

Abstract
A recently developed, real-time spectroscopic technique, burst-integrated fluorescence lifetime (BIFL), is shown to be well suited for monitoring the individual molecular conformational dynamics of a single molecule diffusing through the microscopic, open measurement volume (≈10 fl) of a confocal epi-illuminated set-up. In a highly diluted aqueous solution of 20-mer oligonucleotide strand of DNA duplex labeled with the environment-sensitive fluorescent dye tetramethylrhodamine (TMR), fluorescence bursts indicating traces of individual molecules are registered and further subjected to selective burst analysis. The two-dimensional BIFL data allow the identification and detection of different temporally resolved conformational states. A complementary autocorrelation analysis was performed on the time-dependent fluctuations in fluorescence lifetime and intensity. The consistent results strongly support the hypothesized three-state model of the conformational dynamics of the TMR–DNA duplex with a polar, a nonpolar, and a quenching environment of TMR.
Laser-induced fluorescence detection of single fluorescent molecules represents the ultimate level of sensitivity for fluorescence-based assays in analytical chemistry, biology, and medicine (1–14). Various groups have achieved single-molecule detection in solution (1–6) and at surfaces (7–10). Recently it was possible to characterize single molecules in solution by their fluorescence emission spectra (12) or fluorescence lifetimes (13, 14). Single-molecule spectroscopy eliminates ensemble averaging and allows individual members of a population to be probed, thereby yielding more direct information regarding the distribution of molecular and kinetic properties. This constitutes a major step toward the study of single-molecule dynamics and molecular recognition in solution. The recent improvements in single-molecule detection lead us to expect many diagnostic applications for such measurement techniques in the near future, such as monitoring enzyme function and conformational dynamics of DNA.
An increasing number of publications (15–19) employing fluorophores covalently linked to polynucleotide sites illustrate the widespread interest in investigating thermodynamic, kinetic, and structural characteristics of various DNA molecules. Yet the fluctuations of the conjugated fluorophore between several conformational states, each with a different fluorescence quantum yield (15, 16), introduces a large degree of uncertainty to such evaluations.
Rigler and coworkers recently investigated the structural dynamics measurements of a tetramethylrhodamine (TMR)-labeled DNA duplex in solution (16) and immobilized on a surface (20). They performed classical ensemble and single-molecule measurements using fluorescence correlation and time-resolved fluorescence spectroscopy that were completed by time-integrated fluorescence decay measurements of single-molecule transits. These authors proposed a millisecond equilibrium between two conformational states characterized by different photobleaching lifetimes, fluorescence lifetimes of 0.9 and 3.7 ns, and a widely distributed conformational relaxation rate constant ω obeying a stretched exponential law, which indicates the existence of numerous conformational substates (21).
So far, conventional confocal spectroscopic techniques (1, 16) have been used to analyze several fluorescence bursts caused by individual molecules diffusing through the microscopic, open measurement volume at once, thereby resulting in an averaging of properties over the single-molecule population under analysis. Fluorescence decay curves have been measured for single molecules (16, 22), but the signal was integrated over the entire observation time, resulting in a loss of the kinetic information. Therefore, we have developed an experimental technique for specific burst analysis that avoids the inclusion of extraneous events and registers the macroscopic detection time of each single photon event for subsequent kinetic analysis.
In this paper, we describe the identification and detection of different temporally resolved conformational states of a single TMR-labeled DNA duplex during free diffusion. We applied a combination of far-field confocal detection (23) with a further developed two-dimensional real-time spectroscopic technique (24), denoted burst-integrated fluorescence lifetime (BIFL). Furthermore, it is to our knowledge the first time that the fluorescence lifetime and intensity have been measured simultaneously in single-molecule detection. The dynamics are analyzed in three different ways by direct determination of the relaxation time of the conformational states in fluorescence lifetime traces as well as by autocorrelation analysis of fluorescence lifetime and intensity traces, respectively.
EXPERIMENTAL
Sample Molecules.
