

Abstract
Human Epstein-Barr virus (EBV) and cytomegalovirus (CMV) can cause serious complications in immunocompromised patients. Rapid diagnosis of EBV and CMV infection is critical in the management of the disease so that anti-viral therapy can be started early. Here we describe the development of real-time PCR assays using TaqMan probes and molecular beacons and compare the performance of both assays with a well-established, validated, gel-based PCR method for the quantification of EBV and CMV in patients’ samples. The TaqMan and molecular beacon assays were linear between 10 to 107 viral genomes/reaction. Both assays generated calibration curves with strong correlation and low intra-assay and interassay variation. Results of EBV and CMV viral load determination inpatient samples obtained by the gel-based and real-time PCR were very similar. The real-time PCR assays showed increases in viral load before clinical measures of viral disease and decreases in viral load during anti-viral therapy in two of six pediatric patients. The data indicate that these TaqMan and molecular beacon approaches are accurate, rapid, and reliable assays for the diagnosis and monitoring of EBV and CMV infections in patients.
Epstein-Barr virus (EBV) and cytomegalovirus (CMV) are human herpesviruses that are characterized by a primary infection that generally occurs in a sub-clinical fashion in early childhood, with subsequent lifelong latent infection. At any time following initial infection, reactivation may occur. Reactivation of EBV and CMV may be severe and even life-threatening in immunocompromised individuals such as bone marrow and solid-organ transplant recipients and in AIDS patients. 1, 2, 3 Therefore, early diagnosis of EBV and CMV infections is important in the management of high-risk patients.
Polymerase chain reaction (PCR) amplification is a useful diagnostic method for detecting EBV and CMV infection. Qualitative PCR assays can detect viral genomes; however, they cannot be used to discriminate between latent infection and active disease. 4, 5 Quantitative PCR assays have shown that the amount of herpesvirus in blood could be used to identify patients at risk of developing viral disease and to monitor antiviral therapy. 6, 7, 8, 9, 10, 11, 12, 13 In one study, CMV and EBV viral burden higher than 10,000 viral genomes/ml blood was associated with an increased risk of developing CMV disease or EBV-associated post-transplant lymphoproliferative disorder (PTLD) in pediatric solid organ transplant patients. 11 Similar findings have been reported in other solid organ transplant populations. 12 Quantitative PCR assay can be used to monitor the effectiveness of anti-viral therapy as seen by a rapid drop in viral titer.
Several PCR techniques have been used to quantitate viral burden in immunocompromised individuals. Real-time PCR assays have been recently described to be accurate and rapid tests for the quantification of EBV and CMV that eliminate post-PCR manipulation. The quantitative EBV and CMV real-time PCR assays described so far use TaqMan probes or hybridization probes. 14, 15, 16, 17 We have developed a real-time PCR assay for the detection and quantification of EBV and CMV using molecular beacons or TaqMan probes. TaqMan probes are linear probes that are dual-labeled with a reporter dye and a quencher dye. During the extension phase of the PCR, the TaqMan probe hybridizes to its target. Cleavage of the probe by the 5′ exonuclease activity of the Taq polymerase separates the reporter fluorophore from the 3′ quencher. The fluorescence of the reporter is then increased as it is released from the proximity of the quencher. 18
Molecular beacons are single-stranded oligonucleotide probes that have a stem-loop structure. The stem is formed by two complementary sequences; the loop contains a sequence complementary to the target DNA sequence. A fluorophore is attached to the 5′ end of the stem and a non-fluorescent quencher is attached to other end of the stem. In the absence of a target, the molecular beacons do not fluoresce because the fluorophore is in close proximity to the quencher molecule. When the probe anneals to a target molecule, the probe undergoes a conformational change that forces the arm sequences apart, leading to the separation of the fluorophore from the quencher and restoring fluorescence. 19 In the present study we compared the performance of the TaqMan and molecular beacon assays with a well-established, validated, gel-based PCR method for the quantification of EBV and CMV.
