

Abstract
We investigated the operation of a posttranslational protein translocation pathway to determine whether ions are excluded from the translocase during protein transport. The membrane capacitance during protein translocation across chloroplast thylakoid membranes was monitored via electric-field-indicating carotenoid electrochromic bandshift measurements. Evidence is presented that shows that the membrane ion conductance is not increased during the complete cycle of binding, transport, and substrate release by the ΔpH-dependent translocase; i.e., the membrane remains ion-tight during protein translocation. We further demonstrate that a synthetic targeting peptide that directs proteins across this membrane does not gate translocation pores. We conclude that protein transport across the thylakoid membrane does not compromise its ability to maintain ion gradients and is, thus, unlikely to affect its functions in energy transduction.
Although a number of experiments indicate that many proteins traverse membranes through aqueous pores, passing from N terminus to C terminus in an extended conformation (1), it is becoming increasingly apparent that some translocases are capable of transporting proteins possessing a variety of structures. Mitochondria and bacterial membrane transporters can accommodate branched (2) and disulfide-bridged (3) proteins, respectively. Chloroplasts have recently been shown to import fully folded polypeptides (4, 5) and the import of oligomerized proteins and even gold particles has been demonstrated in peroxisomes (6–8). In addition, it has been shown for several translocases that the same machinery mediates the transport of both soluble and integral membrane proteins (9, 10). These observations indicate that protein translocases are dynamic and versatile in their ability to transport substrates with different structures into diverse environments. Because proteins have irregular surfaces and translocators have broad specificities, the question arises as to whether ion leakage accompanies protein transport through a pore, thereby compromising the ion permeability barrier of the membrane. This issue has been examined by using artificially induced membrane-spanning translocation intermediates, with some researchers describing an attendant ion leakage and others reporting no accompanying change in ion permeability (11–13). In addition, the presumed interactions of different targeting peptides with translocators have been reported to alter ion flux, in some cases by opening ion-conducting channels and in other cases by transiently blocking ion movement through active channels (14–18). In one such report, substrate-dependent proton flux through the bacterial protein secretion machinery was observed, albeit only when some of the translocon components were overproduced (19). In the present study, we used a native precursor to examine the total ion conductance of the energy-transducing chloroplast thylakoid membrane during uninterrupted translocation cycles of a protein transporter.
MATERIALS AND METHODS
Concurrent Assay of Thylakoid Protein Import and Membrane Conductance.
Isolated thylakoids were obtained after osmotic lysis of intact chloroplasts (20). Buffer conditions (50 mM potassium Tricine, pH 8.0/330 mM sorbitol/3 mM MgCl2/20 μM methyl viologen) were typical of those used in plastid protein import experiments; these salt concentrations are approximately half of that measured in intact chloroplasts (21). Low-intensity illumination at 522 nm that was slightly actinic (46 μW/cm2) provided energy for protein translocation. Thylakoids (chlorophyll at 20 μg/ml) were incubated in the presence of a radiolabeled urea-denatured maize precursor form of the 17-kDa subunit of the oxygen-evolving complex (prOE17) obtained from expression in bacteria (5); the concentration of precursor saturated the import capacity of thylakoids under these assay conditions (prOE17, 40 nM; final urea concentration, 20 mM). Stock precursor concentration was determined by bicinchoninic acid assay and verified by densitometric scanning of SDS/polyacrylamide gels after staining with Coomassie brilliant blue.
Spectrophotometric measurements were performed during 30-min protein import reactions to determine the relative ionic conductance of thylakoid membranes. Thylakoids received two closely spaced flashes (8 ms apart) to maximize the initial transmembrane voltage, and the carotenoid absorbance was monitored at 522 nm. Sixteen absorbance transients obtained with a dwell time of 2 ms at a frequency of 0.25 Hz were averaged. The data (traces with noise) were fit to monophasic exponentials (lines without noise) by using igor Software (WaveMetrics, Lake Oswego, OR).
To determine rates of translocation under spectrophotometric measuring conditions, samples withdrawn from the cuvette at various times from the initial addition of precursor were treated with thermolysin (0.05 mg/ml, 10 min on ice, with 2 μM nigericin and 2 μM valinomycin to inhibit further import) to digest untranslocated proteins, then analyzed by SDS/PAGE, and visualized by fluorography.
