

Abstract
An updated genome-scale reconstruction of the metabolic network in Escherichia coli K-12 MG1655 is presented. This updated metabolic reconstruction includes: (1) an alignment with the latest genome annotation and the metabolic content of EcoCyc leading to the inclusion of the activities of 1260 ORFs, (2) characterization and quantification of the biomass components and maintenance requirements associated with growth of E. coli and (3) thermodynamic information for the included chemical reactions. The conversion of this metabolic network reconstruction into an in silico model is detailed. A new step in the metabolic reconstruction process, termed thermodynamic consistency analysis, is introduced, in which reactions were checked for consistency with thermodynamic reversibility estimates. Applications demonstrating the capabilities of the genome-scale metabolic model to predict high-throughput experimental growth and gene deletion phenotypic screens are presented. The increased scope and computational capability using this new reconstruction is expected to broaden the spectrum of both basic biology and applied systems biology studies of E. coli metabolism.
Keywords: computational biology, group contribution method, systems biology, thermodynamics
Introduction
The process of extracting biochemical content from genome annotations and literature sources to computationally catalog and interconnect the metabolic pathways available to the cell (i.e., metabolic reconstruction) is well established and has been carried out for a growing number of organisms on the genome scale (Reed et al, 2006a). This network reconstruction process ultimately results in the generation of a biochemically, genomically and genetically (BiGG) structured database that can be further utilized for both mathematical computation and analysis of high-throughput data sets. Goals of such computation and data integration efforts are to gain a better understanding of the observable phenotypes and coordinated functions of the cell, as well as to apply developed in silico models for biological discovery and engineering applications. For mathematical computation, a number of methods have been developed to characterize models built from a metabolic reconstruction (Price et al, 2004; Stelling, 2004), and reconstructions are becoming increasingly important in understanding high-throughput experimental data (Joyce and Palsson, 2006). Thus, a well-curated metabolic reconstruction has a variety of uses and is of common interest to those studying systems biology relating to cellular metabolism.
The Gram-negative rod-shaped bacterium, Escherichia coli, has been an ideal target for metabolic reconstruction, since it is arguably the most studied and best characterized microorganism in terms of its genome annotation, functional characterization and knowledge of growth behavior (Elena and Lenski, 2003; Janssen et al, 2005). Reconstruction of the metabolic network of E. coli has been progressing since 1990 (reviewed in Reed and Palsson, 2003). This network reconstruction has been through a series of expansions and refinements (Majewski and Domach, 1990; Varma and Palsson, 1993; Varma et al, 1993; Pramanik and Keasling, 1997, 1998; Edwards and Palsson, 2000; Reed et al, 2003; Lee et al, 2005), with each iteration building on previous work while incorporating new knowledge.
Applications utilizing the E. coli reconstruction have had implications in a number of fields (for a list of applications and references, see http://gcrg.ucsd.edu/organisms/ecoli/ecoli_others.html). For metabolic engineering applications, modeling enables examination and simulation of metabolism as a whole, circumventing the possible shortcomings of methods that rely on manual assessment of a limited number of interactions and possibly fail to detect non-intuitive causal interactions (Alper et al, 2005; Fong et al, 2005). For studies of bacterial evolution, a reconstruction serves as a highly curated database and model to examine and simulate evolutionary hypotheses (Pal et al, 2005, 2006). Network analyses have been applied to genome-scale reconstructions of E. coli to identify sets of reactions or metabolites whose activity is interdependent. These studies have obvious implications in aiding therapeutic interventions along with other systemic analyses (Almaas et al, 2004; Nikolaev et al, 2005). Additionally, for the prospective goal of biological discovery, genome-scale reconstructions drive discovery by identifying specific areas where knowledge is lacking, or disagreements with observations, and provide a framework for the integration of high-throughput data (Covert et al, 2004; Reed et al, 2006b).
In this study, we expand and refine the reconstruction of the metabolic network in E. coli. The new additions include: (1) an up to date accounting for open reading frames (ORFs) in E. coli that have metabolic annotations and an alignment of the content in EcoCyc (Keseler et al, 2005), leading to the inclusion of 1260 ORFs (an increase of 356 ORFs over the previous reconstruction (Reed et al, 2003)), (2) an improved breakdown of the biomass composition, the maintenance requirements for growth and sustenance and a sensitivity analysis on the parameters used in computational modeling and (3) thermodynamic information about the chemical transformations accounted for in the reconstruction. The thermodynamic properties estimated for the model reactions and compounds were utilized to test the thermodynamic consistency of the reactions included in the reconstruction (Henry et al, 2006). This expanded version of the E. coli metabolic network will allow for additional and more comprehensive computational and experimental studies of the systems properties of E. coli metabolism. We give several such examples that use the new network reconstruction.
Results
The results of the present study are presented in three parts. First, we describe the new content added to form the updated genome-scale E. coli metabolic reconstruction. Second, we detail the conversion of the metabolic reconstruction into a computational model for physiological studies. Third, we present a series of applications and detailed biochemical studies that the new computational model enables.
Reconstruction content and enhancements
We generated a metabolic reconstruction consisting of the chemical reactions that transport and interconvert metabolites within E. coli K-12 MG1655. This network reconstruction, termed iAF1260, was based on a previous reconstruction, iJR904 (Reed et al, 2003), the current functional annotation of the E. coli genome (Riley et al, 2006), content characterized in the EcoCyc Database (Keseler et al, 2005) and specific biochemical characterization studies on the metabolic machinery and capabilities of E. coli (see Supplementary information for a complete list of references). The general features of iAF1260 are given in Table I. When possible, enzymatically catalyzed reactions were linked to their corresponding ORFs through gene-protein-reaction (GPR) assignments (see Materials and methods and (Reed et al, 2006a)). A complete list of all reactions and metabolites for the reconstruction is given in the Supplementary information in spreadsheet and SBML formats and is also available on the web on the BiGG database (http://bigg.ucsd.edu).
Table 1.
Properties of iAF1260 and iJR904
| iAF1260 this study | iJR904 Reed et al, 2003 | |
|---|---|---|
| Included genes | 1260 (28%)d | 904 (20%)d |
| Experimentally-based function | 1161 (92%) | 838 (93%)e |
| Computationally predicted function | 99 (8%) | 58 (6%)e |
| Unique functional proteins | 1148 | 817 |
| Multigene complexes | 167 | 105 |
| Genes involved in complexes | 415 | 289 |
| Instances of isozymesa | 346 | 149 |
| Reactions | 2077 | 931 |
| Metabolic reactions | 1387 | 747 |
| Unique metabolic reactionsb | 1339 | 745 |
| Cytoplasmic | 1187 | 745 |
| Periplasmic | 192 | 0 |
| Extracellular | 8 | 2 |
| Transport reactions | 690 | 184 |
| Cytoplasm to periplasm | 390 | 0 |
| Periplasm to extracellular | 298 | 0 |
| Cytoplasm to extracellular | 2 | 184 |
| Gene—protein—reaction associations | ||
| Gene associated (metabolic/transport) | 1294/625 | 706/166 |
| Spontaneous/diffusion reactionsc | 16/9 | 2/9 |
| Total (gene associated and no association needed) | 1310/634 (94%) | 708/175 (95%) |
| No gene association (metabolic/transport) | 77/56 (6%) | 37/9 (5%) |
| Exchange reactions | 304 | 143 |
| Metabolites | ||
| Unique metabolitesb | 1039 | 625 |
| Cytoplasmic | 951 | 618 |
| Periplasm | 418 | 0 |
| Extracellular | 299 | 143 |
Tabulated on a reaction basis, not counting outer membrane nonspecific porin transport.
Reactions can occur in or between multiple compartments and metabolites can be present in more than one compartment.
Diffusion reactions do not include facilitated diffusion reactions and are not included in this total if they can also be catalyzed by a gene product at a higher rate.
Overall genome coverage based on 4453 total ORFs in Escherichia coli (Riley et al, 2006); 2403 of these ORFs have been experimentally verified.
Eight ORFs included in iJR904 (1% of the total) have since been removed from the genome annotation (Riley et al, 2006).
The major areas of expansion of iAF1260 over previous E. coli network reconstructions come under the following five categories: (i) increased scope, (ii) compartmentalization, (iii) increased pathway detail, (iv) incorporation of reaction thermodynamics and (v) alignment with EcoCyc.
iAF1260 is significantly larger in scope than iJR904, containing 356 additional ORFs, 1146 additional reactions and 414 additional metabolites (Table I; Figure 1) (Reed et al, 2003). Furthermore, 289 reactions were removed from iJR904, of which 254 were replaced with similar reactions, whereas 35 were totally removed because they were decomposed into more discrete enzymatic steps (27 reactions, see below) or found to be incorrect (8 reactions). Most of the replacements stem from the partition of the model into three distinct subcellular compartments (discussed further below). In order to capture a complete picture of metabolism, certain proteins (e.g., acyl carrier protein) and tRNAs, which function as substrates or products, were also included in the metabolic reconstruction. The ORFs that encode the included proteins were integrated into the GPRs for the reactions in which they participate. It is worthwhile to note that 1161 ORFs (92%) included in iAF1260 have been experimentally verified (Riley et al, 2006). This number (1161) accounts for 48% of the total 2403 ORFs in E. coli that have been experimentally verified.
The reconstruction presented here was separated into three distinct cellular compartments: the cytoplasm, periplasm and extracellular space. Each metabolite in the reaction network was explicitly assigned to one or more of these three compartments (see Table I). This representation allowed the inclusion of transport systems in both the inner and outer membrane and more accurately represented the metabolic machinery available to E. coli in each compartment. Previous E. coli reconstructions have not considered the periplasm as a distinct compartment.
iAF1260 was generated to minimize the number of grouped, or lumped, reactions in the network reconstruction. Previous versions included many lumped reactions, which simply represent a summation of two or more discrete enzymatically catalyzed reactions, in metabolic processes such as membrane lipid and lipopolysaccharide (LPS) biosynthesis. Although iAF1260 includes a smaller total number of lumped reactions than previous reconstructions, some cases remain in which the reaction mechanism(s) has yet to be fully characterized in E. coli (e.g., biotin synthase Lotierzo et al, 2005).