The solutions of the dyes rhodamine B (RB) and rhodamine 6G (R6G) (Radiant Dyes, Wermelskirchen, Germany) were freshly prepared in a solvent mixture (wt % 60/40) of double-distilled water and glycerol (microSelect, Fluka). The 5′-phosphate of a 20-mer oligodeoxynucleotide (CAA AGC GCC ATT CGC CAT TC) (Roth, Karlsruhe, Germany) was labeled with 5-carboxytetramethylrhodamine succinimidyl ester (C-2211, Molecular Probes; the boldface nucleotides may quench the dye’s fluorescence). Purification was performed by spin-column-gel filtration (cartridge 42453, MoBiTec, Göttingen, Germany) and by two preparative HPLC runs. To form a double-stranded molecule, the labeled oligonucleotide was hybridized with the circular plasmid of phage M13ala15 in 15 mM sodium citrate/150 mM NaCl/2 mM MgCl2 at pH 7.3 (15) by slow cooling from 80°C to room temperature. For single-molecule detection this solution was freshly diluted in the aqueous hybridization buffer to obtain a concentration lower than 1 pM.
Single-Molecule Fluorescence Measurements.
Confocal fluorescence detection was performed with a frequency doubled titanium:sapphire laser [excitation wavelength 522 nm; repetition rate 76 MHz (Mira 900F, Coherent, Palo Alto, CA)] in an optical set-up described previously (23) with an oil-immersion objective (Fluar 40×, numerical aperture = 1.3 oil; Zeiss), a beam-splitter at 530 nm, an 80-μm pinhole, and a dichroic band-pass emission filter 582/50 nm (AF Analysentechnik, Tübingen, Germany). The fluorescence signal was detected by an avalanche photodiode (AQ 151, EG & G; Vaudreuil, Quebec, Canada) and registered by the BIFL module and a real-time hardware-correlator card. The BIFL module consists of a time-correlated single-photon counting unit with Nuclear Electronic Instruments (NIM)-modules and an alternating counter-board of our own design, which is triggered by the NIM analog–digital converter output signal, for measuring the time-lag between two detected photons. For each event, both types of temporal information are stored on a computer interfaced with a PC-board (ATDIO32F; National Instruments, Austin, TX).
Using fluorescence correlation spectroscopy, we determined the radial and axial 1/e2 radius of the detection volume, ω0 = 0.6 μm and z0 ≈ 12 μm, respectively, from the characteristic diffusion time τD = 0.32 ms of R6G in water. The characteristic diffusion times of the oligonucleotide τD = 2.2 ± 0.8 ms and of the duplex τD = 25 ± 4 ms correspond closely to reported values (16, 25) when different beam diameters were taken into account.
RESULTS AND DISCUSSION
BIFL: Fluorescence Lifetime Analysis.
Fig. 1 shows the two-dimensional temporal information obtained by pulsed excitation (76 MHz, gray lines) in a typical BIFL measurement. It allows for specific burst selection and yields two different types of information for each recorded photon (black lines): (i) the time lag Δt to the preceding signal photon as a measure of the macroscopic detection time of the events in the experiment (millisecond time scale) and (ii) the arrival time of the signal photon relative to the exciting laser pulse measured by time-correlated single-photon counting (picosecond to nanosecond time scale). In this way, it is possible to monitor the evolution of dynamic molecular processes within a single burst in real time if the fluorescent label used is employed as an environment-sensitive probe whose fluorescence lifetime and intensity are influenced by the molecular dynamics in question. To construct a trace for the evolution of the fluorescence lifetime τ, a burst analysis may be performed either by a sliding-scale or a jump-directed analysis (see below). In a sliding-scale analysis the signal trace of fluorescence arrival times is binned for the τ calculation into a series of fluorescence decay histograms with a constant number of events (throughout this report: 80 photon counts), which are subsequently analyzed by a maximum-likelihood estimator (MLE); i.e., the histogram of the events 1–80 yields the first τ value, the events 2–81 the second, and so on (Fig. 1). Thereby the macroscopic time information of the τ trace is obtained by the time lags Δt.
Figure 1.

Principles of BIFL spectroscopy with a two-dimensional time measurement and techniques for the calculation of τ traces.
For the series of arrival time histograms with k channels and a total time window T (13.3 ns) the fluorescence lifetimes τ are determined by a pattern recognition technique based on a MLE (23, 26–28) with the quality parameter 2I*r (29), which has the lowest misclassification probability for the low number of events used here (26–28). Depending on the macroscopic time, the MLE determines two variables for each signal decay pattern Mi(τ, T, k, γ, B) in the channel i: the appropriate fluorescence lifetime τ of the fluorescence F and the fraction γ of background signal B due to prompt Raman scattering (for details see ref. 23) (Eq. 1).