Materials and Methods
Viral DNA
Quantitated EBV (EBV B95–8) and CMV (AD169) DNA was purchased from Advanced Biotechnologies (Columbia, MD). Viral DNA copy number was confirmed using an internal standard polymerase chain reaction for EBV and CMV. 6
Nucleic Acid Extraction
DNA was extracted from whole blood using the Qiagen Blood Extraction Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol.
Real-Time Quantitative PCR
The sequences of the primers and probes were selected from the EBV-encoded RNA (EBER) gene for EBV and the glycoprotein B gene for CMV. The primers and TaqMan probes were designed using the Primer Express software (Applied Biosystems, Foster City, CA). The molecular beacons were designed according to the guidelines available at http://molecular-beacons.org. A DNA-folding program (http://bioinfo.math.rpi.edu/∼mfold/dna/form1.cgi) was used to estimate the stability of the stem and loop structure of the molecular beacons. The forward and reverse primer sequences for EBV were 5′-AAACCTCAGGACCTACGCTGC-3′ and 5′-AGACACCGTCCTCACCAC-3′ respectively. The forward and reverse primer sequences for CMV were 5′-AAGTACCCCTATCGCGTGTG-3′ and 5′-ATGATGCCCTC(A/G)TCCA(A/G)GTC-3′, respectively. The TaqMan probes for EBV and CMV were labeled with 6-carboxyfluorescein (FAM) at the 5′ end as the reporter fluorophore and with black hole 1 (BHQ1) at the 3′ end as the quencher (Biosearch Technologies, Novato, CA). The molecular beacons were labeled with FAM at the 5′ end as the reporter and with fluorophore 4-dimethylaminophenylazobenzoic acid (DABYCL) at the 3′ end as the quencher (Integrated DNA Technologies, Coralville, IA). The TaqMan probes for EBV and CMV were 5′-TAGAGGTTTTGCTAGGGAGGAGACGTGTG-3′ and 5′-TGGCCCAGGGTACGGATCTTATT-CG-3′, respectively. The molecular beacons were 5′-tcgagcgGCTAGGGAGGAGACGTGTGTGGcgctcga-3′ and cgtcgaGTTCTATGGCCCAGGGTACGGtcgacg-3′ for EBV and CMV, respectively.
PCR was carried out in a 50 μl volume containing 1X PCR buffer (Applied Biosystems), 3 mmol/L MgCl2, 400 nmol/L of each primer, 200 μmol/L each of dATP, dCTP, dGTP, dTTP, and 400 nmol/L of molecular beacon or 200 nmol/L of TaqMan probe, 2.5 units of AmpliTaq Gold DNA polymerase (Applied Biosystems) and DNA from 10 μl of whole blood. For the TaqMan assay, amplification reactions were initially heated to 95°C for 10 minutes and then subjected to 45 cycles of 94°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds in an iCycler iQ PCR Detection System (Bio-Rad, Hercules, CA). Fluorescent data were collected during the 72°C step. For the molecular beacon assay, PCR conditions were 10 minutes at 95°C, followed by 45 cycles of 94°C for 15 seconds, 55°C for 30 seconds, and 72°C for 30 seconds in an iCycler iQ PCR Detection System. Data were collected during the annealing step of 55°C.
Optical data were analyzed by using the default and variable parameters in the iCycler iQ system software. The PCR threshold cycle (Ct), which is defined as the fractional cycle number at which the fluorescence reaches 10 times the standard deviation (SD) of the baseline, was determined by the software. Average Ct for duplicate standards and clinical samples was calculated by the software. Standard curve equations were calculated by regression analysis of average Ct versus the log10 of the standard copy number. The viral copy numbers in the clinical samples were calculated automatically by the data analysis software.