The standard Gibbs free energy change of formation, ΔfG′o, and reaction, ΔrG′o, were estimated for most metabolites and reactions in iAF1260; 872 (84%) and 1996 (96%), respectively. All ΔfG′o and ΔrG′o values were estimated using a new implementation of the group contribution method (MD Jankowski and V Hatzimanikatis, in preparation). The 1 M reference state for the metabolite concentrations on which ΔrG′o is based does not accurately reflect the metabolite concentrations found in the cell (approximately 1 mM). Thus, we computationally adjusted all estimated ΔrG′o to the free energy change of reaction at 1 mM concentrations for all species, ΔrG′m. The distribution of ΔrG′m values for reactions in iAF1260 indicates that 84% of estimated ΔrG′m values are less than or equal to zero in the predicted direction of flux (see below), meaning that most reactions are thermodynamically feasible at 1 mM metabolite concentrations (Figure 2A). Because intracellular metabolite concentrations can differ significantly from 1 mM (typically, 0.00001–0.02 M; Albe et al, 1990), the actual free energy change of a reaction, ΔrG′, can differ significantly from ΔrG′m. This deviation of ΔrG′ from ΔrG′m due to metabolite concentrations is shown in Figure 2B (blue error bars). Uncertainties in the estimated ΔrG′o that arise from the group contribution method were also included in the calculation of the ΔrG′ ranges (purple error bars, Figure 2B). Thermodynamic estimates were further utilized in the reconstruction process (see below).
The content of iAF1260 and the EcoCyc (Keseler et al, 2005), release 10.6 and MetaCyc (Caspi et al, 2006) databases were compared to obtain a more accurate and comprehensive reconstruction. EcoCyc and MetaCyc possess a separate curation history from the database from which iAF1260 was built and are each extensively curated. A detailed comparison of these resources has resulted in a more thorough analysis and inclusion of metabolic content in iAF1260, and in EcoCyc and MetaCyc. A mapping between the reactions and compounds of EcoCyc, MetaCyc and iAF1260 was generated in the course of this process. Overall, 945 metabolites in iAF1260 (91%) were computationally and manually mapped to EcoCyc and Metacyc compounds (Supplementary Table IV). Similarly, 1308 reactions in iAF1260 (63%) were computationally mapped to reactions from EcoCyc and MetaCyc using the compound mappings. The results of these mappings are provided in Supplementary information. A key difference identified from the comparison lies in the usage of generic reactions in which enzymes exhibit broad substrate specificity. In EcoCyc, many reaction equations were obtained from the IUBMB ((NC-IUBMB), 2006). Accordingly, EcoCyc defined compound classes to represent groups of related substrates, and those compound classes were used as reaction substrates to represent the fact that a given enzyme could act on several different substrates (i.e., compounds), without necessarily enumerating all these compounds explicitly. Since iAF1260 was converted into a computational model, all compounds in its reactions need to be explicitly instantiated. Accordingly, no compound classes or generic reactions were included iAF1260.
A breakdown of ORFs, reactions and metabolites included in iAF1260 and earlier reconstructions (Majewski and Domach, 1990; Varma and Palsson, 1993; Varma et al, 1993; Pramanik and Keasling, 1997, 1998; Edwards and Palsson, 2000; Reed et al, 2003) are given in Figure 1 and Supplementary information. Figure 1 was generated using the functional categories assigned through the clusters of orthologous groups (COGs) ontology (http://www.ncbi.nlm.nih.gov/COG/) to classify the reactions included in the E. coli metabolic reconstruction. Figure 1A details the number of ORFs from each COG functional class that were included in iAF1260, as well as five previous versions of the E. coli reconstruction, to indicate the areas in which the network reconstruction has matured with each successive release. The largest increase in coverage compared with iJR904 (Reed et al, 2003) is found in inorganic ion transport and metabolism (26–56%, respectively, 73 ORFs). Overall, amino acid and nucleotide transport and metabolism have the highest number of ORFs and percent coverage in iAF1260 (256 and 89%, respectively). Ion transport and utilization was recognized as an underrepresented area of metabolism in previous reconstructions and was specifically expanded and incorporated into simulations using iAF1260. Figure 1B and C depict the classification of reactions and metabolites in iAF1260 tied to each COG functional class. The largest number of reactions and metabolites associated to ORFs in one COG functional class is in amino-acid transport and metabolism and cell wall/membrane/envelope biosynthesis, respectively; furthermore, lipid transport and metabolism has the highest reaction to ORF ratio (5.8), followed by secondary metabolites biosynthesis, transport and catabolism (4.6) and cell wall/membrane/envelope biogenesis (3.4). The large reaction to ORF ratio highlights the fact that the proteins in these classes act on a large number of molecules that only differ slightly in structure. Furthermore, the highest number of unique metabolites that participate in reactions from one class was from coenzyme transport and metabolism. This finding points out the specialized nature of the proteins in coenzyme transport and metabolism pathways (Figure 1).
Figure 1.

Classification of the ORFs, reactions and metabolites included in iAF1260. (A) Coverage of characterized ORFs from each of the COGs functional classes included in iAF1260 and five previous reconstructions. The percentage given is the total coverage accounted for in iAF1260 for each class. Some ORFs included in the reconstructions did not have a COG functional class assignment (see Supplemental information). (B) The number of reactions (both gene-associated and non-gene associated) that are associated to ORFs from each COG functional class. Since ORFs can belong to multiple classes, the percent unique in each class is listed. Non-gene-associated reactions were assigned to a class manually. (C) The number of metabolites that participate in reactions from each functional class and the percent unique in each class. Other (OT) includes classes J, K, L, O, T, U, V (see Supplementary information). NC, no COG assignment.
Conversion to a computational model
Network reconstructions of the type presented herein effectively represent 2-D genome annotations (Palsson, 2004) defining the metabolic network that is specific to a particular organism. That is, reconstructions describe both the set of components in a network and the respective interactions between them; two layers of information. A reconstruction is easily accessible and transferable once developed and can be queried for content such as genes, proteins, reactions and metabolites. These network reconstructions can further be converted through a defined series of steps into a computational model that can be used for phenotypic simulations.
The following steps are necessary to convert a network reconstruction to a predictive computational model:
Explicit assignment of the metabolites participating in a reaction. Some enzymes can act on a number of different metabolites. For modeling purposes, each of these potential metabolites needs to be explicitly defined as participating in a distinct reaction in order to outline a complete picture of metabolism. This step was incorporated in the reconstruction process of iAF1260, but is necessary for computational use of a reconstruction based on nonspecific metabolites.
Definition of a system boundary. Here, the system boundary was defined around the entire reaction network and an exchange reaction (i.e., a reaction that allows a metabolite to enter and exit the system) was made for each of the metabolites in the extracellular space compartment immediately surrounding the cell. Constraints were assigned to each of these exchange reactions during the modeling simulations to restrict the inputs and outputs of the system, depending on the simulated growth environment.
Conversion of the defined system into a mathematical format that forms the basis for a computational model. After detailing all GPRs and defining a system boundary, the reconstruction was represented in mathematical terms. The system was represented in the form of a stoichiometric matrix (see Materials and methods) and utilized in the available software platforms SimPheny, and LINDO or TOMLAB in conjunction with MATLAB (Becker et al, 2007). The dimension of the stoichiometric matrix for iAF1260 was 1668 × 2381 (# of metabolites × # of reaction species).
Curation: filling gaps in the reconstruction. In order to produce essential biomass components (amino acids, nucleotides, etc) from minimal media components, there needed to be continuous pathways from media substrates to the required metabolites for biosynthesis. In some cases, the biosynthetic pathways to produce these metabolites were incomplete. A good example is in the biosynthetic pathway for the amino acid L-proline. After reconstruction of the enzymatically catalyzed reactions in the pathway, there was no continuous pathway for the de novo generation of L-proline. As a result, the spontaneous reaction L-glutamate 5-semialdehyde dehydratase was needed to complete the pathway and was added to the model with no gene association (Williams and Frank, 1975). In addition to spontaneous reactions, there were also essential reactions for which the catalytic enzyme is yet to be identified (see Supplementary information and http://ecocyc.org/enzymes.shtml). Flux balance analysis (FBA) in conjunction with a biomass objective function (BOF), see below, was used to aid in filling the gaps in iAF1260 and results from this analysis are given in the Supplementary information.
Determining strain specific parameters. In order to examine the networks ability to fulfill the biomass requirements needed for cellular growth, we generated a set of biomass BOFs. The BOFs were linear combinations of experimentally measured metabolites (along with quantities) commonly present in cellular biomass (see Table II and Supplementary information), and the included metabolites and amounts of each were further judged for inclusion in this equation through interpretation of gene essentiality data (see Materials and methods). The process of determining maintenance requirements is outlined in detail below.
After the conversion of the reconstructed network into a computational model, a constraint-based approach was used in the context of generating essential biomass components to predict cellular phenotypes under different genetic and environmental conditions.
Application of iAF1260 to predict cellular phenotypes
A computational model can be used to predict and quantify the active pathways and probable system outputs during growth given a set of inputs that represent growth medium conditions. Analyzing metabolic models in the context of generating maximal amounts of biomass precursors (i.e., simulated optimal growth) from available media substrates using FBA can generate results that are consistent with experimental data (Edwards et al, 2001; Covert and Palsson, 2002; Ibarra et al, 2002; Covert et al, 2004). Thus, we used iAF1260 to predict the physiological state of E. coli in selected growth conditions using this constraint-based approach (see Materials and methods). It is worthwhile to note that the constraint-based computations performed in this section can be readily reproduced utilizing the iAF1260 SBML files (see Supplementary information) and available implemented algorithmic methods (Becker et al, 2007).
Although iAF1260 contains a comprehensive picture of E. coli metabolism, there also are other events that need to be accounted for to computationally predict growth capabilities. Three specific issues arose in computational simulations using iAF1260.
Transcriptional regulatory events. A transcriptional regulatory network can be used to determine which ORFs are being transcribed under a given condition (Covert et al, 2004; Barrett et al, 2005), thus reducing the number of available pathways under a given growth condition. The events can also limit the rate at which certain enzymes are transcribed, therefore, they are important to apply to a given simulation (Covert et al, 2004).
Maintenance costs. Additional energetic requirements exist for growth beyond what is needed to generate the macromolecular content of the cell (beyond the metabolic costs, which are accounted for directly in the reaction network) (Pirt, 1965; Neidhardt et al, 1990). These energetic maintenance requirements are for growth-associated maintenance (GAM, e.g., protein polymerization costs) and non-growth-associated maintenance (NGAM, e.g., membrane leakage) and can be estimated through ATP utilization costs (see Materials and methods).
Reaction kinetic effects. Kinetic issues affect metabolism. A potential result from kinetic limitations is that the cell does not always use the most efficient pathways during growth (Gennis and Stewart, 1996; Helling, 2002). Currently, reaction kinetics are infeasible to incorporate on the genome-scale primarily because of the large number of unknown in vivo kinetic parameters and concentrations. However, we know that kinetic effects can influence the utilization of certain pathways, such as the electron transport system (ETS) in E. coli (Helling, 2002).