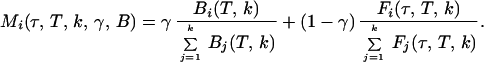 |
1 |
The principal properties of the sliding-scale analysis are shown in Fig. 2 for the BIFL data of 1 pM solutions of a single dye, R6G, and of an equimolar mixture containing two dyes, RB and R6G, in the solvent system water/glycerol (wt % 60/40). A typical multichannel scaler (MCS) trace is shown in Fig. 2A. To compress the signal trace for the subsequent sliding analysis, we used only bursts with more than 100 fluorescence photons (marked by dots in the figure), yielding τ traces of “quasicontinuous” single-molecule events (Fig. 2 B and C). The vertical lines on the x axis indicate excised background signal between analyzed bursts. Fig. 2 B and C also shows the corresponding histograms of the varying fluorescence lifetimes in a τ trace by projection on the y axis.
Figure 2.

(A) Typical MCS trace (bin width 1 ms) of 1 pM R6G in water/glycerol (60/40 wt/wt). (B and C) τ traces and corresponding τ histograms (projection onto the y axis) of selected single-molecule bursts (marked by dots in A) as a function of the macroscopic time for a different number of fluorescent species. The vertical lines on the x axis indicate excised background signal between two bursts. (B) Single dye R6G (1 pM): τ = 3.7 ± 0.6 ns. (C) Equimolar mixture (1 pM) of RB (τ = 2.3 ± 0.3 ns) and R6G (τ = 3.8 ± 0.75 ns).
The τ trace of the single dye R6G has a single fluorescence lifetime, τ = 3.7 ns, and a narrow τ-distribution with a standard deviation σ of 0.6 ns (Fig. 2B). The statistical noise in the τ trace becomes apparent by fast τ fluctuations with a small amplitude.
The τ trace of the equimolar mixture (1 pM) of RB and R6G obtained in the same way exhibits jumps between two fluorescence lifetimes and has a broad τ distribution with two maxima (τ(RB) = 2.3 ns, σ = 0.3 ns and τ(R6G) = 3.8 ns, σ = 0.75 ns)¶ (Fig. 2C). This result with two types of bursts is clear evidence that the lifetime oscillations in Fig. 2C are due to the exchange of one molecule of R6G for a different one (RB) in the excitation volume. We note that the standard deviation of R6G in the mixture agrees well with the results of the pure dye. Furthermore, the τ values of the dyes correspond closely to those obtained in precision measurements individually (τ = 2.3 and 3.6 ns).
Because the photon number N determines the relative standard error (σ/τ) ∼ (1/N1/2), a photon number of N = 80 is sufficient to allow for the identification of single fluorescent dyes by means of their characteristic fluorescence lifetimes, which differ by a factor 1.5. This result is in exact agreement with statistical theory (26–28) and also with recent experimental results (13, 14, 30).
BIFL: Fluorescence Intensity Analysis.
The BIFL technique opens up the possibility of corroborating results in an alternative way, because the two time parameters are measured independently. The time-lag Δt is also a measure of signal intensity (Fig. 1). This allows fluorescence intensity fluctuations to be monitored in a MCS trace with a variable bin width and within a variable time window. By considering only a small time window within the central part of a fluorescence burst, the influence of processes on the fluorescence flow that occur on a faster time-scale than the Brownian motion of the molecule through the Gaussian detection volume can be studied (31).
Here we are interested in the temporal characteristics of the fluorescence flow of a molecule Fc(t) being proportional to the time-dependent fluorescence quantum yield Φ(t), the laser intensity, and the absorption cross section of the dye, which is almost independent of the solvent polarity for TMR. Because the fluorescence quantum yield Φ is defined by the product (Φ = τ × kF) of the fluorescence lifetime τ and the emission rate constant kF, which is assumed to be constant, the observation of fluctuations in fluorescence lifetime τ(t) and intensity Fc(t) are complementary methods to monitor molecular dynamics by a change in the fluorescence properties on a millisecond time scale. The validity of this method can be tested by varying the size of the detection volume V, because, in contrast to rate constants of kinetic processes, characteristic diffusion times are proportional to V (16). It must be pointed out, that in contrast to fluorescence intensity, the lifetime is an absolute parameter to characterize the fluorophore environment, and this parameter cannot be disturbed by the translational motion.