Gel-Based Quantitative PCR
The design and validation of the gel-based quantitative PCR has been described elsewhere in detail. 6 Briefly, each PCR reaction was carried out in a 50 μl volume containing 1X PCR buffer (Applied Biosystems), 1.5 mmol/L MgCl2, 400 nmol/L of each primer, 200 μmol/L each of dATP, dCTP, dGTP, dTTP, 2.5 units of AmpliTaq DNA polymerase (Applied Biosystems), 20 molecules of the HHVQ-1 internal calibration standard, and DNA from 10 μl of whole blood. The forward and reverse primer sequences for EBV were 5′-CCCGCCTACACACCAACTAT-3′ and 5′-AGTCTGGGAAGACAACCACA-3′, respectively. The forward and reverse primer sequences for CMV were 5′-TACCCCTATCGCGTGTGTTC-3′ and 5′-ATAGGAGGCGCCACGTATTC -3′, respectively. PCR conditions were 2 minutes at 95°C, followed by 36 cycles of 94°C for 30 seconds, 65°C for 30 seconds, and 72°C for 1 minute, followed by a final extension of 9 minutes at 72°C in a GeneAmp PCR System 9600 Thermocycler (Applied Biosystems). After amplification, PCR products were separated by agarose gel electrophoresis, identified by staining with SYBR Gold (Molecular Probes, Eugene, OR) and quantified using a Fluorimeter SI (Molecular Dynamics, Sunnyvale, CA).
Results
Molecular beacons and TaqMan probe assays were used to detect EBV and CMV. For both assays the same set of primers was used to amplify EBV or CMV. Serial dilutions of EBV or CMV were used to generate standard curves. The standard curves and amplification plots are shown in Figure 1 . Both assays were able to detect viral DNA over a linear range of 10 to 107 copies/well, which is equivalent to 103 to 109 copies/ml of blood. Statistical analysis of the standard curves over this range show that both assays are linear with a good correlation coefficient (r2 ≥ 0.998) between the Ct and the copy number.
Figure 1.
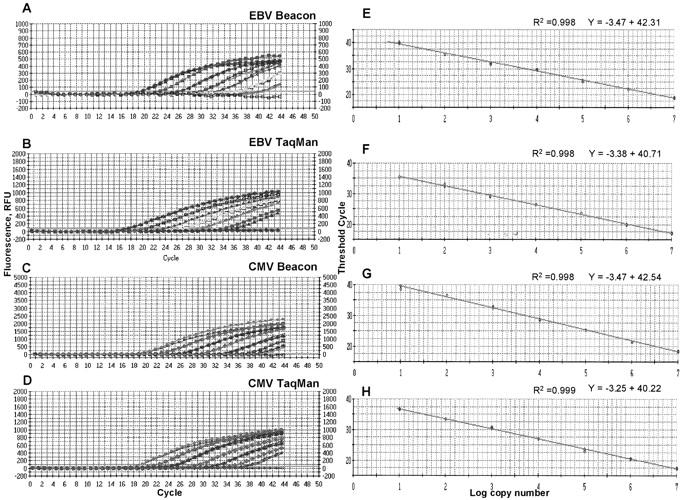
Real-time PCR assays for the detection of EBV and CMV using molecular beacon or TaqMan probes. Amplification plots (A, B, C, D) and standard curves (E, F, G, H) of EBV and CMV real-time PCR. Tenfold serial dilutions of quantitative EBV and CMV DNA ranging from 107 to 101 copies/reaction were amplified in duplicate. Amplification plots show the detection of the serially diluted DNA. PCR cycles are plotted against the fluorescence intensity. The cycle at which the fluorescence reaches a threshold value is called the threshold cycle (Ct). Standard curves were obtained by plotting the Ct values against the copy number. The correlation coefficients and linear regression equations are shown.
To determine whether there is cross-reactivity with the other herpesviruses, all of the eight herpesviruses were subjected to the real-time PCR using either the EBV or CMV primers and probes. All primer pairs and probes were found to detect their specific viral target and did not cross-react with the other viruses (data not shown).