Figure 3 demonstrates how we addressed the three modeling issues outlined above when using FBA with iAF1260 to predict the physiological state of E. coli growing aerobically on glucose. Initially, all of the pathways characterized in iAF1260 were represented in a computational framework (Figure 3A). We then constrained the reactions that correspond to ORFs that are not transcribed under aerobic glucose conditions to zero allowable flux in the network using the Boolean gene regulatory rules based on 104 transcription factors established by Covert et al (2004), Figure 3B, effectively eliminating 152 reactions (see Supplementary information). Using the reduced network, we then constrained the maximum allowable P/O ratio of the ETS by using observations and predictions from previous studies. E. coli possesses two NADH dehydrogenase components (NDH-1 (nuo) and NDH-2 (ndh)) and two terminal oxidases (bo-type (cyo) and bd-type (cyd) oxidase) in the system (Calhoun et al, 1993; Gennis and Stewart, 1996). Different combinations of these respiratory components can result in an overall translocation that can range from 2 H+/2e− to 7 H+/2e− in iAF1260. The specific constraint we placed on the system was to split the flux ratio between the two NADH dehydrogenases 1:1 (NDH-1:NDH-2), allowing a P/O ratio between 0.5 and 1.375 (Figure 3C) (Calhoun et al, 1993; Noguchi et al, 2004).
Figure 2.

Thermodynamic properties of the reactions in iAF1260. (A) The distribution of estimated ΔrG′m values for the reactions in iAF1260. ΔrG′m could be estimated for 1996 reactions (96%) in the reconstruction. 64% of the represented reactions have a negative ΔrG′m, and 20% of the reactions have a ΔrG′m of approximately zero. This distribution of ΔrG′m values indicates that most reactions in the model are thermodynamically favorable at millimolar concentration conditions. (B) The range of possible ΔrG′ values for the reactions in iAF1260. ΔrG′ differs from ΔrG′o (orange diamonds) and ΔrG′m (black diamonds) due to variations in metabolite concentrations from the 1 M and 1 mM reference states, respectively. Metabolite concentrations typically range from 0.02 to 0.00001 M, resulting in a wide range of values for ΔrG′ (blue error bars). Taking uncertainty into account, the range of possible values for ΔrG′ can be extended (purple error bars). The ΔrG′ ranges were used to assess the feasibility and reversibility of the reactions in iAF1260; reactions for which a positive ΔrG′ is not possible are thermodynamically irreversible.
Figure 3.

Utilizing iAF1260 as a predictive model. (A) A drawing of central metabolism and the ETS included in iAF1260. Originally, the entire network is unconstrained. (B) Application of transcriptional regulatory effects restricts the total number of pathways, or routes, flux can pass through in the network. (C) Further application of known reaction capacities can result in more accurate predictions. For example, the flux through the NADH dehydrogenase enzymes is split in a 1:1 ratio during a simulation to produce an optimal P/O ratio of approximately 1.4 (Gennis and Stewart, 1996; Noguchi et al, 2004). (D) The non-metabolic activity of the cell can be accounted for through maintenance parameters and these were approximated using experimental data under known media conditions. Chemostat data (see Materials and methods) was used (triangles) and the dotted line shows the modeling predictions with the appropriate maintenance parameters. (E) After the parameters are approximated, the model can then be used to predict the GR (circles), product formation (acetate, squares) and additional uptake rates (oxygen, triangles) under different environmental conditions (for succinate growth in this case).
Using chemostat data for E. coli growing aerobically on glucose (see Supplementary information), we estimated the GAM and NGAM costs (Figure 3D). We found that an NGAM value of 8.39 mmol ATP gDW−1 h−1 and a GAM value of 59.81 mmol ATP gDW−1 best fit the experimental data. Using these values and no restriction on pathway choice for the ETS, we calculated the line of optimal growth using FBA for aerobic growth on succinate. This line was plotted against the measured values for wild-type batch growth determined by Edwards et al (2001). The calculated line of optimality corresponds to the conditions (substrate uptake and product generation rates), which can maximize the biomass yield. The results show that most of the measured values lie very near the line of optimality in the experimental range examined (see Figure 3E).
To further examine the agreement between modeling simulations and experimental data, computationally predicted flux values, product formation rates and growth rates (GRs) were compared with experimentally determined values derived from 13C labeling experiments (Fischer et al, 2004). Using measured glucose and oxygen uptake rates (OURs) as modeling constraints (Fischer et al, 2004), FBA was used to examine the predicted network flux distribution when optimizing for flux through the BOFCORE reaction (see Materials and methods). The produced flux distribution accurately predicted both the growth and acetate secretion rate using the measured average glucose and OURs from triplicate 13C-labeled experiments. Additionally, the CO2 production rate was accurately predicted when considering the standard deviation on the reported uptake values. Both the experimental and computational results suggest that no other carbon containing products were generated in measurable amounts. Examining the flux distribution in central metabolism, there was complete agreement in the direction of flux through the glycolytic, pentose phosphate, TCA and pyruvate metabolism pathways between modeling and experimental results. For the Entner–Doudoroff pathway, the experimentally determined flux was equal to or less than 4% of the total glucose flux entering the system, whereas no flux was predicted for this pathway for an optimal growth solution using iAF1260. Looking at the quantitative values for 22 individual central metabolism fluxes (Fischer et al, 2004), the experimentally reported and computationally predicted values were in good agreement (mean of the difference=8±1.4% (s.e.), R2=0.96, where fluxes were normalized to glucose uptake rates (GURs) being 100%). The most notable discrepancy when comparing the computational and experimentally reported values was in the pentose phosphate pathway, where 26% of the total glucose flux entering the system was calculated to be shuttled through this pathway when analyzing the 13C labeling data, and the model predicted a value of 46% during an optimal growth solution. All of the flux values predicted for other central metabolism pathways agreed well with the experimental flux data.
Thermodynamic consistency analysis
Previous metabolic network reconstructions have focused on the chemistry of the reactions that take place and their genetic basis. The physicochemical characteristics of the reactions, namely thermodynamic and kinetic properties, have not been incorporated. The kinetics are hard to obtain and change with organism adaptation and evolution (Herring et al, 2006). Conversely, the thermodynamic properties represent physicochemical limitations that can be estimated and taken into account (Beard et al, 2002; Kümmel et al, 2006a; Henry et al, 2007). In forming iAF1260, we incorporated thermodynamic information (see Figure 2) to provide another means of assessing reaction reversibility beyond what is stated in the primary literature and assignments made using general heuristic rules (see Materials and methods).
Through a process termed thermodynamic consistency analysis, the thermodynamic estimates were utilized to evaluate the reversibility and directionality assigned to the reactions in the reconstruction based on the primary literature and heuristic rules. First, flux variability analysis (FVA) (Mahadevan and Schilling, 2003) was utilized in combination with the calculated ΔrG′ ranges to identify reactions that operated in a thermodynamically infeasible direction during near optimal growth on at least one carbon source (see Materials and methods). The co-substrates and cofactors involved in these inconsistent reactions were adjusted (either the participants or stoichiometries) with guidance from the literature so that the reactions in the final version of the reconstruction were not thermodynamically infeasible in any of the directions in which they must operate for near optimal growth on 174 carbon sources.
One example of an initially thermodynamically inconsistent reaction that was altered is the hydrogenase 3 catalyzed reaction, formate-hydrogen lyase. This reaction initially powered the transport of 1.3 protons across the cell membrane while oxidizing formate to hydrogen and carbon dioxide (Hakobyan et al, 2005). Thermodynamic analysis of this reaction indicated that the intracellular portion of this reaction is already unfavorable, with a ΔrG′o and ΔrG′m of 2.1±1.7 kcal/mol, in agreement with reported values (Thauer et al, 1977; Alberty, 2003). Given the concentration gradients achievable in vivo, it was found to be highly improbable that this already unfavorable reaction could power the transport of 1.3 protons across the cell membrane. As a result, the transmembrane transport portion of this reaction was removed.
Some of the other thermodynamically inconsistent reactions identified prompted the adjustment of the reversibility for network reactions and also, expansion of the reconstruction content. For example, we identified thermodynamic infeasibilities in the reactions involving transport of the inorganic ions (i.e., Fe2+, Cu2+, etc). Initially, the only reactions in the model allowing for the transport of these ions across the cytoplasmic membrane were reversible diffusion reactions of the form:
According to FVA, during growth on some carbon sources, these metal ions could be exported from the cell. However, based on our thermodynamic calculations, we determined that these reactions are only thermodynamically feasible in the direction of import where the transmembrane electrochemical potential contributes energy to the transport process (Henry et al, 2006). The literature confirms that whereas import of these ions proceeds via diffusion through a regulated ion channel, export of these ions requires a separate mechanism that utilizes ATP hydrolysis or proton antiport as a source of energy to drive the reaction (Silver, 1996; Grass et al, 2005). These alternative export reactions were consequently added to the reconstruction.
The FVA performed as part of the thermodynamic consistency analysis further allowed the reactions in the reconstruction to be functionally classified as essential (requiring a nonzero flux), substitutable (capable of carrying zero or non-zero flux) or blocked (zero flux) during growth on each of the 174 carbon source studied. Interestingly, a large number of the reactions in the reconstruction behaved uniformly regardless of the carbon source being utilized (Table III). Many reversible reactions only operated in a single direction despite being reversible, whereas many other reactions were predicted not to operate in any of the FVA studies performed. These reactions are potentially involved dead-ends in the reconstruction or conversely, were limited because of the BOF used to examine the network.
Table 2.
The biomass composition of the average wild type E. coli cell
| Typical ‘wild-type' composition | ||||||
|---|---|---|---|---|---|---|
| Protein (55.0%) | Lipid (9.1%) | |||||
| L-alanine | L-arginine | L-asparagine | structure | |||
| L-aspartate | L-cysteine | L-glutamine | phosphatidylethanolamine | phosphatidylglycerola | cardiolipin | |
| L-glutamate | glycine | L-histidine | acyl chain length: number of unsaturated bonds | |||
| L-isoleucine | L-leucine | L-lysine | 16:0 | 16:1 | 18:1 | |
| L-methionine | L-phenylalanine | L-proline | ||||
| L-serine | L-threonine | L-tryptophan | LPS (3.4%) | |||
| L-tyrosine | L-valine | inner/outer core KDO2 lipid A | ||||
| RNA (20.5%) | Cofactors, Prosthetic Groups and Other (<2.9%) | |||||
| ATP UTP | CTP | GTP | S-adenosylmethionine thiamine diphosphate pyridoxal 5′-phosphateb | FAD riboflavin folates | coenzyme A undecaprenyl pyrophosphate quinones | NAD(P) hemes |
| DNA (3.1%) | chorismate | enterobactin | glutathione | putrescine | ||
| dATP | dCTP | dGTP | spermidine | vitamin B12 | ||
| dTTP | ||||||
| Murein (2.5%) | ||||||
| Inorganic ions (1.0%) | structure | |||||
| ammonium | calcium | chlorine | murein disaccharide | |||
| cobalt | copper | iron | peptide chain length | |||
| magnesium | manganese | molybdate | pentapeptide | |||
| phosphorous | potassium | sulfate | tetrapeptide | tripeptide | ||
| zinc | Glycogen (2.5%) | |||||
| glycogen | ||||||
|
‘Core' biomass composition substitutes | ||||||
| inner/outer core KDO2 lipid A: substituted with KDO2 lipid (IV) A | ||||||
| quinones: substituted with 2-octaprenyl-6-hydroxyphenol | ||||||
| hemes: protoheme; siroheme included | ||||||
| folates: tetrahydrofolate; 10-formyltetrahydrofolate; 5,10-methylenetetrahydrofolate included | ||||||
The average E. coli wild-type macromolecules (and the weight percentage for each) are listed along with their corresponding network metabolites or metabolic precursors. The non-essential wild-type metabolites were determined using gene essentiality data (Baba et al, 2006; Joyce et al, 2006) and are shown in red. Metabolites listed in blue were determined to have a reduced ‘core' structure different from the wild-type metabolite(s) and these are listed in the ‘core' biomass composition substitutes.