Single-Molecule Dynamics: τ Traces.
We used BIFL to study the dynamics of a TMR-labeled oligodeoxynucleotide (CAA AGC GCC ATT CGC CAT TC) in aqueous solution. TMR is ideal for an environmental probe because of two properties: (i) the polarity of the environment influences its fluorescence lifetime, which ranges from 2.4 ns in aqueous solution to 3.6 ns in nonpolar solvents (15, 22, 32); (ii) the dye characterizes the binding site on the DNA, because its fluorescence is specifically quenched by guanine (33).
The dwell time of the labeled oligonucleotide in the detection volume was increased by a factor of 13 by the formation of a hybrid (16) with a complementary M+ strand of the circular plasmid of phage M13ala15 (7,250 bases). We used a very low concentration (<1 pM) of the TMR-labeled DNA duplex combined with a small open detection volume (approx. 10 fl) and performed a selective analysis of the BIFL data to study the spectroscopy of individual molecules. Because we are interested in kinetic studies in the millisecond time range, we selected for subsequent analysis only those bursts that contain at least 1,000 consecutive photon events with a restricted time lag Δt (average Δt of 50 events ≤ 250 μs; see Fig. 3A). Thirty single-molecule fluorescence bursts (approximately 15% of all bursts) with a minimum transit time of approximately 70 ms and an average count rate of 11 kHz were analyzed.
Figure 3.

Two equivalent BIFL representations of a time-dependent signal trace of a single TMR-DNA duplex. (A) Plot of the time delay, Δt (averaged for 20 events), between consecutive photons versus the signal event number. (Inset) Fluorescence fluctuations within the burst. (B) MCS trace with a bin width of 50 ms calculated from the Δt trace in A.
Two equivalent representations of a typical BIFL measurement of a TMR-labeled DNA duplex with two fluorescence bursts are shown in Fig. 3. Fig. 3A displays the time lag, Δt, and the event number of the signal. Using this Δt trace, we calculated a MCS trace (Fig. 3B) with a bin width of 50 ms. A single molecule transit causes a fluorescence burst in the MCS trace, corresponding to a drop in the Δt trace because of the high count rate. The Inset of Fig. 3 shows detail of the fluorescence fluctuations in the Δt trace.
By using the sliding-scale analysis for a single burst, molecule dynamics can be investigated on the basis of a change of an absolute parameter, the fluorescence lifetime τ. Two typical τ traces, giving a snapshot of the conformational states and kinetics of individual molecules, are shown in Fig. 4 A and B, wherein the τ trace in A is calculated from the event numbers 28,075 to 30,243 in Fig. 3. The three histograms I, II ,and III (Fig. 4C), obtained from the bursts indicated by arrows in Fig. 4 A and B, represent typical fluorescence decay data and the corresponding MLE fit (Eq. 1), yielding the characteristic fluorescence lifetimes of 2.2, 3.7, and 1.3 ns, respectively. Both τ traces have a broad τ distribution with slow, large amplitude oscillations of the τ values with a frequency in the 5-ms range, which are much slower than for a single dye molecule (see Fig. 2). The center values of the distributions τ = 2.8 ± 0.7 ns (A) and τ = 2.3 ± 0.6 ns (B) differ significantly and the standard deviations are significantly larger than the expected statistical limit (28).
Figure 4.

Plot of τ traces with the corresponding τ histogram as a function of the macroscopic time of two individual TMR-DNA duplex molecules. (A) τ trace of events 28,075 to 30,243 in Fig. 3. The horizontal lines represent a lower and an upper τ level (2.3 and 3.6 ns). (B) τ trace with considerable shorter fluorescence lifetimes and three τ levels (1.5, 2.3, and 3.6 ns). (C) Three typical fluorescence decays, I, II, and III, (indicated by arrows in A and B). Instrument response function (dotted line) and fit (bold line) by MLE (Eq. 1): τ(I) = 2.2 ns (background γ = 0.28), τ(II) = 3.7 ns (γ = 0.14), and τ(III) = 1.3 ns (γ = 0.20).
Three-State Model.
