977;5:482–486. [PubMed] [Google Scholar]
- 66.Fassnacht M, Hahner S. Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs. 2005;6:386–394. [PubMed] [Google Scholar]
- 67.Allolio B, Hahner S, Weismann D, Fassnacht Management of adrenocortical carcinoma. Clin Endocrinol. 2004;60:273–287. doi: 10.1046/j.1365-2265.2003.01881.x. [DOI] [PubMed] [Google Scholar]
- 68.Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol. 2003;169:5–11. doi: 10.1016/S0022-5347(05)64023-2. [DOI] [PubMed] [Google Scholar]
- 69.Haak HR, Hermans J, Van de Velde CJH, Lentjes EGWM, Goslings BM, Fleuren GJ, et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer. 1994;69:947–951. doi: 10.1038/bjc.1994.183. (fill the names???) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baudin E, Pellegriti G, Bonnay M, Penfornis A, Laplanche A, Vassal G, Schlumberger M. Impact of monitoring plasma 1,1-dichlorodiphenil dichloroethane (o,p’DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer. 2001;92:1385–1392. doi: 10.1002/1097-0142(20010915)92:6<1385::aid-cncr1461>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 71.Van Slooten H, Moolenaar AJ, Van Seters AP, Smeenk D. The treatment of adrenocortical carcinoma with o,p’-DDD: prognostic implications of serum level monitoring. Eur J Clin Oncol. 1984;20:47–53. doi: 10.1016/0277-5379(84)90033-6. [DOI] [PubMed] [Google Scholar]
- 72.Berruti A, Terzolo M, Sperone P, Pia A, Della Casa S, Gross DJ, Carnaghi C, Casali P, Porpiglia F, Mantero F, Reimondo G, Angeli A, Dogliotti L. Etoposide, doxorubin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocr Relat Cancer. 2005;12:657–666. doi: 10.1677/erc.1.01025. [DOI] [PubMed] [Google Scholar]
- 73.Allolio B, Fassnacht M. Adrenocortical carcinoma: clinical update. J Clin Endocrin Metab. 2006 doi: 10.1210/jc.2005-2639. epub ahead of print, PMID:16551738. [DOI] [PubMed] [Google Scholar]