Was determined to be non-essential from (Kikuchi et al, 2000).
Determined to be essential under minimal media conditions and was not essential under the rich media condition examined.
Table 3.
Classification of iAF1260 reactions based on an FVA for 174 different carbon sourcesa
| Number of reactions (All thermodynamically feasible in all directions of flux) | |
|---|---|
| Essential for all 174 carbon sources, with flux always in the same direction | 183 |
| Essential for all 174 carbon sources, with flux in different directions depending on the carbon source | 3 |
| Substitutable for all 174 carbon sources, with flux always in the same direction | 863 |
| Substitutable for all 174 carbon sources, with flux in different directions depending on the carbon source | 41 |
| Essential, substitutable or blocked depending on the carbon source with flux always in the same direction whenever flux is present | 502 |
| Essential, substitutable or blocked depending on the carbon source, with flux in different directions also depending on the carbon source | 182 |
| Irreversible reactions blocked for all 174 carbon sources | 227 |
| Reversible reactions blocked for all 174 carbon sources | 76 |
See text for a definition of essential, substitutable and blocked. Exchange and demand reactions were not considered.
Once the reactions that operated in thermodynamically infeasible directions according to the FVA were identified and adjusted to remove all thermodynamic inconsistencies, we examined the ΔrG′ ranges calculated for all of the reactions defined as reversible based on the primary literature or heuristic rules. Through comparison with predicted ΔrG′ values, we identified many reactions that were originally specified as reversible and were actually thermodynamically irreversible (i.e., reactions being incapable of achieving both negative and positive values of ΔrG′ under physiological conditions). We corrected these reversible reactions to be consistent with our thermodynamic estimates (i.e., made them irreversible). In total, after checking for consistency, 553 reactions in iAF1260 were assigned as reversible and 1524 reactions were assigned as irreversible.
Looking at the reversibility of the reactions predicted using thermodynamic estimates alone, 1673 (84%) of the reactions for which ΔrG′o could be estimated were predicted to be reversible, whereas 323 (16%) were predicted to be irreversible. This finding indicated that reaction reversibility specified in the reconstruction was more restrictive than what is called for by thermodynamic analysis alone. The primary reason for this more restrictive property is that the reversibility set forth for the reactions in the reconstruction is often based on the physiological behavior of the reactions in the cell, not using the relatively broad concentration range achievable for metabolites along with the uncertainty inherent in the utilized method. Comparing these values with another approached, the method used in this work recognized 323 (16%) reactions as being irreversible, whereas Kümmel et al (2006b) recognized 130 (14%) reactions as being irreversible in the iJR904 network (Reed et al, 2003), utilizing a similar thermodynamic-based assignment and an additional assignment through heuristic rules.
Sensitivity analysis
To determine the sensitivity of the computational results (e.g., optimal product formation rate) generated using FBA with iAF1260, we varied independently the: (i) constraints imposed by transcriptional regulation on the network, (ii) metabolites included in the BOF, (iii) macromolecular content of the cell, (iv) effective P/O ratio in aerobic growth and (v) the maintenance costs associated with growth (i.e., NGAM and GAM). This analysis was performed using simulations of optimal growth under aerobic glucose-limited conditions.
In order to examine the overall regulatory effects on the computational results, we performed simulations both with the constraints outlined earlier (Covert et al, 2004) and with no transcriptional regulatory constraints (see Materials and methods and Figure 3). The resulting optimal GR and OUR for a given GUR (i.e., the line of optimality) predicted using FBA was found to be insensitive to the regulatory constraints placed on the system under these conditions (Supplementary Figure 1). However, the regulated network was less flexible in terms of the number of reactions that could possess a non-zero flux for an optimally predicted GR (33 less reactions). This result is not altogether surprising given that glucose is a preferred substrate for growth on one carbon source and thus, regulation had likely evolved to limit the uptake of additional carbon sources (Covert and Palsson, 2002). Similarly, comparing the use of the BOFWT and the BOFCORE for predicted optimal growth using FBA, the line of optimality produced was essentially identical in both cases (Supplementary Figure 1). However, there were 95 more reactions that required a non-zero flux value for an optimal solution in the network using the BOFWT in FBA simulations. This result is expected since the BOFWT is comprised of more metabolites requiring more active fluxes for their synthesis (see Table II).
To examine the effect of changing the macromolecular composition represented in the BOF, we varied the weight percentage of the three largest macromolecules in the cell (Table II and Figure 4A–C) in FBA simulations. We thus generated new BOFs varying the protein content from 50–80 wt%, the RNA content from 10–25 wt% and the lipid content from 7–15 wt% of the cell based on recorded experimental values (Pramanik and Keasling, 1997). While the macromolecular composition of the BOFCORE had some effect on the overall optimal GR and OUR, the most extreme variance was, at most, 5 and 8% at the median GUR in the range examined, respectively. Previously, Pramanik and Keasling (1997) evaluated a BOF that was GR dependant and determined that the building blocks that make up the macromolecular content of the cell (e.g., the amino acids that make up the protein content) are essentially constant when E. coli is grown under different growth conditions and any small changes in these compositions do not significantly affect calculated reaction flux values (Pramanik and Keasling, 1998). Therefore, this variable was not examined.
Figure 4.

Sensitivity analysis on the modeling parameters used in analyzing iAF1260. The relationship between the GUR (mmol gDW−1 h−1) (bottom axes, the dependant variable) and the resulting (1) GR (h−1) (left axes) and (2) OUR (mmol gDW−1 h−1) (right axes) produced during the sensitivity analysis using iAF1260. Using FBA and iAF1260, optimal growth was simulated under glucose aerobic conditions while varying (A) the dry weight percentage of protein (50–80%), (B) RNA (10–25%) and (C) lipid (7–15%) in the BOFCORE using physiologically measured values (Pramanik and Keasling, 1997). Also analyzed was (D) potential P/O ratios (1.0–2.7) in the network, as well as the (E) NGAM (±50%) and (F) GAM (±50%) that were determined for these conditions.
The P/O ratio of the ETS in E. coli was varied to determine its effect on optimal solutions produced using iAF1260 and FBA. The maximum value that the P/O ratio can achieve under aerobic conditions is 1.75, based on the stoichiometry of the ETS enzymes in iAF1260. Since there is some debate on the possible overall stoichiometries of the ETS in E. coli (see above), we further increased the potential maximum to 2.7 in our analysis and tested the effect of a P/O ratio ranging from 1.0 to 2.7. Specifically, a P/O ratio of 2.7 could be achieved if the most energy efficient pathway was used exclusively and the ETS possessed an ATP synthase with a stoichiometry of 3 H+/ATP and an NDH-1 with a stoichiometry of 4 H+/2e−. A P/O ratio of 1.0 is an estimated low-end value for aerobic growth. The analysis indicated that the modeling results are most sensitive to the P/O ratio than any other variable examined in this analysis. Optimal GR and OUR predictions varied, at most, 37 and 71% at the median GUR in the range examined, respectively.
Finally, we analyzed the effects of maintenance energy on optimal growth predictions. We varied the values of the NGAM and GAM±50% of the most consistent values of 8.39 mmol ATP gDW−1 h−1 and 59.81 mmol ATP gDW−1, respectively. The region that the line of optimality could posses for the varying maintenance energies was plotted in Figure 4E and F. The NGAM can affect the optimal GR and OUR predictions, at most, 8 and 15% and the GAM 16 and 31% at the median GUR in the range examined, respectively. Thus, these also are important variables to consider in FBA simulations of optimal growth under these conditions. Looking at the specific effect that each variable inflicts on the system; the NGAM shifts the intercept of the line of optimality with the GUR and OUR axes, whereas the GAM values change the slope of the line of optimality. The impact of the sensitivity analysis is addressed in the discussion.
Context for content
As high-throughput data become available for a number of organisms (Joyce and Palsson, 2006), there is a need for an underlying platform to analyze these data by placing them in a biological context. Genome-scale metabolic reconstructions, such as iAF1260, offer such a basis, since they are biochemically and genetically structured databases. As a result, they can be utilized to interpret high-throughput data in analyses looking at specific reactions, pathways or even genome-wide trends.
Context for content: analysis of alternate growth conditions
Similar to our previously described application of iAF1260 to predict the physiological state of E. coli growing under an aerobic glucose or succinate limiting condition, we also performed a broader analysis to determine all of the additional carbon, nitrogen, phosphorus and sulfur sources that could support simulated growth in minimal medium and compared this with findings using iJR904 (Reed et al, 2003). Overall, there were 174 carbon, 78 nitrogen, 49 phosphorous and 11 sulfur sources that were predicted to support growth using FBA and iAF1260 (see Table IV and Supplementary information); an increase over iJR904 by 84 carbon, 44 nitrogen, 45 phosphorous and nine sulfur sources. We compared the computational results to a high-throughput experimental screen using the Biolog platform (http://www.biolog.com). Table IV details the comparison between the computational and experimental predictions. The overall agreement is approximately 76% using iAF1260, compared with 60% for iJR904. This result reflects the increased scope of iAF1260 to analyze a wider range of growth conditions and helps validate the content of iAF1260.
Table 4.
Growth condition analysis
| Computational |
Experimental |
Agreement (iAF1260/iJR904) |
Disagreement (iAF1260/iJR904) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Source | Potential substrates | Support growtha | Total possible comparisons | E-G C-G | E-NG C-NG | % Total | E-NG C-G | E-G C-NG | % Total |
| Carbon | 262 | 174/90 | 87 | 54/46 | 11/15 | 75%/70% | 22/18 | 0/8 | 25%/30% |
| Nitrogen | 163 | 78/34 | 51 | 28/24 | 8/12 | 71%/71% | 8/4 | 7/11 | 29%/29% |
| Phosphorous | 63 | 49/4 | 20 | 20/3 | 0/0 | 100%/15% | 0/0 | 0/17 | 0%/85% |
| Sulfur | 25 | 11/2 | 12 | 8/2 | 0/0 | 67%/17% | 0/0 | 4/10 | 33%/83% |
Results using the iAF1260/iJR904 computational model; G, growth; NG, no growth; E, experimental; C, computational.
