

Abstract
Single-base lesions in DNA are repaired predominantly by base excision repair (BER). DNA polymerase β (pol β) is the polymerase of choice in the preferred single-nucleotide BER pathway. The characteristic phenotype of mouse fibroblasts with a deletion of the pol β gene is moderate hypersensitivity to monofunctional alkylating agents, e.g., methyl methanesulfonate (MMS). Increased sensitivity to MMS is also seen in the absence of pol β partner proteins XRCC1 and PARP-1, and under conditions where BER efficiency is reduced by synthetic inhibitors. PARP activity plays a major role in protection against MMS-induced cytotoxicity, and cells treated with a combination of non-toxic concentrations of MMS and a PARP inhibitor undergo cell cycle arrest and die by a Chk1-dependent apoptotic pathway. Since BER-deficient cells and tumors are similarly hypersensitive to the clinically used chemotherapeutic methylating agent temozolomide, modulation of DNA damage-induced cell signaling pathways, as well as BER, are attractive targets for potentiating chemotherapy.
Keywords: Base excision repair, DNA polymerase β, XRCC1, PARP-1, PARP inhibitor
1. Introduction
1.1. Mammalian base excision repair (BER)
There are multiple and overlapping DNA repair pathways essential for maintaining the integrity of genomic DNA. The base excision repair (BER) pathway removes damaged base residues, nucleotides and abasic sites arising from a variety of exogenous and endogenous stressors [1]. These include spontaneous and enzymatic removal of bases through N-glycosidic bond cleavage, deamination of cytosine and adenine, oxidation of all four bases, and base alkylation by metabolites (e.g., S-adenosylmethionine) or environmental/therapeutic alkylating agents (e.g., nitrosamines and temozolomide (TMZ)). In addition, perturbations of nucleotide metabolism can lead to incorporation of unnatural bases, such as uracil and 8-oxoguanine, into DNA. The BER pathway is distinguished from other DNA repair pathways by the relatively short excision patch generated in double-stranded DNA after removal of the base lesion. In addition, DNA lesions repaired by BER are generally limited to base modifications that are similar in size and shape to the normal bases. The 6 core steps of BER are (i) base removal, (ii) strand incision, (iii) incised strand processing to enable DNA synthesis, (iv) DNA synthesis to fill the gap, (v) flap removal, and (vi) ligation.
Recent studies have shown that BER can occur by several sub-pathways. Repair may be initiated by a damage-specific DNA glycosylase belonging to one of two distinct classes. In the case of a monofunctional glycosylase (e.g., N-methylpurine- or uracil-DNA glycosylase), the damaged base is removed leading to formation of an abasic or apurinic/apyrimidinic (AP) site that can be cleaved on the 5′ side by AP endonuclease (APE). The resulting one nucleotide gap with 3′-OH and 5′-deoxyribose phosphate (dRP) termini is a suitable primer for single-nucleotide DNA synthesis. The dRP group is subsequently removed prior to DNA ligation. Bifunctional glycosylases (e.g., 8-oxoguanine- or thymine glycol-DNA glycosylase) have an associated lyase activity that nicks the DNA strand 3′ to the abasic site following base removal. Now polynucleotide kinase, or the 3′ diesterase activity of APE, is required to remove the 3′ blocking sugar phosphate group prior to DNA synthesis. When strand processing is not efficient, for example during repair of a reduced abasic site, an alternate sub-pathway of BER can generate repair patches greater than one nucleotide (long-patch BER). For BER studied in vitro, short and long-patch pathways can repair the same type of DNA lesion and operate in the same extract [2,3].