The inspection of all 30 bursts supports the above findings of statistically significant τ fluctuations with the occurrence of recurrent characteristic τ levels. The broad τ distributions suggest the existence of at least three conformational states. To prove this hypothesis, the true τ distribution has to be constructed by a jump-directed τ analysis (Fig. 1), because a sliding-scale analysis, which uses a fixed number of events, cannot recover in principle sharp steps in a τ trace. However, the jump-directed analysis is an appropriate tool provided that the τ fluctuations are statistically significant. It is performed in three steps (Fig. 1): (i) A τ trace is generated by the sliding-scale analysis to identify recurrent τ levels (Fig. 4). (ii) Assuming three conformational states of the TMR-DNA duplex with characteristic fluorescence lifetime levels of 1.5, 2.3, and 3.6 ns (horizontal lines in Fig. 4), the fluorescence lifetimes in a trace are assigned to one of the three τ levels on the basis of a minimum 2I*r (Eq. 1), being equivalent to pattern recognition algorithms (26, 27, 30, 34). Thereby, jumps between two τ levels are located at equal 2I*r minimum values for two of the three τ levels. These 2I*r crossing points allow the detection of turning points in the τ trace of step 1. (iii) Finally, a fluorescence lifetime of the detected τ state is calculated by the MLE for the events between two jumps.
Clear evidence for the existence of three states is obtained by the jump-directed τ analysis of the 30 bursts yielding 948 τ levels (Fig. 5A). The structured τ distribution indicates the superposition of two major populated τ levels [τ(A) = 3.5 ± 0.6 ns and τ(B) = 2.3 ± 0.2 ns] and one minor level [τ(C) = 1.5 ± 0.2 ns].
Figure 5.

(A) Fluorescence lifetime distribution obtained by jump-directed analysis of 30 bursts containing 70,300 photons in 948 τ levels. (B) Direct correlation of three data sets of a single burst: τ traces (right axis): jump-directed (bold line) and sliding-scale (dotted line); and MCS-trace (left axis, gray lines).
Fig. 5B gives the evidence for a direct correlation between the fluorescence intensity and lifetimes measured by BIFL. The MCS trace of the fluorescence intensity is compared (left axis, gray lines) with τ traces generated by jump-directed (bold line) as well as sliding-scale analysis (dotted line). The good agreement between the fluctuations of fluorescence lifetime and intensity is a direct proof that the dynamics can be independently monitored by τ(t) and Fc(t).
Average Kinetics.
The average kinetics of the dynamics can be studied directly by analyzing the duration of τ levels. According to our hypothesized three-state model, 948 τ values determined by jump-directed analysis (Fig. 5A) were assigned to three τ level classes defined by a certain range of τ values: level A(τ = 3.6 ns) = 2.7–5.4 ns, level B(τ = 2.3 ns) = 1.8–2.6 ns, and level C(τ = 1.5 ns) = 0.7–1.7 ns. For each state Fig. 6 gives a plot of the frequency in a state versus its τ level duration (ms). The curves can be well fit with a single exponential decay, yielding level relaxation times ρ of 5.1, 6.7, and 3.1 ms for the three states A, B, and C, respectively. Thereby, the limited time resolution of this method for the detection of a τ level, which is in the order of 1.5 ms, is taken into account by convolution of the exponential with a recovery response function (RRF) based on a calculation of the probabilities for the occurrence of no, one, or more τ level jumps within 1.5 ms.
Figure 6.

Frequency of τ level duration and calculated average relaxation times ρ of the three levels A, B, and C (ρA = 5.1 ms, ρB = 6.7 ms; ρC = 3.1 ms) obtained by convolution of a single-exponential decay with a recovery response function (RRF width 1.5 ms).
Single-Molecule Dynamics: Selective Correlation.
As an alternative method without any assumptions, the observed temporal fluctuations in fluorescence lifetime [α(t) = τ(t)] and intensity [α(t) = Fc(t)] of a single burst can be statistically compared by calculating the normalized autocorrelation functions Gα(tc) (Eq. 2),
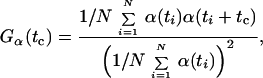 |
2 |
where N is the number of product terms obtained for a time difference tc (correlation time). The value of Gα(tc = 0) depends on the amplitude of the observed fluctuations, which corresponds to the signal contrast due to different fluorescence lifetime or intensity levels. The dependence of Gα(tc) on time tc allows for an analysis of the equilibrium between these states by determining a relaxation correlation time ω and the corresponding equilibrium constant.