Disagreements between the computational and experimental data fall into two main categories and, going forward, will be resolved by different approaches. Cases in which computational growth is predicted and not observed experimentally indicate possible areas where there are either errors in the reconstruction or alternatively, where regulation limits the utilization of pathways needed for growth. This type of false positive for growth increased with iAF1260 since the network increased in total reactions available to support growth. In contrast, instances where experimental growth is observed and no growth is predicted computationally point to areas where further biochemical characterization is needed for E. coli and define targeted areas for biological discovery (Reed et al, 2006b). These false negatives for growth were significantly reduced from iJR904 to iAF1260 and clearly demonstrate the effect of the expanded content on computational simulations. Additional targets for model-driven expansion can be found in Supplementary information.
Context for content: gene essentiality analysis in iAF1260
We used the reconstruction as a framework to analyze the conditionally essential ORFs identified for E. coli K-12 (Baba et al, 2006; Joyce et al, 2006). A comparison between the computationally predicted essential ORFs and the experimental data (Baba et al, 2006; Joyce et al, 2006) is provided in Table V and Figure 5. Gene essentiality predictions under glucose aerobic conditions using iAF1260 show an overall increase in the number of ORFs that can be examined and correctly predicted when compared with iJR904 (an increase of 356 and 357 ORFs, respectively). There also is a modest improvement in overall accuracy (92% compared with 88%, see Table V; Supplementary Table III). These findings provide confidence for using iAF1260 to investigate previously unstudied conditions and examining the specific functionality that an essential (or non-essential) ORF provides for the system under that given condition. The agreement between the experimental and computational results, on the whole, validates the content of the reconstruction and the modeling procedure (assuming a low error rate in the experimental data).
Table 5.
Computational essentiality predictions
| Experimental |
||
|---|---|---|
| Essential | Non-essential | |
| Computational | ||
| Essential | 159 (13%) | 29 (2%) |
| Non-essential | 79 (6%) | 993 (79%) |
Figure 5.

ORF essentiality predictions using iAF1260. This heat map characterizes the agreement between ORFs predicted to be essential using iAF1260 and those experimentally determined from Baba et al (2006) and Joyce et al (2006). The enlarged region details how each a row corresponds to a computationally predicted essential ORF (188 total). The overall agreement between iAF1260 predictions and those found to be experimentally essential (overall, column 1) is shown along with a breakdown for ORFs found to be essential under rich media conditions (rich, column 2), under both glucose and glycerol minimal media conditions (shared, column 3) and under just glucose minimal medium conditions (glucose, column 4). ORFs are further grouped by their COG functional class (see Figure 1 for abbreviations; MU-ORF belongs to multiple COG classes). Dark blue indicates the condition under which each ORF was found to be essential. For example, folP was predicted to be an essential ORF for the biosynthesis of folate in iAF1260 under these conditions, but was not identified as essential by Baba et al (2006). This suggested the possibility of an alternative pathway for this step in E. coli that has yet to be characterized.
Disagreements between the experimental and computational data point to further areas of refinement and expansion of the metabolic and regulatory networks know for E. coli, as well as possible errors in the experimental data and model. The disagreements where ORFs were found to be computationally essential, but experimentally non-essential, point to specific areas where additional intracellular and transport reactions can be examined to rectify the disagreements (29 cases, see Table V). For example, the ubiC gene was predicted to be essential for its involvement in the ubiquinone biosynthesis pathway. This finding points to the fact that additional work is needed to characterize the full complement of genes responsible for the aerobic and anaerobic production of ubiquinone (Alexander and Young, 1978). Additionally, eight of the 29 cases were predicted to be essential for thiamine biosynthesis, an essential cofactor in E. coli. This result suggests a likely error in the experimental data and is supported by Vander Horn et al (1993). ORFs that are found to be experimentally essential but computationally non-essential suggest potential regulatory effects on the system and possible inaccuracies in the metabolic network (79 cases, see Table V). Transcriptional regulation limits network pathways under a given condition; therefore, computational disagreement could arise if such pathways are computationally utilized. Disagreements in this class also identify a current limitation of the model. The action of 18 tRNA charging reactions are contained in the reconstruction, but are not currently accounted for in the modeling scheme. The resulting computational disagreements will likely be resolved through expansion of the network to include transcription and translation processes in the cell. For a complete list of the computational and experimental disagreements, see Supplementary information.
Discussion
Metabolic reconstruction and subsequent mathematical computation has become a useful tool in the post-genomic era by aiding both biological computation and experimentation. In this work, we present, characterize and utilize the iAF1260 metabolic reconstruction of E. coli K-12 MG1655. The reconstruction serves as both a BiGG database containing the current knowledge of E. coli metabolism, as well as a framework for mathematical analysis. Accordingly, the major contributions from this work are: (1) an expansion in size, scope and detail of the metabolic network of E. coli, effectively exhausting the available literature, (2) an enumeration and description of the parameters and methods needed to utilize the reconstruction as a predictive model; examples of simulation results compared with high-throughput experimental data are presented and (3) the inclusion of thermodynamic information and a novel thermodynamic consistency analysis for chemical transformations accounted for in the reconstruction.
iAF1260 represents the largest metabolic reconstruction of any unicellular organism and accounts for 1260 ORFs (28%) in the current E. coli genome annotation (Riley et al, 2006). Furthermore, 1161 of the included ORFs (92%) have experimentally-based functions, conferring a high degree of confidence in the corresponding interactions. Just as gene annotation and sequence databases are used to identify and characterize genes in newly sequenced genomes, iAF1260 will similarly serve as a primary reference for future metabolic reconstructions. Because of its curation history and size, future reconstructions, especially those for closely related organisms, will draw directly from this content. This process will further be aided by the synchronization and mapping with the EcoCyc database. The next step in the expansion of the E. coli metabolic network will require further discovery of metabolic functions and computational methods are needed that can facilitate this process (Reed et al, 2006b).
In addition to expanded content, significant advancements in reconstruction techniques and methods used to determine network capabilities were presented. Thermodynamic consistency analysis represents a novel way to flag or highlight highly improbable intracellular and transport reactions for further evaluation. This approach can be added to future metabolic reconstruction and modeling projects. It effectively constitutes a QC/QA test that should improve the utility and scope of modeling predictions. Additionally, the use of a core biomass BOF (BOFCORE) has identified an improved strategy to probe gene essentiality for growth. Previous analyses examining gene essentiality have utilized a BOF, which is based on measurements from a specific growth environment and is also constant in the type and relative proportion of metabolites. A common problem that arises when using a BOF based on wild-type measurements is that potential false positives can be generated when conditionally essential metabolites are inappropriately included in the BOF (Ghim et al, 2005; Imielinski et al, 2005). The BOFCORE presented here, with continual refinement guided by experimentation, should increase the accuracy and utility of computational predictions with respect to mutant phenotype predictions.
The approach taken to evaluate reaction reversibility in iAF1260 was to prevent the inclusion of reactions that were highly unlikely to be reversible. This approach was carried out by using the thermodynamic consistency analysis and subsequent analysis of reaction thermodynamic estimates. Due to the thermodynamic coupling of reactions operating simultaneously, reactions that are individually thermodynamically reversible under physiological conditions will not necessarily be reversible when operating in concert with the other reactions in the cell. In line with this, using reaction reversibility determined from the thermodynamic analysis of individual reactions alone with FBA will result in improper model behavior due to the operation of thermodynamically infeasible pathways and cycles. Only if thermodynamic constraints are used in conjunction with the mass balance constraints of FBA to prevent the operation of these thermodynamically infeasible pathways (for example, Henry et al, (2007)) can the reaction reversibility determined for individual reactions be used. Therefore, utilization of the thermodynamic information presented to fully assign reversibility and irreversibility in modeling simulations automatically requires additional implementation of methods, which consider thermodynamics on the systems level.
With the increasing use of network reconstruction and the constraint-based modeling approach, a need has emerged to clearly define and demonstrate the steps required to computationally utilize a reconstruction. By outlining these steps and examining the sensitivity of modeling parameters used in computations, we have both explicitly defined the protocol and revealed the impact of modeling parameters on predictions. A computational software package is also available to efficiently implement such metabolic modeling (Becker et al, 2007). A sensitivity analysis of key strain-specific parameters, using an early version of the reconstructed E. coli network (Varma and Palsson, 1995), found that the P/O ratio significantly affects the GR and flux predictions, whereas varying the BOF had relatively little effect. However, our analysis shows a greater dependence on the maintenance parameters calculated for these conditions. This result is primarily due to our testing of a broader maintenance value range (±50% of the calculated values as opposed to 20% by Varma and Palsson (1995)). This larger value was selected because it is approximately the amount of the GAM that is difficult to quantify (i.e., unknown maintenance that accounts for gradient maintenance, protein turnovers and so forth; Neidhardt et al, 1990) and produced a range that can be justified by examining different E. coli growth data (results not shown). Future projects should take into account the impact of the influential parameters (i.e., P/O ratio, growth maintenance) when designing their computational studies.
The culmination of the increased size and expanded coverage of the reconstruction, in combination with the improved reconstruction techniques, has broadened the scope and accuracy of computational predictions. Comparisons of iAF1260 simulations with experimental data for gene-essentiality and growth phenotypes showed an overall increase of 4 and 16% over iJR904 predictions, respectively. Specifically, iAF1260 is markedly improved in analyzing and predicting a wider range of minimal media growth conditions (see Table IV). It can also better predict and screen the essential genes needed for viability in E. coli (see Table V). The one area where it appears that the model's ability to match experimental data decreased was where ORFs were found to be experimentally essential but computationally non-essential. This area can be addressed through further expansion of the reconstruction's scope (e.g., by including the transcriptional and translation machinery in E. coli as well as transcriptional regulatory effects) and targeted experimentation (e.g., elucidating the entry step into the de novo biosynthesis of biotin).
Future directions for improvement of the metabolic reconstruction of E. coli remain. As previously stated, the scope of the reconstruction will continually increase. Dead-ends and lumped reaction in the reconstruction point to specific areas of E. coli metabolism that can be further characterized in this expansion effort. A computational approach to resolve these dead-ends that utilizes constraint-based methods can be used in this effort. Additionally, an area for further compartmentalization of metabolites in the reconstruction is for metabolites located in the lipid bilayers. For example, a lipid on the inner leaflet of the outer membrane is different than one on the outer leaflet of the inner membrane, but currently in the reconstruction, they are both located in the periplasm. Further advancements in modeling will also be achieved through acquisition of additional experimental gene essentiality studies under different minimal media conditions to better define the core metabolites needed for viability and improve overall computational accuracy. Advancements are also likely to arise from additional incorporation of reaction and system thermodynamics.