To obtain an objective estimate of the minimal time required for the appearance of transported mature OE17 (mOE17), the import data were fit by using kaleidagraph software (Synergy Software, Reading, PA) to an equation describing thylakoid protein transport as an irreversible two-step first-order reaction, i.e., A → B → C, with rate constants k1 and k2 governing the first and second steps, respectively (22). In this scheme, A(t), B(t), and C(t) correspond to the free prOE17, bound prOE17, and the imported mOE17, respectively. The solid line in Fig. 2A, representing
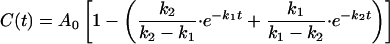 |
was drawn using A0 = 23.5, k1 = 0.1051 min−1, and k2 = 0.1046 min−1 (rate constants are given to four decimal places solely to indicate that they are not identical; correlation coefficient = 0.985). Although a single experiment is shown in Fig. 2, we found similar rate constants when combined kinetic data from seven independent experiments were fit to this mechanism (data not shown). The maximal rate of protein transport (906 proteins transported per min per thylakoid) was calculated as the slope of the tangent line at the curve’s inflection point (dC(t)/dt at the time where d2C(t)/dt2 = 0), and the minimum protein crossing time was taken as the time at which this tangent line crossed the abscissa at t = 2.7 min.
Figure 2.

Simultaneous measurement of protein translocation and carotenoid absorbance change transients demonstrates that proteins are transported by ion-tight translocators. (A) Time course of prOE17 import. Saturating concentrations of transport substrate (40 nM) imported at a rate of approximately 900 precursors per thylakoid per min under spectrophotometric measurement conditions. The required ΔpH was established by 522-nm illumination. The solid line is a fit of the data to an irreversible two-step first-order reaction mechanism. The dashed line is the slope of the tangent at the inflection point of the solid curve and serves to define the maximal rate of protein import (906 proteins per min per thylakoid) and the minimal crossing time for prOE17, where the line intersects the abscissa (2.7 min). (B) Electric-field-induced absorbance change transients recorded between 10 and 11 min after precursor addition (horizontal bar in A) display identical decay rates to those transients recorded with thylakoids not engaged in protein translocation. Traces were recorded before addition of either prOE17 (import sample) or urea alone (control) and thereafter for a full 30 min to assess the electrical conductivity of the membrane during protein translocation. Urea alone had no effect on conductance (data not shown). After 30 min, protein transport in the experimental sample was assessed as in A; this is shown in the last lane in the gel in the Inset. The decay traces were fit to monophasic exponentials (solid lines); half-time of decay was 0.44 s for the protein import trace and was 0.47 s for the control trace. The lower trace is the difference between the two above and indicates that they are essentially identical.
Gramicidin Calibration of Electrochromic Absorbance Changes.
Gramicidin additions were made from ethanolic stocks (final ethanol concentration, 0.5%) with rapid stirring. The maximum number of channels formed by gramicidin per thylakoid was calculated from the known number of dimers added (6.6 pM gramicidin corresponds to 100 channels per thylakoid vesicle). We estimated 1 × 109 chloroplasts per mg of chlorophyll and assumed one thylakoid vesicle per chloroplast (23). It is important to note that these assumptions do not enter into the final conclusions of our study, because potential translocase conductivities are discussed in relation to the conductance of gramicidin channels that are calibrated by using the same assumed numbers. Carotenoid absorbance changes were measured under buffer and spectrophotometer conditions identical to those used for measuring conductance during protein translocation reactions. The unit conductance of gramicidin under our assay conditions was calculated as outlined in ref. 30.
Transmembrane ΔpH Assay.
The 9-aminoacridine (5 μM) fluorescence (λex = 420 nm; λem = 520 nm) was measured at the onset of actinic illumination (>590 nm, provided with a Schott KL1500 halogen lamp) to monitor the magnitude of the ΔpH that could be maintained in the presence of different concentrations of gramicidin (24). In this assay, gramicidin was added from ethanolic stock solutions with vigorous stirring, and buffer conditions were identical to those used for the concurrent import and conductance assays.