To determine interassay reproducibility of both the molecular beacon and the TaqMan assays, control EBV and CMV DNA were serially diluted to create standard curves with a range from 107 to 10 copies/well. These standard curves were run in duplicate for 5 consecutive days and the interassay variance of the mean Ct was evaluated. The interassay variance for both molecular beacon and TaqMan assays was less than 4% (Table 1) . To determine intra-assay variance, the same serial dilutions were run using six replicates of each dilution. The intra-assay variance was less than 3% for both assays (Table 2) .
Table 1.
EBV and CMV Inter-Assay Variation (%CV)
| Copies/well | 107 | 106 | 105 | 104 | 103 | 102 | 10 | |
|---|---|---|---|---|---|---|---|---|
| Beacon | EBV | 0.82 | 0.97 | 0.75 | 0.49 | 1.94 | 1.6 | 1.26 |
| CMV | 0.30 | 1.81 | 1.89 | 1.52 | 1.58 | 2.01 | 2.55 | |
| TaqMan | EBV | 0.68 | 1.80 | 0.63 | 1.11 | 1.16 | 1.26 | 1.23 |
| CMV | 0.54 | 0.87 | 0.23 | 0.67 | 0.95 | 0.82 | 1.32 |
CV, coefficient of variation.
Table 2.
EBV and CMV Intra-Assay Variation (%CV)
| Copies/well | 107 | 106 | 105 | 104 | 103 | 102 | 10 | |
|---|---|---|---|---|---|---|---|---|
| Beacon | EBV | 0.44 | 0.59 | 0.98 | 0.85 | 1.23 | 0.7 | 2.66 |
| CMV | 1.02 | 1.62 | 1.06 | 1.32 | 1.81 | 2.21 | 3.14 | |
| TaqMan | EBV | 1.08 | 1.33 | 1.64 | 1.87 | 1.66 | 1.45 | 1.68 |
| CMV | 1.22 | 0.78 | 1.37 | 2.94 | 0.82 | 0.98 | 1.79 |
To determine whether human DNA could inhibit the real-time PCR assay, 10 patient samples that were EBV- and CMV-negative were spiked with 104 copies of control EBV or CMV per ml of blood. Quantification of EBV or CMV viral loads on those samples was performed with the molecular beacon real-time assay. The viral loads obtained from the 10 samples were similar to the control EBV and CMV (data not shown). Therefore, no inhibition of the EBV and CMV was seen among the 10 spiked patient samples.
Correlation studies of the TaqMan and molecular beacon real-time assays with the gel-based quantitative PCR assay previously described 6 were performed. Twenty-three clinical samples were selected from a panel of samples from pediatric transplant recipients 11 to cover the observed clinical range in viral titers. As shown in Figure 2 , good correlation was observed between the molecular beacon assay and the gel-based quantitative PCR assay (r2 = 0.9539) and the TaqMan assay and the gel-based quantitative PCR assay (r2 = 0.94).
Figure 2.

Correlation between the gel-based quantitative PCR and molecular beacon real-time assay or TaqMan real-time assay for EBV (A, B) and CMV (C, D). DNA from 23 patients’ samples was subjected to the conventional quantitative PCR and to real-time PCR and their EBV or CMV viral load quantitated. The copy numbers of EBV DNA or CMV DNA measured by the conventional quantitative PCR and the real-time PCR were plotted. A regression analysis was used for the comparison. The linear regression plots and corresponding values are shown.