In summary, iAF1260 represents a significantly expanded and comprehensively verified reconstruction of the E. coli metabolic network with broadened and enhanced predictive capabilities. With the growing number of studies based on previous versions of this reconstruction appearing, this work will enable a wider spectrum of studies focused on both proximal (i.e., immediate) and distal (i.e., over time) causation in biology. As the field of systems biology expands to incorporate cellular interactions from multiple core functions (e.g., regulation, signaling, etc.) on the genome scale, iAF1260 will serve as a key component for the study of E. coli by providing an extensive picture of cellular metabolism.
Materials and methods
Network reconstruction
The reconstruction process has also been previously outlined (Feist et al, 2006; Reed et al, 2006a). Here, we provide certain details specific to this work. Starting from the metabolic network for iJR904 (Reed et al, 2003), additional reactions were added to the network based on E. coli-specific biochemical characterization studies (see Supplementary information for a complete list of references) and other reactions were removed (see Results). This process was aided by comparing the content of iJR904 with the EcoCyc database (see below). The E. coli genome annotation (Riley et al, 2006) was used as a citation source for biochemical characterization studies and a framework upon which translated metabolic proteins, and subsequently reactions, were assigned to form gene to protein to reaction (GPR) assignments. The SimPheny™ (Genomatica Inc., San Diego, CA) software platform was used to build the reconstruction. For each reaction entered into the reconstruction, the involved metabolites were characterized according to their chemical formula and charge determined using their pKa value for a pH of 7.2. Metabolite charge was determined using its pKa value(s). When the metabolite pKa was not available, charge was determined using the pKa of ionizable groups present in a metabolite (http://www.chemaxon.com/product/pka.html). All of the reactions entered into the network were designated as enzymatically catalyzed reactions or spontaneous reactions, were both elementally and charged balanced and are either reversible or irreversible. Reversibility was determined first from primary literature for each particular enzyme/reaction, if available (see Supplementary information for references). Additionally, general heuristic rules, like those applied by Kümmel et al (2006b), were used to enter reversibility using knowledge about the physiological direction of a reaction in a pathway (sometimes including regulatory knowledge) and/or basic thermodynamic information (such as reactions hydrolyzing high-energy phosphate bonds are almost always irreversible). Furthermore, a thermodynamic analysis of reversibility was utilized to assign the directionality of some reactions (see above).
Comparison of iAF1260 and the EcoCyc and MetaCyc databases
The comparison between the content of the iAF1260 and the EcoCyc (Keseler et al, 2005) and MetaCyc (Caspi et al, 2006) databases was performed in three phases. Initially, a list of metabolic ORFs contained in EcoCyc and not in iJR904 (the previous reconstruction) was manually evaluated for inclusion in iAF1260 in an effort to merge content. A total of 176 out of 308 ORFs from this list were included into iAF1260 from manual analysis of this list or were included before this analysis from primary literature in a separate effort. Many of the inclusions in this phase were transporter encoding ORFs. A common type of ORF that was not included were those acting on nonspecific metabolites (e.g., nonspecific drugs), proteins or RNA molecules.
The second phase of the comparison consisted of generating a complete mapping of the metabolites contained in iAF1260 and EcoCyc or MetaCyc. This phase permitted the inclusion of compounds in each database that were missing from the other and identified possible errors in enzyme substrate specificity and metabolite structure. It also provided a future reference for linking of the metabolite content between the two resources. In an initial automated effort, mappings between metabolites in iAF1260 and EcoCyc/MetaCyc were established computationally using textual matching between the official name in iAF1260 to the common name and/or synonyms of metabolites in EcoCyc/MetaCyc, version 10.6. In addition, when available in both data sets, KEGG identifiers and CAS numbers were used to double-check matches or to make additional matches. After this computational step, 871 out of 1039 metabolites in iAF1260 were mapped to EcoCyc/MetaCyc. The remaining metabolites were mapped manually and changes to the content of iAF1260 made during this mapping process were facilitated by cross-referencing the ORFs that encoded for the proteins that acted on specific metabolites in iAF1260 with their annotation in EcoCyc (see Results for findings and Supplementary information for the mapping).
The final phase of the comparison was an automated mapping between reactions contained in iAF1260 and EcoCyc/MetaCyc. This phase generated a list of high-confidence reactions that both iAF1260 and EcoCyc contained, and provides a future reference for a full merging of the reaction content between the two resources. The automated reaction mapping was performed with software written specifically for this task, to accommodate frequently occurring types of differences between the models. The matcher parses the equations of every reaction R in iAF1260 and uses the previously described metabolite mappings to find the reaction object in EcoCyc/MetaCyc that contains the same set of metabolites as does R. Numerous reactions in iAF1260 contain protons in the equation that do not appear in EcoCyc, and the matcher can take into account this and other similar differences. The matcher also tries to find a generic reaction in EcoCyc that is specified in terms of compound classes, if the metabolite instances used in the equation in iAF1260 did not yield a direct match.
Generation of the biomass BOF
The biomass BOF was generated by defining all of the major and essential constituents that make up the cellular biomass content of E. coli. To determine these metabolites and their quantity, we used the dry weight composition data for an average E. coli B/r cell growing exponentially at 37°C under aerobic conditions in glucose minimal medium, with an approximate doubling time of 40 min having a dry cell weight of 2.8 × 10−13 grams (Neidhardt et al, 1990) (Table II; Supplementary information). Each cellular biomass macromolecule (i.e., protein, RNA, DNA, etc.) was divided into its corresponding metabolic precursors present in the reconstruction (for example, L-alanine, UTP or dTTP, respectively). Each of the precursor metabolites was assigned a value that it contributes to the total percentage of the macromolecule, except for the soluble pool metabolites (e.g., thiamine diphosphate). This process was followed so that if the overall quantities of macromolecules were changed, the corresponding precursor metabolite would be scaled appropriately (see Sensitivity analysis section). The quantity of soluble pool metabolites (approximately 2.9% of the total biomass) was taken from experimentally measured values or alternatively, it was estimated as a 0.1 mM intracellular concentration (see Supplementary information for a complete list of references). From this data, a linear biomass BOF was formulated based on the wild-type cell composition for E. coli and an ATP maintenance approximation to account for non-metabolic processes (see Supplementary information). Using FBA, the model was analyzed to determine if each BOF metabolite could be generated from the defined minimal medium under both aerobic and anaerobic conditions with D-glucose, D-ribose and glycerol as the carbon and energy source. Only metabolites identified as cofactors could not be generated from the glucose minimal medium (discussed in Supplementary information).
Using the BOFWT, gene essentiality and published data, a ‘core' BOF was formulated that was consistent with the minimal set of macromolecular molecules needed for cell viability. The 20 common amino acids, inorganic ions and nucleotide metabolites were all considered essential (Neidhardt et al, 1990). For the other BOFWT metabolites, each metabolite was evaluated individually to determine if the genes that were necessary to synthesize the metabolite from minimal media substrates (see Supplementary Table II) were essential (Baba et al, 2006; Joyce et al, 2006). One macromolecule, glycogen, was not essential for cell viability because there were no essential ORFs encoding for enzymes in the synthesis or breakdown of glycogen. The essential metabolites were defined by identifying the end product from the closest essential reaction to the BOFWT metabolite (Table II) in the possible de novo pathway(s) for biosynthesis. Molecules in this group, such as riboflavin, were determined to be essential, whereas the wild-type outer membrane E. coli K-12 LPS molecule was not found to be essential. However, a precursor of the common wild-type LPS molecule, KDO2-Lipid A, was found to be essential for cell viability (Raetz, 1996). Alternatively, ‘core' metabolites were also determined from specific published studies. For example, thiamine diphosphate was found to be essential (Vander Horn et al, 1993), whereas phosphatidylglycerol was determined not to be essential (Kikuchi et al, 2000).
The ATP maintenance approximation in the BOFs, which account for non-metabolic processes were approximated with the ATP utilization equation, 
where the number of ATP equivalents hydrolyzed is characterized in the GAM variable. The entire BOF is given in mathematical terms in Supplementary information.
Aside from the BOF maintenance, an NGAM (mmol ATP gDW−1 h−1) value was used as an energy ‘drain' on the system during the linear programming calculations and accounts for non-growth cellular activities (Pirt, 1965). The NGAM was represented as a defined flux in the reaction flux vector, vNGAM (see below and Supplementary information).
Modeling simulations
A stoichiometric matrix, S (m × n), was constructed for iAF1260, where m is the number of metabolites and n is the number of reactions. The corresponding entry in the stoichiometric matrix, Sij, represents the stoichiometric coefficient for the participation of the ith metabolite in the jth reaction. FBA was then used to solve the linear programming problem under steady-state criteria (Price et al, 2004) represented by the equation:
where v (n × 1) is a vector of reaction fluxes. Since the linear problem is normally an underdetermined system for genome-scale metabolic models, there exist multiple solutions for v that satisfy equation 2. To find a particular solution for v, the cellular objective of producing the maximal amount of biomass constituents, represented by the ratio of metabolites in the BOF, is optimized for in the linear system. Additionally, constraints that are imposed on the system are in the form of:
where α and β are the lower and upper limits placed on each reaction flux, vi, respectively. For reversible reactions, −∞⩽vi⩽∞, and for irreversible reactions, 0⩽vi⩽∞. The constraints on the reactions that allow metabolite entry into the extracellular space were set to 0⩽vi⩽∞ if the metabolite was not present in the medium, meaning that the compounds could leave, but not enter the system. For the metabolites that were in the medium, the constraints were set to −∞⩽vi⩽∞ for all except the limiting substrate(s) (e.g., glucose and/or oxygen). The reaction flux through the BOF was constrained from 0⩽vBOF⩽∞.
Linear programming calculations were performed using SimPheny™ (Genomatica, San Diego, CA) and the LINDO (Lindo Systems Inc., Chicago, IL) or TOMLAB (Tomlab Optimization Inc., San Diego, CA) solvers in MATLAB® (The MathWorks Inc., Natick, MA) with the COBRA Toolbox (Becker et al, 2007).
When comparing the flux distribution in central metabolism to experimentally reported values (Fischer et al, 2004), all of the comparisons were performed using computational results when optimal growth is predicted using the BOFCORE, the 152 regulated reactions under these conditions constrained to zero (see above), a split in the flux ratio between the two NADH dehydrogenases of 1:1, an NGAM value of 8.39 mmol ATP gDW−1 h−1, a GAM value of 59.81 mmol ATP gDW−1 and iAF1260. An FVA on the optimal flux distribution yielded no flexibility in the central metabolism pathways examined in this study. From the Fischer et al (2004) study, data from E. coli growth in reactor conditions were used because the oxygen uptake and CO2 secretion rates were reported, and the flux values that were used were based off 13C-constrained flux balancing.