1.2. Role of DNA polymerase β (pol β) in BER
DNA polymerase β (pol β) is an X-family polymerase found in all vertebrate species as a ~39-kDa DNA polymerase lacking intrinsic nuclease activities, such as 3′- or 5′-exonuclease and endonuclease, but containing 5′-dRP lyase and AP lyase activities. Controlled proteolysis and structural studies have revealed that pol β is organized into two distinct domains (Fig. 1). The 31-kDa polymerase domain can be functionally and structurally separated into three subdomains (D, C, and N). The polymerase active site resides in the catalytic C–subdomain. The 8-kDa domain contributes dRP lyase activity and gapped DNA targeting through 5′-phosphate recognition [4,5]. Early inhibitor-based in vitro studies first implicated pol β in BER as a short gap repair DNA polymerase [6–8], and in vitro studies with purified pol β showed that it can completely fill short gaps like those found during BER [9–11]. Cell extract-based experiments further implicated pol β in single-nucleotide BER [12,13]. Later, a crude nuclear extract from bovine testis was found to be extremely efficient in uracil-initiated BER, removing U and inserting C opposite G. Here, the single nucleotide gap-filling DNA synthesis was catalyzed by pol β in the extract; only weak gap-filling DNA synthesis capacity was remaining when a neutralizing pol β antibody was added [14].
Fig. 1.

Schematic of domain organization of human pol β, XRCC1 and PARP-1 proteins. Pol β comprises an 8-kDa lyase domain, and a 31-kDa polymerase domain contributing DNA binding (D), catalytic (C) and nucleoside triphosphate binding (N) activities or subdomains as shown. Pol β interacts with the N-terminal domain (NTD) of XRCC1 as indicated by the dotted line. XRCC1 also interacts with DNA repair proteins AP endonuclease (APE) and polynucleotide kinase (PNK), and with DNA ligase III (lig IIIα) and PARP-1 via its C-terminal BRCT II and its centrally located BRCT I domains, respectively, as indicated. PARP-1 comprises an N-terminal DNA binding domain, an automodification (BRCT) domain, and a C-terminal catalytic domain. PARP-1 has two zinc finger (FI and FII) DNA nick detection motifs, and a nuclear localization (NLS) signal within the DNA binding domain. PARP-1 interacts with XRCC1 as indicated by the dotted line.
Now, it is known that pol β is the polymerase of choice, and contributes both DNA synthesis and dRP lyase activities to the overall single-nucleotide BER reaction. In a reconstituted system, removal of the dRP group has been shown to be the rate-limiting step [15]. However, it should be noted that pol β-deficient cell extracts are able to perform single-nucleotide BER of abasic sites and oxidative DNA lesions, therefore other polymerases can substitute for pol β [16–18]. Under conditions where a modified dRP group is not a substrate for the lyase activity of pol β, pol β-dependent strand displacement synthesis, in conjunction with flap endonuclease-1 (FEN1)-mediated flap cleavage, can conduct long-patch BER [19,20]. Pol β-independent long-patch repair has also been well characterized [2,21,22].
2. Mouse cellular models for BER deficiency
2.1. DNA polymerase β
Deletion of the pol β gene in the mouse is embryonic lethal indicating an essential role for pol β in embryonic development. However, pol β null (−/−) mouse embryonic fibroblasts are viable, and the characteristic phenotype of cells with this genotype is moderate hypersensitivity to monofunctional DNA alkylating agents such as methyl methanesulfonate (MMS) (Fig. 2) [23]. In general, methyl or ethyl substitution of DNA can involve first order “SN1-type” or second order “SN2-type” reactions. Compounds reacting by an SN1 mechanism show a greater preference for substitution at oxygen atoms (e.g., O6 of guanine), whereas those reacting by an SN2 mechanism show preference for nucleophilic nitrogens (e.g., N7 of guanine and N3 of adenine). In addition to MMS, pol β −/− fibroblasts are hypersensitive to other methylating (e.g., dimethyl sulfate; DMS) and ethylating agents (e.g., ethyl methanesulfonate; EMS) that react with DNA through an SN2 mechanism [24] (Table 1). Pol β −/− cells are generally more hypersensitive to methylating agents (e.g., methyl nitrosourea; MNU and N′-nitro-N-nitrosoguanidine; MNNG) than ethylating agents (e.g., ethyl nitrosourea; ENU) that react through an SN1 mechanism [24]. The lesser hypersensitivity of pol β-deficient cells to SN1 ethylating agents may be related to the comparatively lower level of 7-ethylguanine formation compared with 7-methylguanine (around 14% of total adduct vs. 70% [25]). In addition, it is clear that pol β −/− cells are less hypersensitive to a minor groove-specific 3-methyladenine-producing agent, methyl lexitropsin (Me-lex), than to MMS (Table 1) suggesting that the hypersensitivity is not due to inefficient repair of this particular methylated base [24]. Taken together, the results are consistent with alkylation at N7 guanine as the key cytotoxic N-alkyl adduct formed following exposure to monofunctional alkylating agents.