Thylakoid Luminal Targeting Peptide Competition Assay.
A 33-residue peptide was synthesized (Chiron, >87% purity) corresponding to the predicted luminal targeting domain of the maize OE17 presequence. Peptide was added to isolated thylakoids in buffer conditions described above from stocks in 2 M urea/1 mM acetic acid. Import of in vitro-translated prOE17 (20) in the presence of various concentrations of peptide competitors (all samples received equivalent amounts of urea and acetic acid, 50 mM and 25 μM, respectively) was assessed after 10 min of actinic illumination. SDS/PAGE and fluorography followed by densitometry of mature bands was used to quantify the extent of import. We measured carotenoid absorbance changes (buffer and spectrophotometer conditions given above) in the presence of peptide concentrations that completely inhibited import of the full-length precursor (180 nM in measuring cuvette; 5.4 × 106 peptides per thylakoid). Control samples contained equivalent amounts of urea and acetic acid.
RESULTS
Thylakoid membranes are uniquely suited for concomitantly monitoring both protein translocation and membrane resistance to ion flux. Carotenoids within this membrane undergo a well-characterized electrochromic bandshift in response to the imposition of an electric field (Stark effect) by single-turnover flashes delivered to photosynthetic reaction centers (25) (Fig. 1 A–C). Absorbance transients subsequent to the flash directly report the total membrane ionic conductance (including, but not limited to, proton flux), with faster transients corresponding to greater ion permeability (Fig. 1D). These absorbance measurements allowed us to nonintrusively monitor membrane ion flux during in vitro protein import in thylakoids.
Figure 1.

Carotenoid electrochromic absorbance changes report the transmembrane electric field generated by the light-induced charge separation in photosystem reaction centers. (A) Flashes of light generate a membrane potential through the primary photochemical charge separation in photosynthetic reaction centers. A simplified scheme of the mechanism of electrochromism is shown (B and C), adapted from ref. 26. (B) Carotenoids in the membrane respond to the electrical field with an altered energy transition between the ground and the first excited singlet states, which results in a red shift in the visible spectrum (C). (D) Measurement of the rate of dissipation of flash-generated carotenoid absorbance shift indicates the relative ion conductance of the thylakoid membrane.
As a transport substrate, we used prOE17 that had been isolated after high-level radioactively labeled expression in bacteria (5). This protein traverses the membrane posttranslationally, using the energy contained in the transmembrane pH gradient, without the hydrolysis of ATP (20). In our experiments, energy required for the in vitro translocation of this precursor into the thylakoid lumen was provided by low-intensity illumination (46 μW/cm2) at 522 nm; this also served as the measuring beam for the electrochromic absorption changes.
Fig. 2A shows a time course of the translocation of prOE17 into the thylakoid lumen under the conditions of the spectrophotometric measurements. Maturation of prOE17 to mOE17 by the lumen-resident thylakoid processing protease demonstrates that the measuring beam was sufficiently energetic to establish a pH gradient that supported protein transport. The extent of translocation in these samples was determined by treating the thylakoids with exogenous protease subsequent to the import reaction to degrade proteins that had not completely traversed the thylakoid membrane. Quantitation of the results presented in Fig. 2A Inset revealed a translocation rate of approximately 900 proteins per thylakoid vesicle per min during the linear phase of the reaction. Electric-field-indicating absorption measurements were made before the addition of precursor and during the entire course of the import reaction. These traces were compared with those measured during control reactions, which were set up identically but without the addition of the prOE17 substrate. Fig. 2B shows the average of 16 traces obtained during the linear phase of the import reaction between 10 and 11 min. The rates of decay of flash-induced membrane potentials were identical in experimental and control samples at all times throughout the import reaction, as emphasized by the flat difference curve in Fig. 2B. Thus, protein translocation across the thylakoid membrane does not result in detectable changes in ionic conductance.