Sensitivity analysis
The sensitivity analysis was performed under aerobic glucose-limiting minimal medium conditions. For each analysis, the parameter being examined was varied while the GUR was sequentially set between 0 and 10 mmol gDW−1 h−1 for a series of simulations, with the maximum OUR set to 18.5 mmol gDW−1 h−1. This maximum uptake rate was chosen, since it closely matched the maximum uptake rate of oxygen observed in vivo (e.g., see Edwards et al, 2001; Fischer et al, 2004). All other modeling parameters were set to those determined in the application of iAF1260 to predict cellular phenotypes section. The BOFCORE was used in all simulations (except those stated otherwise), since the predicted GR and OUR were found to be insensitive to the use of either the BOFCORE or BOFWT.
Alternate growth condition analysis
To determine the carbon, nitrogen, phosphorus and sulfur sources that could support simulated growth, we screened all of the metabolites that could be exchanged with the environment (i.e., exchange reactions) in the iAF1260 and iJR904 models. The identified metabolites formed the potential substrate sets (Table IV). Through subsequent simulations, we set an arbitrary maximum flux of 20 mmol substrate gDW−1 h−1 for each potential substrate tested (consistent with maximum observed substrate uptake rates in vivo) and optimized for flux through the BOFCORE using FBA and either iAF1260 or iJR904. An OUR of 18.5 mmol gDW−1 h−1, the BOFCORE, a NGAM of 8.39 mmol ATP gDW−1 h−1, a GAM of 59.81 mmol ATP gDW−1 and no regulatory constrains were used during the growth condition analysis of iAF1260 (for iJR904, see Reed et al, 2003). During the analysis, the reactions CAT, SPODM and SPODMpp were constrained to zero to prevent generation of cellular energy equivalents through reactions involved in E. coli's response to oxidative stress. If a positive flux could be generated through the BOFCORE reaction (vBOFcore>0), then the substrate was considered a viable source. Experimental data used in the comparison were provided by Biolog (http://www.biolog.com) and both ‘weak' and ‘positive' readings from the biolog data were considered as a positive growth condition.
Gene essentiality analysis
To determine the effect of a gene deletion, the reaction(s) associated with each gene in iAF1260 were individually deleted from S and FBA was used to predict the mutation growth phenotype. The simulations were performed using glucose minimal medium conditions with a GUR of 10 mmol gDW−1 h−1, an OUR of 20 mmol gDW−1 h−1, the BOFCORE, an NGAM of 8.39 mmol ATP gDW−1 h−1, a GAM of 59.81 mmol ATP gDW−1 and zero flux through the 152 reactions regulated under glucose aerobic conditions (see Supplementary information). The flux through the BOFCORE was optimized in the mutated network, S′, and a positive flux through the BOF (vBOFcore>0) was considered non-essential (equation 2). Experimental criteria for gene essentiality are described in detail in Joyce et al (2006).
Standard conditions for all estimated ΔrG′o and ΔfG′o
All ΔfGest′o and ΔrGest′o calculated for the reconstruction using the group contribution method are based upon the standard condition of aqueous solution with pH equal to 7, temperature equal to 298.15 K, zero ionic strength and 1 M concentrations of all species except H+, and water. In the cases where multiple charged forms of a molecule exist at pH 7 (i.e., ATP4− and HATP3−), the most abundant form is used. This is consistent with the form of the molecules used in the fitting of the group contribution energy values (MD Jankowski and V Hatzimanikatis, in preparation).
The charges of the molecules and the proton balances for the reactions included in the reconstruction are based on a reference pH of 7.2. In order for the ΔrGest′o values included with the reconstruction to match the reference pH of the reconstruction, all ΔrGest′o calculated using the group contribution method (based on a reference pH of 7) were adjusted to a reference pH of 7.2 using the method described in Alberty (2003). The adjusted ΔrGest′o values were used in the calculation of ΔfG′m and for all other thermodynamic analysis performed on the reconstruction. The pKa values for the compounds in the reconstruction used in the transformation of ΔrGest′o to a reference pH of 7.2 were estimated from the molecular structures of the compounds using the MarvinBeans software developed by ChemAxon.
Adjustment of ΔrG′o to ΔfG′m
The ΔrG′m calculated for all reactions contained in the reconstruction is based on the reference state of 1 mM concentrations for all species except H+, water, H2 and O2. The reference concentrations for H2 and O2 are the saturation concentrations for these species in water at 1 atm and 298.15 K. All ΔrG′m values reported in this work also include the energy contribution of the transmembrane electrochemical potential and proton gradient for all reactions involving transport across the cytoplasmic membrane assuming a periplasmic pH of 7.7 and a cytoplasmic pH of 7.2. All ΔrG′m calculated for reactions in the iAF1260 model are listed in Supplementary information.
We also determined the direction of flux required in the reactions contained in iAF1260 to achieve near optimal growth (90–100%) on each of 174 carbon sources using FVA (Mahadevan and Schilling, 2003) and the BOFCORE. It is worthwhile to note that the same set of reactions can or cannot be utilized in FVA simulations when examining approximately 5–95% of the optimal flux value achievable for the BOFCORE under glucose aerobic conditions (one exception is the cytochrome oxidase bo and oxygen transport reactions, which are needed for generating the necessary energy to achieve approximately 80% or greater of the BOFCORE flux). During the FVA of conditions corresponding to glucose aerobic growth, the reactions CAT, SPODM and SPODMpp were constrained to zero to prevent generation of cellular energy equivalents through reactions involved in E. coli's response to oxidative stress, and the reaction formate hydrogenlyase, which appears to be involved in regulating cytosolic pH (Mnatsakanyan et al, 2004), was also constrained to zero to prevent the production of significant amounts of hydrogen gas that is not typically observed for most buffered experiments around pH 7. The results of the FVA indicated that some of the reactions in the reconstruction consistently operated in the reverse direction. During the calculation of ΔrG′m for these reactions, the forward direction of each reaction was redefined to be in the direction of flux required for near optimal growth to occur. Because of this adjustment, all negative ΔrG′m and ΔrG′ values reported (see Figure 2) indicate reactions that are thermodynamically feasible in the direction of flux while positive values indicate thermodynamically infeasible reactions.
Estimation of achievable range of values for ΔrG′
The range of possible values for the ΔrG′ of a reaction depends not only on ΔrG′o but also on the uncertainty in the estimated ΔrG′o (Ur,est), the activities of the metabolites involved in the reaction and for transport reactions, the energy contribution of the electrochemical potential and proton gradient across the cytoplasmic membrane (ΔGTransport) (Henry et al, 2006). ΔrG′ can deviate from ΔrG′m because the activity of a metabolite can deviate from the reference value of 1 mM. The maximum and minimum values for ΔrG′ were calculated using the following equations.
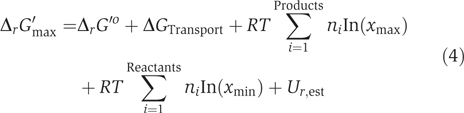
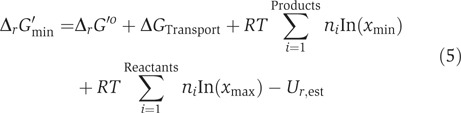
where xmin is the minimal metabolite activity assumed to be 0.00001 M, and xmax is the maximum metabolite activity assumed to be 0.02 M. The physiological range of activities for the dissolved gasses H2, O2 and CO2 is much lower than the range of activities for other metabolites involved in metabolism. For this reason all of the xmin values for H2, O2 and CO2 were set to 10−8 M, which is approximately equivalent to one molecule per cell, and the xmax values for H2, O2 and CO2 were set to the saturation concentrations for these gasses in water at 298.15 K and 1 atm, 0.000034, 0.000055 and 0.0014 M, respectively. The activity terms for H+ and H2O were left out of equations 4 and 5 because these activities have already been lumped into the ΔrG′o.
The ΔrG′ ranges encompassed by ΔrGmin′o and ΔrGmax′o calculated for the reactions in the reconstruction were used to assign reversibility and directionality to the reactions based on the thermodynamic estimates. Reactions with exclusively negative ΔrG′ values were identified as thermodynamically irreversible in the forward direction, reactions with exclusively positive ΔrG′ values were identified as thermodynamically irreversible in the reverse direction and reactions with both positive and negative ΔrG′ values were identified as thermodynamically reversible. FVA was then utilized to determine the directions in which each of the reactions in the reconstruction operated during near optimal growth on 174 carbon sources. In this way, reactions for which the direction of operation indicated by FVA conflicted with the direction of thermodynamic feasibility indicated by the ΔrG′ ranges were identified.
Supplementary Material
Legend to Supplementary Material
Supplementary Text, Figures and Tables
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Supplementary Information 4
Supplementary Information 5
Supplementary Information 6
Acknowledgments
We thank Kenyon Applebee, Edward Chuong, Ingrid Keseler, Sean Nihalani, Alan Ruttenberg, Milton Saier, Jan Schellenberger and Jeremy Zucker for their help in the generation and analysis of the reconstruction. Studies performed at UCSD were supported by National Institutes of Health Grant GM057089. Bernhard Palsson and UCSD have a financial interest in Genomatica Inc. Although the NIH R01 GM057089 grant has been identified for conflict of interest management based on the overall scope of the project and its potential to benefit Genomatica Inc., the research findings included in this publication do not necessarily directly relate to the interests of Genomatica Inc.