To determine the utility of the real-time assays in the clinical setting, blood samples from six pediatric patients, including transplant patients, were tested for the presence of EBV over a period of 6 months to a year, and their viral loads were determined by the molecular beacon or TaqMan assay. Four patients had a steady viral load (< 1000 to 4000 copies/ml). Two patients had an increase in EBV viral load. The first patient was a pediatric liver transplant patient suspected of having post-transplant lymphoproliferative disorder (Figure 3A) . He had an elevated EBV level 7 weeks after transplantation (180,000 copies/ml). He was placed on anti-viral therapy and the EBV viral load dropped in response to the therapy. The second patient was diagnosed as having an acute EBV infection. The real-time PCR test showed a high EBV viral titer the next day (> 2 × 106 copies/ml) and 5 days after admission. The patient was placed on acyclovir and intravenous immunoglobulin therapy and the levels of EBV dropped and were not detectable thereafter (Figure 3B) .
Figure 3.

EBV viral levels in two pediatric patients. A: EBV levels in sequential samples from pediatric transplant recipient were determined using the Taqman real-time PCR assay. B: EBV levels in sequential samples from a pediatric patient with acute EBV infection were determined using the molecular beacon real-time PCR assay.
Discussion
Quantitative PCR is an important tool in the diagnosis and monitoring of viral infections especially in immunocompromised individuals. This study reports the development of real-time PCR assays for the detection and quantification of EBV and CMV using TaqMan probes and molecular beacons. Real-time PCR is a technique that provides a rapid and accurate quantification of PCR product. 20 Real-time PCR employs a closed-tube system that eliminates the need for post-PCR analysis and thus reduces the risk of contamination. Real-time PCR assays have been previously described for the detection of EBV and CMV using TaqMan or hybridization probes. 2, 14, 15, 16, 17 Here we describe the development of the TaqMan and the molecular beacon real-time assays and compare their performance with well-established, validated, gel-based quantitative PCR assay. We have shown that both assays are linear, between 10 to 107 copies/well, which is in concordance with other published EBV and CMV real-time PCR assays. The previous studies used different real-time instruments for the detection of PCR products. Some have used the ABI Prism 7700 Sequence Detection System or LightCycler, 2, 14, 15, 16, 17 whereas we used the iCycler iQ PCR Detection System. The iCycler iQ PCR Detection System and the ABI Prism 7700 Sequence Detection System have the advantage of analyzing 96 samples in one run; the LightCycler can analyze 32 samples per run. The advantage of the LightCycler is that it provides fast run time because of its rapid cycling conditions and could be better for small numbers of samples. It should be noted that the assay developed here should be easily adaptable to any of these detection systems.
In this study false-negative results, due to the presence of inhibitors, did not occur. The presence of inhibitors should be evaluated by amplifying a control gene, especially in the absence of an internal control in the PCR reaction. The extraction methods (eg, gel-silica membrane spin columns) that are currently in use in the clinical laboratory appear to be quite effective at removing PCR inhibitors such as divalent cations and proteins.
The Taqman and molecular beacon assays were highly reproducible and reliable; both assays generated calibration curves with strong correlation and low intra-assay and interassay variation. Both methods are suitable for routine diagnostic testing. In our experience, the design of the molecular beacons was slightly more time-consuming than the TaqMan probes.
In this study we compared the real time assays to the conventional gel-based quantitative PCR assay. The comparison of the EBV and CMV viral loads in patients’ samples obtained by the gel-based and the real-time PCR were very similar. Previous studies have also compared real-time PCR assays with conventional PCR assays for the detection of EBV 21 and other herpesviruses. 22 Again, comparison of the two quantitative assays showed good correlation in these clinical samples. Real-time PCR assays have advantages over gel-based quantitative PCR. Real-time assays have a wide dynamic range of quantification of viral DNA, thus eliminating the need for diluting samples with high viral titers. The other advantage is the elimination of post-PCR steps thus reducing the likelihood for sample cross-contamination. The latter is of special value in the clinical setting leading to a reduction in cost and turnaround time. Taken together, real-time PCR assays are rapid, accurate, and reliable and could replace conventional quantitative PCR methods.