We currently do not have an estimate of the number of transport sites for prOE17 in thylakoids and so cannot precisely calculate the turnover time of the translocator. However, we can estimate the transport time for this protein from the data in Fig. 2A. The solid line represents a computer-generated best fit to the amount of mature OE17 accumulating as a function of time by using a two-step kinetic mechanism. Mathematical extrapolation from the linear portion of this curve through the inflection point to the abscissa reveals that the first appearance of the mature transported protein was at approximately 2.5 min. Accordingly, we estimate this to be the approximate turnover time of the thylakoid ΔpH-dependent translocation machinery with this precursor and under our experimental conditions. Because the maximal rate of protein transport observed when thylakoids were illuminated with a higher-intensity actinic light was roughly twice that shown in Fig. 2A, we expect the minimum turnover time of the protein translocation apparatus in vitro to be on the order of 1 min, at least with this bacterially expressed precursor. This number is in approximately the same range as estimates for the translocation times for proteins crossing other biological membranes [3–60 s (27)], including the chloroplast envelope membrane (28).
The data presented in Fig. 2 could be explained if the ion conductance of the active protein translocase is below the detection limit of the measuring technique we used. Reports of protein translocation pores in other membrane systems have described single-channel conductivities from 10 pS to 1.5 nS (14–18, 29), and it remained to be established that the carotenoid electrochromic shift could detect pores with similar conductivities in thylakoid membranes. To address this issue, we evaluated the intrinsic sensitivity of the electrochromic measurement by using the pore-forming ionophore gramicidin D. This compound is the most extensively characterized pore to date, and its single-channel conductivity in thylakoids has been determined empirically by using the carotenoid electrochromic shift and confirmed with theoretical calculations (30). From this work, we estimated the time-averaged unit conductance of gramicidin under our assay conditions to be 8.2 pS, corresponding to a flux of approximately 5 × 106 ions per s at a 100-mV membrane potential. When we assume one pore per gramicidin dimer and one thylakoid per chloroplast equivalent, we calculated the maximum number of pores per thylakoid from the concentrations of gramicidin and chlorophyll in our experiments. Fig. 3A reveals that our electrochromic measurements readily detected increased current through as few as 100 gramicidin channels per thylakoid. In comparison, we did not observe increased ion flux through putative protein translocation channels when approximately 900 proteins per min were translocated across the membrane of a thylakoid vesicle (Fig. 2A). By using the estimate for the transit time for prOE17 of approximately 1 min (from our kinetic analysis above), these experiments set a conservative upper limit of a few picosiemens on the leak conductance of putative translocation channels operating on the ΔpH-dependent protein transport pathway in thylakoids. It is noteworthy that gramicidin channels fluctuate between open and closed states, with an average open time (dependent on many factors) of approximately 1 s (31). From this fact and the data of Figs. 1 and 2, we believe that ion flux through a leaky translocation pore would not have gone undetected in our experiments even if the transit time were on the order of 5–10 s, rather than 60 s.
Figure 3.

Effects of gramicidin on thylakoid membrane ionic conductance and on ΔpH. (A) Gramicidin channels increase the decay rate of the flash-induced electrochromic absorbance changes. Thylakoid vesicles in the presence of the indicated number of gramicidin channels per thylakoid were analyzed spectrophotometrically after dual-flash excitation under the same conditions used in Fig. 2. Monophasic exponential fits of the decay traces were used to determine half-times of decay: control, 0.37 s; 100 channels, 0.30 s; 500 channels, 0.18 s). Difference curves (control minus + gramicidin) accentuate changes in decay rates induced by increased ionic conductance through gramicidin channels. (B) Increased ion conductance observed with these low concentrations of gramicidin does not alter the transmembrane ΔpH formed under steady-state illumination. The fluorescent amine 9-aminoacridine reports the extent of acidification of the lumen after the onset of illumination (upward open arrow). The mean ΔpH of control thylakoids in the presence of ethanol alone (solid line) was 3.60 (SD = 0.16, n = 5); in the presence of 500 pores per thylakoid (dotted line), ΔpH = 3.61 (SD = 0.23, n = 5). Increased conductance provided by 500 nM gramicidin (∼7.5 × 105 pores per thylakoid, dashed line) resulted in the inability of thylakoids to maintain a substantial pH gradient.