References
- (NC-IUBMB) NCotIUoBaMB (2006) Enzyme nomenclature. Moss, GP
- Albe KR, Butler MH, Wright BE (1990) Cellular concentrations of enzymes and their substrates. J Theor Biol 143: 163–195 [DOI] [PubMed] [Google Scholar]
- Alberty RA (2003) Thermodynamics of Biochemical Reactions. Massachusetts Institute of Technology: Cambridge, MA [Google Scholar]
- Alexander K, Young IG (1978) Alternative hydroxylases for the aerobic and anaerobic biosynthesis of ubiquinone in Escherichia coli. Biochemistry 17: 4750–4755 [DOI] [PubMed] [Google Scholar]
- Almaas E, Kovacs B, Vicsek T, Oltvai ZN, Barabasi AL (2004) Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 427: 839–843 [DOI] [PubMed] [Google Scholar]
- Alper H, Miyaoku K, Stephanopoulos G (2005) Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol 23: 612–616 [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CL, Herring CD, Reed JL, Palsson BO (2005) The global transcriptional regulatory network for metabolism in Escherichia coli attains few dominant functional states. Proc Natl Acad Sci USA 102: 19103–19108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard DA, Liang SD, Qian H (2002) Energy balance for analysis of complex metabolic networks. Biophys J 83: 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, Herrgard MJ (2007) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA toolbox. Nat Protoc 2: 727–738 [DOI] [PubMed] [Google Scholar]
- Calhoun MW, Oden KL, Gennis RB, de Mattos MJ, Neijssel OM (1993) Energetic efficiency of Escherichia coli: effects of mutations in components of the aerobic respiratory chain. J Bacteriol 175: 3020–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Foerster H, Fulcher CA, Hopkinson R, Ingraham J, Kaipa P, Krummenacker M, Paley S, Pick J, Rhee SY, Tissier C, Zhang P, Karp PD (2006) MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res 34: D511–D516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO (2004) Integrating high-throughput and computational data elucidates bacterial networks. Nature 429: 92–96 [DOI] [PubMed] [Google Scholar]
- Covert MW, Palsson BO (2002) Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem 277: 28058–28064 [DOI] [PubMed] [Google Scholar]
- Edwards JS, Ibarra RU, Palsson BO (2001) In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat Biotechnol 19: 125–130 [DOI] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO (2000) The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc Natl Acad Sci USA 97: 5528–5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena SF, Lenski RE (2003) Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4: 457–469 [DOI] [PubMed] [Google Scholar]
- Feist AM, Scholten JCM, Palsson BO, Brockman FJ, Ideker T (2006) Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Mol Syst Biol 2: 2006.0004 10.1038/msb4100046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E, Zamboni N, Sauer U (2004) High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem 325: 308–316 [DOI] [PubMed] [Google Scholar]
- Fong SS, Burgard AP, Herring CD, Knight EM, Blattner FR, Maranas CD, Palsson BO (2005) In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng 91: 643–648 [DOI] [PubMed] [Google Scholar]
- Gennis RB, Stewart V (1996) Respiration. In Escherichia coli and Salmonella, Neidhardt FC (ed), pp 217–261. ASM Press: Washington, DC [Google Scholar]
- Ghim CM, Goh KI, Kahng B (2005) Lethality and synthetic lethality in the genome-wide metabolic network of Escherichia coli. J Theor Biol 237: 401–411 [DOI] [PubMed] [Google Scholar]
- Grass G, Otto M, Fricke B, Haney CJ, Rensing C, Nies DH, Munkelt D (2005) FieF (YiiP) from Escherichia coli mediates decreased cellular accumulation of iron and relieves iron stress. Arch Microbiol 183: 9–18 [DOI] [PubMed] [Google Scholar]
- Hakobyan M, Sargsyan H, Bagramyan K (2005) Proton translocation coupled to formate oxidation in anaerobically grown fermenting Escherichia coli. Biophys Chem 115: 55–61 [DOI] [PubMed] [Google Scholar]
- Helling RB (2002) Speed versus efficiency in microbial growth and the role of parallel pathways. J Bacteriol 184: 1041–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS, Broadbelt LJ, Hatzimanikatis V (2007) Thermodynamics-based metabolic flux analysis. Biophys J 92: 1792–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS, Jankowski MD, Broadbelt LJ, Hatzimanikatis V (2006) Genome-scale thermodynamic analysis of Escherichia coli metabolism. Biophys J 90: 1453–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring CD, Raghunathan A, Honisch C, Patel T, Applebee MK, Joyce AR, Albert TJ, Blattner FR, van den Boom D, Cantor CR, Palsson BO (2006) Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet 38: 1406–1412 [DOI] [PubMed] [Google Scholar]
- Ibarra RU, Edwards JS, Palsson BO (2002) Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420: 186–189 [DOI] [PubMed] [Google Scholar]
- Imielinski M, Belta C, Halasz A, Rubin H (2005) Investigating metabolite essentiality through genome-scale analysis of Escherichia coli production capabilities. Bioinformatics 21: 2008–2016 [DOI] [PubMed] [Google Scholar]
- Janssen P, Goldovsky L, Kunin V, Darzentas N, Ouzounis CA (2005) Genome coverage, literally speaking. The challenge of annotating 200 genomes with 4 million publications. EMBO Rep 6: 397–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce AR, Palsson BO (2006) The model organism as a system: integrating ‘omics' data sets. Nat Rev Mol Cell Biol 7: 198–210 [DOI] [PubMed] [Google Scholar]
- Joyce AR, Reed JL, White A, Edwards R, Osterman A, Baba T, Mori H, Lesely SA, Palsson BO, Agarwalla S (2006) Experimental and computational assessment of conditionally essential genes in Escherichia coli. J Bacteriol 188: 8259–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseler IM, Collado-Vides J, Gama-Castro S, Ingraham J, Paley S, Paulsen IT, Peralta-Gil M, Karp PD (2005) EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res 33: D334–D337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Shibuya I, Matsumoto K (2000) Viability of an Escherichia coli pgsA null mutant lacking detectable phosphatidylglycerol and cardiolipin. J Bacteriol 182: 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümmel A, Panke S, Heinemann M (2006) Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol Syst Biol 2: 2006.0034 10.1038/msb4100074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümmel A, Panke S, Heinemann M (2006b) Systematic assignment of thermodynamic constraints in metabolic network models. BMC Bioinformatics 7: 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Woo HM, Lee D-Y, Choi HS, Kim TY, Yun H (2005) Systems-level analysis of genome-scale in silico metabolic models using MetaFluxNet. Biotechnol Bioproc Eng 10: 425–431 [Google Scholar]
- Lotierzo M, Tse Sum Bui B, Florentin D, Escalettes F, Marquet A (2005) Biotin synthase mechanism: an overview. Biochem Soc Transac 33: 820–823 [DOI] [PubMed] [Google Scholar]
- Mahadevan R, Schilling CH (2003) The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng 5: 264–276 [DOI] [PubMed] [Google Scholar]
- Majewski RA, Domach MM (1990) Simple constrained optimization view of acetate overflow in E. coli. Biotechnol Bioeng 35: 732–738 [DOI] [PubMed] [Google Scholar]
- Mnatsakanyan N, Bagramyan K, Trchounian A (2004) Hydrogenase 3 but not hydrogenase 4 is major in hydrogen gas production by Escherichia coli formate hydrogenlyase at acidic pH and in the presence of external formate. Cell Biochem Biophys 41: 357–366 [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Ingraham JL, Schaechter M (1990) Physiology of the Bacterial Cell: a Molecular Approach. Sinauer Associates: Sunderland, Mass [Google Scholar]
- Nikolaev EV, Burgard AP, Maranas CD (2005) Elucidation and structural analysis of conserved pools for genome-scale metabolic reconstructions. Biophys J 88: 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi Y, Nakai Y, Shimba N, Toyosaki H, Kawahara Y, Sugimoto S, Suzuki E (2004) The energetic conversion competence of Escherichia coli during aerobic respiration studied by 31P NMR using a circulating fermentation system. J Biochem (Tokyo) 136: 509–515 [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Lercher MJ (2005) Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat Genet 37: 1372–1375 [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Lercher MJ, Csermely P, Oliver SG, Hurst LD (2006) Chance and necessity in the evolution of minimal metabolic networks. Nature 440: 667–670 [DOI] [PubMed] [Google Scholar]
- Palsson BO (2004) Two-dimensional annotation of genomes. Nat Biotechnol 22: 1218–1219 [DOI] [PubMed] [Google Scholar]
- Pirt SJ (1965) The maintenance energy of bacteria in growing cultures. Proc R Soc Lond B Biol Sci 163: 224–231 [DOI] [PubMed] [Google Scholar]
- Pramanik J, Keasling JD (1997) Stoichiometric model of Escherichia coli metabolism: incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol Bioeng 56: 398–421 [DOI] [PubMed] [Google Scholar]
- Pramanik J, Keasling JD (1998) Effect of Escherichia coli biomass composition on central metabolic fluxes predicted by a stoichiometric model. Biotechnol Bioeng 60: 230–238 [DOI] [PubMed] [Google Scholar]
- Price ND, Reed JL, Palsson BO (2004) Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol 2: 886–897 [DOI] [PubMed] [Google Scholar]
- Raetz CRH (1996) Bacterial lipopolysaccharides: A remarkable family of bioactive macroamphiphiles. In Escherichia coli and Salmonella, Neidhardt FC (ed), pp 1035–1063. ASM Press: Washington, DC [Google Scholar]
- Reed JL, Famili I, Thiele I, Palsson BO (2006a) Towards multidimensional genome annotation. Nat Rev Genet 7: 130–141 [DOI] [PubMed] [Google Scholar]
- Reed JL, Palsson BO (2003) Thirteen years of building constraint-based in silico models of Escherichia coli. J Bacteriol 185: 2692–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JL, Patel TR, Chen KH, Joyce AR, Applebee MK, Herring CD, Bui OT, Knight EM, Fong SS, Palsson BO (2006b) Systems approach to refining genome annotation. Proc Natl Acad Sci USA 103: 17480–17484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JL, Vo TD, Schilling CH, Palsson BO (2003) An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol 4: R54.1–R54.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, Mori H, Perna NT, Plunkett G III, Rudd KE, Serres MH, Thomas GH, Thomson NR, Wishart D, Wanner BL (2006) Escherichia coli K-12: a cooperatively developed annotation snapshot-2005. Nucleic Acids Res 34: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver S (1996) Bacterial resistances to toxic metal ions—a review. Gene 179: 9–19 [DOI] [PubMed] [Google Scholar]
- Stelling J (2004) Mathematical models in microbial systems biology. Curr Opin Microbiol 7: 513–518 [DOI] [PubMed] [Google Scholar]
- Thauer RK, Jungermann K, Decker K (1977) Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41: 100–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Horn PB, Backstrom AD, Stewart V, Begley TP (1993) Structural genes for thiamine biosynthetic enzymes (thiCEFGH) in Escherichia coli K-12. J Bacteriol 175: 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A, Boesch BW, Palsson BO (1993) Biochemical production capabilities of Escherichia coli. Biotechnol Bioeng 42: 59–73 [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BO (1993) Metabolic capabilities of Escherichia coli: I. Synthesis of biosynthetic precursors and cofactors. J Theor Biol 165: 477–502 [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BO (1995) Parametric sensitivity of stoichiometric flux balance models applied to wild-type Escherichia coli metabolism. Biotechnol Bioeng 45: 69–79 [DOI] [PubMed] [Google Scholar]
- Williams I, Frank L (1975) Improved chemical synthesis and enzymatic assay of delta-1-pyrroline-5-carboxylic acid. Anal Biochem 64: 85–97 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legend to Supplementary Material
Supplementary Text, Figures and Tables
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Supplementary Information 4
Supplementary Information 5
Supplementary Information 6