Quantitative PCR for the detection of CMV or EBV in whole blood appears to be an effective approach, not only for the diagnosis of disease once it has developed, but also for predicting which patients will develop disease and for monitoring antiviral therapy. Patients with CMV or EBV disease or patients at risk of developing disease will show significantly higher viral titers than patients that harbor latent virus. 10, 11, 23 Therefore, sequential monitoring of circulating EBV or CMV DNA by quantitative real-time PCR is a useful technique. In this study two patients had EBV infection confirmed by real-time PCR. The patients were treated with anti-viral therapy and their EBV viral levels were monitored with the real-time assay.
In summary, we report the development of real-time PCR assays using Taqman probes and molecular beacons. Both assays are equally accurate, rapid and reliable for the quantitative analysis of EBV and CMV and can be routinely used in the diagnosis of EBV and CMV infections.
Address reprint requests to Rana Domiati-Saad, Ph.D., Department of Pathology, Baylor University Medical Center, 3500 Gaston Avenue, Dallas, TX 75246. E-mail: ranas@baylorhealth.edu.
References
- 1.Bowen EF: Cytomegalovirus reactivation in patients infected with HIV: the use of polymerase chain reaction in prediction and management. Drugs 1999, 57:735-741 [DOI] [PubMed] [Google Scholar]
- 2.Machida U, Kami M, Fukui T, Kazuyama Y, Kinoshita M, Tanaka Y, Kanda Y, Ogawa S, Honda H, Chiba S, Mitani K, Muto Y, Osumi K, Kimura S, Hirai H: Real-time automated PCR for early diagnosis and monitoring of cytomegalovirus infection after bone marrow transplantation. J Clin Microbiol 2000, 38:2536-2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orii T, Ohkohchi N, Kikuchi H, Koyamada N, Chubachi S, Satomi S, Kimura H, Hoshino Y, Morita M: Usefulness of quantitative real-time polymerase chain reaction in following up patients with Epstein-Barr virus infection after liver transplantation. Clin Transpl 2000, 14:308-317 [DOI] [PubMed] [Google Scholar]
- 4.Delgado R, Lumbreras C, Alba C, Pedraza MA, Otero JR, Gomez R, Moreno R, Noriega AR, Paya CV: Low predictive value of polymerase chain reaction for diagnosis of cytomegalovirus disease in liver transplant recipients. J Clin Microbiol 1992, 30:1876-1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riddler SA, Breinig MC, McKnight JL: Increased levels of circulating Epstein-Barr virus (EBV)-infected lymphocytes and decreased EBV nuclear antigen antibody responses are associated with the development of posttransplant lymphoproliferative disease in solid-organ transplant recipients. Blood 1994, 84:972-984 [PubMed] [Google Scholar]
- 6.Bai X, Hosler G, Rogers BB, Dawson DB, Scheuermann RH: Quantitative polymerase chain reaction for human herpesvirus diagnosis and measurement of Epstein-Barr virus burden in posttransplant lymphoproliferative disorder. Clin Chem 1997, 43:1843-1849 [PubMed] [Google Scholar]
- 7.Hoshino Y, Kimura H, Kuzushima K, Tsurumi T, Nemoto K, Kikuta A, Nishiyama Y, Kojima S, Matsuyama T, Morishima T: Early intervention in post-transplant lymphoproliferative disorders based on Epstein-Barr viral load. Bone Marrow Transplant 2000, 26:199-201 [DOI] [PubMed] [Google Scholar]
- 8.Jabs WJ, Hennig H, Kittel M, Pethig K, Smets F, Bucsky P, Kirchner H, Wagner HJ: Normalized quantification by real-time PCR of Epstein-Barr virus load in patients at risk for posttransplant lymphoproliferative disorders. J Clin Microbiol 2001, 39:564-569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohga S, Kubo E, Nomura A, Takada H, Suga N, Ishii E, Suminoe A, Inamitsu T, Matsuzaki A, Kasuga N, Hara T: Quantitative monitoring of circulating Epstein-Barr virus DNA for predicting the development of posttransplantation lymphoproliferative disease. Int J Hematol 2001, 7:323-326 [DOI] [PubMed] [Google Scholar]