The electrochromic shift assay used in our experiments reports all electrogenic ion movement, including that of protons. Our results showing no change in membrane conductance during protein translocation suggest that dissipation of a proton gradient by leakage through an active thylakoid translocase does not occur. To examine this more directly, we used 9-aminoacridine (24) to measure the effect of an imposed ion conductance on the ability of thylakoids to maintain a proton gradient. The experiment in Fig. 3B shows that gramicidin had no effect on the pH gradient developed by thylakoids in the light when added at 500 pores per thylakoid vesicle (33 pM; see Fig. 3A), a concentration yielding a leak conductance well above that developed during protein transport. It is clear, therefore, that the transport of proteins across this membrane, at least at the rates achieved in our experiments, does not diminish the membrane’s capacity for efficient energy transduction.
Electrophysiological experiments have led to reports of putative transport channel activities that are gated by synthetic peptides that target proteins across the bacterial plasma membrane (14) and the chloroplast envelope membranes (15). Targeting peptides have been shown to transiently block ion conductance through channels in mitochondrial membranes (16–18). Although our results revealed no leak conductance through the ΔpH-dependent thylakoid translocators that are engaged in complete protein translocation cycles, this did not rule out the possibility that the transporter might similarly be gated open or blocked by targeting peptides. To examine this possibility, we measured thylakoid membrane conductance in the presence of a 33-residue synthetic peptide corresponding to the thylakoid targeting domain of prOE17 (tpOE17). Fig. 4A demonstrates that the peptide interacted productively with sites on the receptor/translocation complex, because it effectively inhibited the transport of the full-length prOE17. In the presence of 5.4 × 106 tpOE17 molecules per thylakoid vesicle (180 nM in the spectrophotometric cuvette, a concentration that completely blocked transport of the full-length prOE17 protein in Fig. 4A), we detected no change in the membrane conductance (Fig. 4B). These data demonstrate that the tpOE17 targeting peptide does not act as a gating ligand to alter ion conductance through translocation pores in the thylakoid membrane.
Figure 4.

tpOE17 competitively engages the ΔpH-dependent translocator but does not alter membrane conductance. (A) tpOE17 peptide inhibited the import of in vitro-translated prOE17. The extent of translocation is plotted as percentage of mOE17 appearing in control samples; values reported are means for five experiments. (Inset) Representative experiment. Lanes: 1. import in the absence of tpOE17; 2–9, import in the presence of 0.018, 0.052, 0.18, 0.52, 1.8, 5.2, 18, and 182 × 106 tpOE17 per thylakoid, respectively. (B) The presence of 5.4 × 106 peptides (180 nM in measuring cuvette) per thylakoid does not increase the electric field decay rate. Fitting to monophasic exponentials indicated half-times of decay for the control and tpOE17-containing samples of 0.43 and 0.42 s, respectively. The difference curve emphasizes the identical conductance characteristics in these samples.
DISCUSSION
According to the principles set out in Mitchell’s chemiosmotic coupling hypothesis (32), biological energy-transducing membranes convert the energy contained in transmembrane ion and electrical gradients into chemical energy, usually in the form of ATP. As such, any process that renders these membranes unable to maintain an ion gradient would compromise their energy-transducing function. We show herein that protein transport across the energy-transducing thylakoid membrane, at least on the ΔpH-dependent pathway, is not accompanied by a large ion leak, and as such, would not compromise plastid energy metabolism. Where investigated, similar findings were reported for mitochondrial and bacterial membranes, although the latter membrane did develop a halide-specific permeability in response to arrested protein transport (13), and there is some evidence that this translocator may be able to conduct protons under certain conditions (19). It may be relevant that many of these experiments were performed with translocation intermediates that were halted in the membranes, raising the possibility that the mechanism of arrest contributed to the lack of membrane permeability observed. Our experiments differed from those in these previous studies in that we monitored the membrane conductance during complete cycles of binding, transport, and release of the targeted polypeptide. This allows us to rule out the possibility that a bulky arresting group sterically blocked the entrance to the putative translocation pore, thereby lowering or eliminating electrogenic leakage.