The autocorrelation curves GF(tc) for the Fc(t) trace and Gτ(tc) for the τ trace were calculated for all bursts (Fig. 7 Insets), yielding ω values in a wide range of 0.5 to 10 ms. Fig. 7 demonstrates the good linear correlation between the obtained F relaxation and τ relaxation correlation times (dotted line with slope unity).
Figure 7.

Comparison of the relaxation times obtained by F- and τ-autocorrelation analysis of the various bursts. The arrow on the y axis gives an estimate for the achievable time resolution of Gτ(tc) because of correlated statistical noise. (Insets) Normalized autocorrelation curves Gτ(tc) (A) and GF(tc) (B). Fit to Eq. 3 with a single relaxation correlation time ω. (A) ωτ = 4 ms, Gτ(0) = 0.04. (B) ωF = 5 ms, GF(0) = 0.22.
To exclude that the analysis of the intensity correlation was influenced by diffusion, the detection volume was varied by a factor of 2. We obtained the same ω values as expected for a pure kinetic process.
The values of the amplitudes of the intensity correlation GF(0) of different bursts vary between 0.04 and 0.3. If only a two-level system would be assumed, which has an equilibrium constant of 1 and τ levels of 3.6 and 2.3 ns, an amplitude GF(0) = 0.049 is obtained (equation 7 of ref. 35). To explain all observed data, there must be a third level yielding a higher contrast ratio, which agrees with the three observed fluorescence lifetime levels (Figs. 4 and 5).
Considering a three-level system, the states A, B, and C with level relaxation times ρn = 1/kn (n = A, B, C) being in the same order of magnitude (36), the time-dependent part of the fluorescence autocorrelation function GF(tc) may be described by a single exponential with relaxation correlation time ω = 2/(kA + kB + kC) (Eq. 3). The autocorrelation amplitude GF(0) of three states depends on the fluorescence quantum yields ΦA, ΦB, and ΦC of the levels A, B, and C and the ratio of the relaxation rate constants kn and predicts the range of amplitudes observed above.
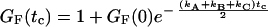 |
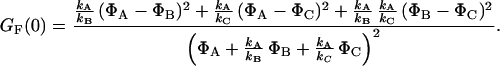 |
3 |
We note that there is good agreement for the average relaxation correlation times ωav determined by the three different methods: F correlation, ωav(GF) = 4.0 ms; τ correlation, ωav(Gτ) = 4.3 ms; and direct τ level analysis, ωav(ρ) = 3.0 ms. This result is additional evidence that the same conformational equilibrium is observed between states characterized by different fluorescence lifetimes and quantum yields.
Differing from Rigler et al. (16, 20), we propose for our DNA sequence a scheme with three primary characteristic conformational states based on well established characteristic fluorescence lifetimes τ and quantum yields Φ of TMR (Fig. 8): state A, nonpolar environment (32) of a DNA complex: τDNA = 3.6 ns, ΦDNA = 0.95; state B, polar environment: τaq = 2.4 ns, Φaq = 0.6 (identical to TMR in water); state C, quenching environment by guanine (33, 37): τG = 0.9–1.7 ns, ΦG ≤ 0.33. With regard to state C, it is noteworthy that photo-induced electron transfer is an important reaction step of nucleobase-specific quenching. Depending on the local conformation of the DNA, the quenching efficiency of guanine is determined by its distance from and geometric orientation relative to the dye. Hence, a fast equilibrium between several intermediate states C1, C2, and C3 (Fig. 8) may exist: collisional quenching, significantly quenched ground-state complex, and reversible excited-state equilibrium.
Figure 8.

Model of three conformational states of the TMR-DNA duplex consistent with the results of BIFL spectroscopy.
CONCLUSIONS
The present report demonstrates that BIFL spectroscopy combines the advantages and complementary features of two techniques: (i) selective time-resolved fluorescence spectroscopy, enabling direct monitoring and absolute characterization of molecular states; and (ii) selective fluorescence correlation spectroscopy, allowing for a quantitative statistical analysis of the system with respect to kinetic parameters and fluorophore properties.