Fig. 2.

MMS hypersensitivity phenotype of BER-deficient mouse fibroblasts. The sensitivity of pol β (circles), XRCC1 (diamonds) and PARP-1 (triangles) −/− cells (open symbols) to a 1 h exposure to MMS is compared with that of their respective isogenic wild-type (+/+) cell lines (closed symbols). Cell sensitivity was determined by growth inhibition assays as described [3]. The sensitivity ratio of the isogenically matched +/+ and −/− cells is given in the table to the right. The cell variants have been described previously [23,32,49].
Table 1.
Hypersensitivity phenotypes associated with pol β gene deletion and inhibition of PARP activity
| DNA damaging agent | Sensitivity ratioa | References | Sensitization by PARP inhibitor?b | References |
|---|---|---|---|---|
| MMS (SN2) | 2.8 | [23,24,31,117] | extreme | [60,66] |
| DMS (SN2) | 3.1 | [24] | intermediate | [59] |
| EMS (SN2) | 2.3 | [23,24] | ?c | |
| MNU (SN1) | 3.8 | [23,24] | extreme | [66] |
| MNNG (SN1) | 4.1 | [23,24,117] | extreme | (unpublished) |
| TMZ (SN1) | 4.5 | [24] | extreme | [66,72,93,95] |
| ENU (SN1) | 1.8 | [24] | ? | |
| Me-lex | 1.4 | [24] | low | [74] |
| hmdUrd | 3.8 | [24] | extreme | [66,118] |
| hydrogen peroxide | 1.6d | [30,31] | low | [30,119] |
| peroxynitrite | 1.4d | [30] | low | [66] |
| bleomycin | ~2.0d | [30] | low | [66,120,121] |
| IR | 1.0 | [23,34] | low | [70,121–123] |
| UV | 1.0 | [3,23,117] | low | [66] |
| cisplatin | 1.0 | [30] | none | [60] |
| BCNU | 1.1 | [24] | none | [124,125] |
| HeCNU | 1.0 | [117] | ? |
Ratio of IC90 in pol β +/+ and −/− cells. Values here are those reported in the first reference listed.
Results utilizing repair-proficient cell lines.
Unknown.
In late-passage cells; no hypersensitivity observed in early cultures [30].
Toxicity of the clinically utilized methylating agent TMZ, as well as other SN1 methylators, is generally attributed to mismatch repair (MMR) of O6-methylguanine [26,27]. However, hypersensitivity to TMZ has been observed in pol β null BER-deficient cells [24] (Table 1). Clearly, in these cell lines with low methyl transferase activity, as well as in others deficient in MMR, the repair of N-methyl DNA adducts by BER is an important determinant of TMZ-mediated cytotoxicity.