Although we did not detect an electrogenic ion leak through the active protein translocase, our experiments do not address the mechanism by which energy from the proton gradient is used for protein transport. If this mechanism involves proton movement, it must either use a relatively small number of protons or occur on a time scale much longer than the few seconds during which the flash-induced membrane potential decays.
From a functional point of view, it is perhaps not surprising that the thylakoid translocation machinery has evolved with the capacity to remain ion-tight during protein transport; this feature allows energy transduction to continue unabated even when proteins are in transit across the membrane. However, it might be expected that some ion leakage through protein translocases could be tolerated, simply by increasing expenditure of energy to maintain ion gradients across membranes (33). It is not obvious how a translocation machinery could nonselectively allow transport of a variety of substrates while selectively excluding small ions. A translocase must, at the least, accommodate the various sizes and irregular shapes of the 20 different amino acid side chains, and recent experiments indicate that this particular thylakoid translocase can transport fully folded proteins (5, 34). The Sec61 translocon of the endoplasmic reticulum allows diffusion of large ions, such as NAD+, while nascent chains occupy the pore; such experiments demonstrate that the pore diameter is between 40 and 60 Å (35). In the case of this cotranslationally active transporter, the membrane permeability barrier is maintained by a tight ribosome–membrane junction at the mouth of the pore (36). Because thylakoid protein transport is inherently posttranslational, the translocase studied herein must function to maintain ion impermeability by some other mechanism. Whether this occurs via a pore with a dynamic diaphragm-like structure or through a more exotic (perhaps endocytosis-like) mechanism remains to be elucidated.
Acknowledgments
We thank Dr. Ellen Leheny for critical comments on the manuscript, Craig West for computer programming, and Marc Tormey for assistance with the figures. This work was supported in part by grants from the Department of Energy and National Science Foundation to S.M.T. and by an National Science Foundation training grant fellowship to S.A.T.
ABBREVIATIONS
- OE17
17-kDa subunit of the oxygen-evolving complex
- prOE17 and mOE17
precursor and mature forms of OE17, respectively
- tpOE17
thylakoid targeting domain of prOE17
References
- 1.Schatz G, Dobberstein B. Science. 1996;271:1519–1525. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 2.Vestweber D, Schatz G. J Cell Biol. 1988;107:2045–2049. doi: 10.1083/jcb.107.6.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tani K, Tokuda H, Mizushima S. J Biol Chem. 1990;265:17341–17347. [PubMed] [Google Scholar]
- 4.Ceccarelli E A, Krapp A R, Serra E C, Carrillo N. Eur J Biochem. 1996;238:192–197. doi: 10.1111/j.1432-1033.1996.0192q.x. [DOI] [PubMed] [Google Scholar]
- 5.Clark S A, Theg S M. Mol Biol Cell. 1997;8:923–934. doi: 10.1091/mbc.8.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glover J R, Andrews R A, Rachubinski R A. Proc Natl Acad Sci USA. 1994;91:10541–10545. doi: 10.1073/pnas.91.22.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNew J A, Goodman J M. J Cell Biol. 1994;127:1245–1257. doi: 10.1083/jcb.127.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee M S, Mullen R T, Trelease R N. Plant Cell. 1997;9:185–197. doi: 10.1105/tpc.9.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rapoport T A, Jungnickel B, Kutay U. Annu Rev Biochem. 1996;65:271–303. doi: 10.1146/annurev.bi.65.070196.001415. [DOI] [PubMed] [Google Scholar]