Under the experimental conditions of this work, fluorescence lifetime analysis allowed the unequivocal detection of three environments in the τ window of 1–4 ns. The width of statistical errors prevents giving more detailed information. However, the heterogeneity of the individual TMR-DNA duplex molecules investigated by BIFL is evidenced by a wide range of additional results: (i) the fluorescence relaxation times ω = 0.5–10 ms; (ii) the amplitudes of the normalized intensity autocorrelation function GF(0) = 0.04–0.3; (iii) the τ distributions with maxima between 2.0 and 3.5 ns and variable widths and asymmetric shapes. The heterogeneity of kinetics in a millisecond time scale shows that the three detected environments of the TMR-DNA duplex must be seen in the context of a complex isoenergetic energy landscape resulting in a variety of equal polar conformational states of the flexibly linked TMR and of the DNA (e.g., nucleotide stacking dynamics and chain torsion) and that not all of them can be explored within the finite dwell time of the molecule in the detection volume of a few hundred milliseconds. Whether this result is evidence of an inhomogeneous distribution of molecules with individual properties or of a homogeneous distribution, where the reaction rate of each molecule is time dependent, cannot yet be decided. However, the found occurrence of conformational states with heterogeneous relaxation properties on biological relevant time scales may be valuable information to understand molecular reactivity determined by certain conformations. The limitation of the observation time by the dwell time of a freely diffusing molecule may be circumvented if the molecule under investigation is immobilized (20, 22, 38). In this respect BIFL may be especially valuable in studying protein function or receptor–ligand equilibria on membranes, as well as DNA and RNA dynamics, by using the cross-correlation of the signals from two fluorescent labels (39).
Acknowledgments
We are grateful to J. Troe, K. H. Drexhage, and J. Wolfrum for support of this work. We thank T. Heimburg, P. Schwille, R. Turner, J. Schaffer, and T. Jovin for stimulating discussions and H. Olbrich and H.-J. Fritz (Institut für Molekulare Genetik, Universität Göttingen, Germany) for the generous gift of the plasmid of the phage M13ala15. This work was supported by the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie, Grant 0310806.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: TMR, tetramethylrhodamine; BIFL, burst-integrated fluorescence lifetime; MLE, maximum-likelihood estimator; MCS, multichannel scaler; RB, rhodamine B; R6G, rhodamine 6G.
References
- 1.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nie S, Chiu D T, Zare R N. Anal Chem. 1995;67:2849–2857. [Google Scholar]
- 3.Keller R A, Ambrose W P, Goodwin P M, Jett J H, Martin J C, Ming W. Appl Spectrosc. 1996;50:12–32. [Google Scholar]
- 4.Goodwin P M, Ambrose W P, Keller R A. Acc Chem Res. 1996;29:607–613. [Google Scholar]
- 5.Affleck R L, Ambrose W P, Demas J N, Goodwin P M, Schecker J A, Wu M, Keller R A. Anal Chem. 1996;68:2270–2276. doi: 10.1021/ac9512517. [DOI] [PubMed] [Google Scholar]
- 6.Mets Ü, Rigler R. J Fluoresc. 1994;4:259–264. doi: 10.1007/BF01878461. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt T, Schütz G H, Baumgartner W, Gruber H J, Schindler H. Proc Natl Acad Sci USA. 1996;93:2926–2929. doi: 10.1073/pnas.93.7.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ha T, Enderle T, Chemla D S, Selvin P R, Weiss S. Phys Rev Lett. 1996;77:3979–3982. doi: 10.1103/PhysRevLett.77.3979. [DOI] [PubMed] [Google Scholar]