Hypersensitivity to MMS can be rescued by transfection of cells with a pol β expression vector indicating the importance of BER in general, and more specifically of pol β, in the protection of cells against MMS-induced cytotoxicity [23]. The defect in uracil-initiated BER in cell extracts deficient in pol β is similarly reversed [23]. Variants of the pol β minigene were constructed where either the DNA polymerase activity or the dRP lyase activity was perturbed. Unexpectedly, it was found that the pol β DNA synthesis activity was not essential for resistance to MMS, whereas the dRP lyase activity was [28]. It was proposed that hypersensitivity of pol β −/− cells does not result from increased formation or removal of 7-methylguanine following MMS exposure [24], but rather reflects accumulation of cytotoxic repair intermediates, such as the 5′-dRP group, following removal of 7-methylguanine from DNA. In support of this conclusion, pol β-deficient cells are also hypersensitive to the thymidine analog, 5-hydroxymethyl-2′-deoxyuridine (hmdUrd) [24]. This agent is incorporated into cellular DNA and elicits cytotoxicity only once removed by glycosylase-initiated BER [29].
Initial studies showed that pol β −/− cells were not hypersensitive to oxidative stress agents that result in oxidative DNA damage [23]. This suggested that pol β is not of primary importance in BER of oxidized DNA, or that its activity can be fully complemented by another DNA polymerase. However, more recently it was discovered that pol β −/− mouse fibroblasts become increasingly sensitive to oxidative damaging agents (e.g., hydrogen peroxide and peroxynitrite) after time in culture. Late-passage cells demonstrate hypersensitivity when compared with the early-passage pol β null cells, and both early and late-passage wild-type fibroblasts [30] (Table 1). There are independent reports of pol β null cells demonstrating either hypersensitivity or a lack of hypersensitivity to hydrogen peroxide, but the age of the cells was not addressed [31,32]. It is likely that pol β is important in oxidative DNA repair but that, in the early-passage null cells, this is masked by an alternate detoxification or repair pathway that becomes down regulated as the cells age. Transfection of the late-passage pol β −/− cells with a pol β minigene reversed the hypersensitivity phenotype [30]. An in vitro assay system for oxidative (8-oxoguanine) DNA damage was used to compare the ability of early- and late-passage pol β null cell extracts to support the repair of this damage. Both types of extract were deficient in BER of the lesion, again indicating that the BER deficiency in the early-passage cells is masked. The low efficiency of single-nucleotide repair of 8-oxoguanine by pol β-deficient extracts had been reported previously [17,33].
Finally, pol β −/− cells demonstrate minimal or no hypersensitivity to ionizing radiation and UV exposure [23,34], or to cisplatin and chloroethylating nitrosoureas (Table 1). These agents produce cytotoxic DNA damage that is primarily repaired by pathways other than BER.
2.2. Other enzymes and partner proteins associated with BER
DNA glycosylases, as well as APE, are key enzymes in the initial steps of most BER sub-pathways. It has been suggested that the sequential steps of the BER process may be coordinated via specific interactions among component and other proteins [35]. For example, there is evidence for a protein-protein interaction between the oxidative base glycosylase NEIL1 and pol β [36]. An interaction between APE and pol β, resulting in stimulation of pol β-dependent dRP lyase activity, has also been reported [37]. Wild-type p53 interacts directly with APE and pol β, and stimulates BER in vitro by stabilizing the interaction between pol β and abasic DNA [38]. The Werner syndrome protein (WRN), heat shock protein 70, and the Rad9/Rad1/Hus1 (9-1-1) complex have all been reported to have a stimulatory effect on pol β activity [39–41]. NEIL1 also interacts with XRCC1 [36], and biochemical and NMR experiments have demonstrated a protein-protein interaction between pol β and the N-terminal domain of XRCC1 [42,43] (Fig. 1). Other pol β interactive proteins include ligase I, PCNA, APC and DNA-PKcs [44–47].