- 10.Ferreira-Gonzalez A, Fisher RA, Weymouth LA, Langley MR, Wolfe L, Wilkinson DS, Garrett CT: Clinical utility of a quantitative polymerase chain reaction for diagnosis of cytomegalovirus disease in solid organ transplant patients. Transplantation 1999, 68:991-996 [DOI] [PubMed] [Google Scholar]
- 11.Bai X, Rogers BB, Harkins PC, Sommerauer J, Squires R, Rotondo K, Quan A, Dawson DB, Scheuermann RH: Predictive value of quantitative PCR-based viral burden analysis for eight human herpesviruses in pediatric solid organ transplant patients. J Mol Diagn 2000, 2:191-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldanti F, Grossi P, Furione M, Simoncini L, Sarasini A, Comoli P, Maccario R, Fiocchi R, Gerna G: High levels of Epstein-Barr virus DNA in blood of solid-organ transplant recipients and their value in predicting post-transplant lymphoproliferative disorders. J Clin Microbiol 2002, 38:613-619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humar A, Gregson D, Caliendo AM, McGeer A, Malkan G, Krajden M, Corey P, Greig P, Walmsley S, Levy G, Mazzulli T: Clinical utility of quantitative cytomegalovirus viral load determination for predicting cytomegalovirus disease in liver transplant recipients. Transplantation 1999, 68:1305-1311 [DOI] [PubMed] [Google Scholar]
- 14.Kearns AM, Guiver M, James V, King J: Development and evaluation of a real-time quantitative PCR for the detection of human cytomegalovirus. J Virol Methods 2001, 95:121-131 [DOI] [PubMed] [Google Scholar]
- 15.Kimura H, Morita M, Yabuta Y, Kuzushima K, Kato K, Kojima S, Matsuyama T, Morishima T: Quantitative analysis of Epstein-Barr virus load by using a real-time PCR assay. J Clin Microbiol 1999, 37:132-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niesters HG, van Esser J, Fries E, Wolthers KC, Cornelissen J, Osterhaus AD: Development of a real-time quantitative assay for detection of Epstein-Barr virus. J Clin Microbiol 2000, 38:712-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitsche A, Steuer N, Schmidt CA, Landt O, Siegert W: Different real-time PCR formats compared for the quantitative detection of human cytomegalovirus DNA. Clin Chem 1999, 45:1932-1937 [PubMed] [Google Scholar]
- 18.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K: Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl 1995, 4:357-362 [DOI] [PubMed] [Google Scholar]
- 19.Tyagi S, Kramer FR: Molecular beacons: probes that fluoresce upon hybridization. Nature Biotechnol 1996, 14:303-308 [DOI] [PubMed] [Google Scholar]
- 20.Heid CA, Stevens J, Livak KL, Williams PM: Real time quantitative PCR. Genome Res 1992, 6:986-994 [DOI] [PubMed] [Google Scholar]
- 21.Brengel-Pessce K, Morand P, Schmuck A, Bourgeat M-J, Buisson M, Bargues G, Bouzid M, Seigneurin J-M: Routine use of real-time quantitative PCR for the laboratory diagnosis of Epstein-Barr virus infections. J Clin Med Virol 2002, 66:360-369 [DOI] [PubMed] [Google Scholar]
- 22.Ryncarz AJ, Goddard J, Wald A, Huang ML, Roizman B, Corey L: Development of a high-throughput quantitative assay for the detection of herpes simplex virus DNA in clinical samples. J Clin Microbiol 1999, 37:1941-1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drouet E, Colimon R, Michelson S, Fourcade N, Niveleau A, Ducerf C, Boibieux A, Chevallier M, Denoyel G: Monitoring levels of human cytomegalovirus DNA in blood after liver transplantation. J Clin Microbiol 1995, 33:389-394 [DOI] [PMC free article] [PubMed] [Google Scholar]