- 9.Ha T, Enderle T, Ogletree D F, Chemla D S, Selvin P R, Weiss S. Proc Natl Acad Sci USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu X-H, Yeung E S. Science. 1997;275:1106–1109. doi: 10.1126/science.275.5303.1106. [DOI] [PubMed] [Google Scholar]
- 11.Moerner W E. Acc Chem Res. 1996;29:563–571. [Google Scholar]
- 12.Soper S A, Davis L M, Shera E B. J Opt Soc Am B. 1992;9:1761–1769. [Google Scholar]
- 13.Zander C, Sauer M, Drexhage K H, Ko D-S, Schulz A, Wolfrum J, Brand L, Eggeling C, Seidel C A M. Appl Phys B. 1996;63:517–523. [Google Scholar]
- 14.Müller R, Zander C, Sauer M, Deimel M, Ko D-S, Siebert S, Arden-Jacob J, Deltau G, Marx N J, Drexhage K H, Wolfrum J. Chem Phys Lett. 1996;262:716–722. [Google Scholar]
- 15.Vámosi G, Gohlke C, Clegg R M. Biophys J. 1996;71:972–994. doi: 10.1016/S0006-3495(96)79300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edman L, Mets Ü, Rigler R. Proc Natl Acad Sci USA. 1996;93:6710–6715. doi: 10.1073/pnas.93.13.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selvin P R. Methods Enzymol. 1995;246:300–334. doi: 10.1016/0076-6879(95)46015-2. [DOI] [PubMed] [Google Scholar]
- 18.Jares-Erijman E A, Jovin T M. J Mol Biol. 1996;257:597–616. doi: 10.1006/jmbi.1996.0188. [DOI] [PubMed] [Google Scholar]
- 19.Clegg R M, Merchie A I H, Zechel A, Lilley D M J. Proc Natl Acad Sci USA. 1993;90:2994–2998. doi: 10.1073/pnas.90.7.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wennmalm S, Edman L, Rigler R. Proc Natl Acad Sci USA. 1997;94:10641–10646. doi: 10.1073/pnas.94.20.10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frauenfelder H, Parak F, Young R D. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 22.Jia Y, Sytnik A, Li L, Vladimirov S, Cooperman B S, Hochstrasser R M. Proc Natl Acad Sci USA. 1997;94:7932–7936. doi: 10.1073/pnas.94.15.7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brand L, Eggeling C, Zander C, Drexhage K H, Seidel C A M. J Phys Chem A. 1997;101:4313–4321. [Google Scholar]
- 24.Tellinghuisen J, Goodwin P M, Ambrose W P, Martin J C, Keller R A. Anal Chem. 1994;66:64–72. [Google Scholar]
- 25.Kinjo M, Rigler R. Nucleic Acids Res. 1995;23:1795–1799. doi: 10.1093/nar/23.10.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Köllner M, Wolfrum J. Chem Phys Lett. 1992;200:199–204. [Google Scholar]
- 27.Köllner M. Appl Optics. 1993;32:806–820. doi: 10.1364/AO.32.000806. [DOI] [PubMed] [Google Scholar]
- 28.Hall P, Selinger P J. J Chem Phys. 1981;85:2941–2946. [Google Scholar]
- 29.Kullback S. Information Theory and Statistics. New York: Wiley; 1959. [Google Scholar]
- 30.Enderlein J, Goodwin P M, van Orden A, Ambrose W P, Erdmann R, Keller R A. Chem Phys Lett. 1997;270:464–470. [Google Scholar]
- 31.Widengren J, Mets Ü, Rigler R. J Phys Chem. 1995;99:13368–13379. [Google Scholar]
- 32.Vogel M, Rettig W, Sens R, Drexhage K H. Chem Phys Lett. 1988;147:452–460. [Google Scholar]
- 33.Seidel C A M, Schulz A, Sauer M H. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 34.Köllner M, Fischer A, Arden-Jacob J, Drexhage K-H, Müller R, Seeger S, Wolfrum J. Chem Phys Lett. 1996;250:355–360. [Google Scholar]
- 35.Fleury L, Zumbusch A, Orrit M, Brown R, Bernard J. J Lumin. 1993;56:15–28. doi: 10.1103/PhysRevLett.70.3584. [DOI] [PubMed] [Google Scholar]
- 36.Bamford C H, Tipper C F H. Chemical Kinetics. Vol. 2. Amsterdam: Elsevier; 1969. p. 31. [Google Scholar]
- 37.Sauer M, Han K-T, Müller R, Nord S, Schulz A, Wolfrum J, Arden-Jacob J, Deltau G, Marx N J, Zander C, Drexhage K H. J Fluoresc. 1995;5:247–261. doi: 10.1007/BF00723896. [DOI] [PubMed] [Google Scholar]
- 38.Dickson R M, Cubitt A B, Tsien R Y, Moerner W E. Nature (London) 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 39.Schwille P, Meyer-Almes F-J, Rigler R. Biophys J. 1997;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]