- 10.Walter P A, Johnson A E. Annu Rev Cell Biol. 1994;10:87–119. doi: 10.1146/annurev.cb.10.110194.000511. [DOI] [PubMed] [Google Scholar]
- 11.Vestweber D, Schatz G. J Cell Biol. 1988;107:2037–2043. doi: 10.1083/jcb.107.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wienhues U, Becker K, Schleyer M, Guiard B, Tropschug M, Horowich A L, Pfanner N, Neupert W. J Cell Biol. 1991;115:1601–1609. doi: 10.1083/jcb.115.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiebel E, Wickner W. J Biol Chem. 1992;267:7505–7510. [PubMed] [Google Scholar]
- 14.Simon S M, Blobel G. Cell. 1992;69:677–684. doi: 10.1016/0092-8674(92)90231-z. [DOI] [PubMed] [Google Scholar]
- 15.Bulychev A, Pilon M, Dassen H, van ’t Hof R, Vredenberg W, de Kruijff B. FEBS Lett. 1994;356:204–206. doi: 10.1016/0014-5793(94)01259-8. [DOI] [PubMed] [Google Scholar]
- 16.Lohret T A, Kinnally K W. J Biol Chem. 1995;270:15950–15953. doi: 10.1074/jbc.270.27.15950. [DOI] [PubMed] [Google Scholar]
- 17.Juin P, Thieffry M, Henry J-P, Vallette F M. J Biol Chem. 1997;272:6044–6050. doi: 10.1074/jbc.272.9.6044. [DOI] [PubMed] [Google Scholar]
- 18.Jensen R E, Kinnally K W. J Bioenerg Biomembr. 1997;29:3–10. doi: 10.1023/a:1022470303365. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki S, Mizushima S, Tokuda H. J Biol Chem. 1993;268:8193–8198. [PubMed] [Google Scholar]
- 20.Cline K, Ettinger W F, Theg S M. J Biol Chem. 1992;267:2688–2696. [PubMed] [Google Scholar]
- 21.Nakatani H Y, Barber J, Minski M J. Biochim Biophys Acta. 1979;545:24. doi: 10.1016/0005-2728(79)90110-5. [DOI] [PubMed] [Google Scholar]
- 22.Capellos C, Bielski B H J. Kinetic Systems. Huntington, NY: Robert E. Krieger; 1980. [Google Scholar]
- 23.Schoenknecht G, Althoff G, Junge W. FEBS Lett. 1990;277:65–68. doi: 10.1016/0014-5793(90)80810-6. [DOI] [PubMed] [Google Scholar]
- 24.Schuldiner S, Rottenberg H, Avron M. Eur J Biochem. 1972;25:64–70. doi: 10.1111/j.1432-1033.1972.tb01667.x. [DOI] [PubMed] [Google Scholar]
- 25.Junge W. In: Current Topics in Membranes and Transport. Slayman C L, editor. Vol. 16. New York: Academic; 1982. pp. 431–464. [Google Scholar]
- 26.Witt H T. Biochim Biophys Acta. 1979;505:355–427. doi: 10.1016/0304-4173(79)90008-9. [DOI] [PubMed] [Google Scholar]
- 27.Wickner W. Science. 1994;266:1197–1198. doi: 10.1126/science.7973701. [DOI] [PubMed] [Google Scholar]
- 28.Pilon M, Weisbeek P J, de Kruijff B. FEBS Lett. 1992;302:65–68. doi: 10.1016/0014-5793(92)80286-p. [DOI] [PubMed] [Google Scholar]
- 29.Simon S M, Blobel G. Cell. 1991;65:371–380. doi: 10.1016/0092-8674(91)90455-8. [DOI] [PubMed] [Google Scholar]
- 30.Lill H, Althoff G, Junge W. J Membr Biol. 1987;98:69–78. [Google Scholar]
- 31.Woolley G A, Wallace B A. J Membr Biol. 1992;129:109–136. doi: 10.1007/BF00219508. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell P. Nature (London) 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 33.Isenman L, Liebow C, Rothman S. Biochem Biophys Acta. 1995;1241:341–370. doi: 10.1016/0304-4157(95)00009-7. [DOI] [PubMed] [Google Scholar]
- 34.Roffey R A, Theg S M. Plant Physiol. 1996;111:1329–1338. doi: 10.1104/pp.111.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamman B D, Chen J-C, Johnson E E, Johnson A E. Cell. 1997;89:535–544. doi: 10.1016/s0092-8674(00)80235-4. [DOI] [PubMed] [Google Scholar]
- 36.Crowley K S, Liao S, Worrell V E, Reinhart G D, Johnson A E. Cell. 1994;78:461–471. doi: 10.1016/0092-8674(94)90424-3. [DOI] [PubMed] [Google Scholar]