Mouse strains with targeted mutations in DNA glycosylases (e.g., 8-oxoguanine- or thymine glycol-DNA glycosylase) have been developed (see mouse strain data base [48]). However, those resulting in deletions of proteins involved in later stages of BER (e.g., APE, ligase I, FEN1) are embryonic lethal [48]. Similar to pol β and other late stage proteins, XRCC1 is essential for mouse embryo development [49]. Initial attempts to establish cell lines from the embryos were unsuccessful, however, it was possible to culture XRCC1 −/− mouse embryonic fibroblasts if they were also nullizygous for p53 [49]. XRCC1 genetic deficiency in mouse fibroblasts is associated with a greater cellular hypersensitivity to monofunctional alkylating agents than that observed in the absence of pol β (Fig. 2) [49]. This extreme hypersensitivity points to a requirement of XRCC1 protein for efficient BER, yet a biochemical role of XRCC1 in BER is not at all apparent since XRCC1 lacks enzymatic activity. It has been proposed that XRCC1 serves as a scaffold protein during BER reactions through its interactions with pol β, as well as those with PARP-1 and ligase III via is two BRCT domains [50–52] (Fig. 1), and APE and polynucleotide kinase (PNK) [53,54]. XRCC1−/− cells also exhibit a low level hypersensitivity to ionizing radiation [49] implicating its involvement in single-strand break, as well as base excision, repair [55].
PARP-1 is the first described member of a family of at least eighteen proteins with poly(ADP-ribosyl)ating activity [56]. It is an abundant nuclear enzyme, comprising an N-terminal DNA binding domain, an automodification domain and a C-terminal catalytic domain (Fig. 1). PARP-1 can detect and bind to DNA nicks and strand breaks. Upon binding to DNA, its activity is rapidly stimulated by as much as 500-fold and, using NAD+ as substrate, it poly(ADP-ribos)ylates numerous proteins including those involved in chromatin architecture and DNA metabolism. Of the family members, only PARP-1 and PARP-2 are known to be activated in this way in response to DNA damage, with PARP-1 being responsible for 90% of the activity. As a consequence of self poly(ADP-ribosyl)ation, PARP-1 loses its affinity for DNA, and is released from its binding site thus allowing repair to continue [57,58]. As long ago as 1980, it was proposed that PARP-1 plays a role in BER [59], but the exact mechanism of its involvement remains unclear. PARP-1 activity is induced by specific strand break intermediates of monofunctional glycosylase-initiated BER. Photoaffinity cross-linking studies with an oligonucleotide representing a BER intermediate demonstrated that PARP-1 was the most abundant labeling product [60]. In addition, this labeling of PARP-1 depended on the presence of the 5′-sugar phosphate group implicated in MMS-induced cytotoxicity. In its poly(ADP-ribosyl)ated state, PARP-1 recruits XRCC1 to the site of DNA damage suggesting that formation of repair foci is mediated by poly(ADP-ribose) [61,62].
PARP-1 −/− mouse fibroblasts are moderately hypersensitive to the methylating agents MMS (Fig. 2) and MNU, as well as to hydrogen peroxide and ionizing radiation [32,63–66], indicating the importance of PARP-1 protein in cellular resistance to these agents. In the case of MMS, the sensitivity ratio between PARP-1 +/+ and −/− cells is similar to that seen in the pol β cells (Fig. 2). It is interesting to note that mouse fibroblasts with a double knockout of PARP-1 and pol β are reported to be no more hypersensitive to MNU than cells with a single gene deletion [32]. The observed hypersensitivity of PARP-1 −/− cells to hmdUrd [66] is consistent with the idea that PARP-1 is activated in response to DNA strand breaks and with the proposal that sensitivity to hmdUrd results from accumulation of repair intermediates in cells with a deficiency in BER.
3. Role of PARP activity in protection against the cytotoxicity of DNA-damaging agents
Chemical inhibitors of PARP activity can enhance cell killing by agents that produce cytotoxic repair intermediates following initiation of BER. PARP-1 activity can also be inhibited in cells by overexpression of the trans-dominant PARP-1 DNA binding domain [67]. Many synthetic inhibitors are analogs of the nicotinamide moiety of NAD+. The crystal structure of the catalytic domain of PARP-1 complexed with the inhibitor 4-amino-1,8-naphthalimide (4-AN) shows that the inhibitor binds to the well-defined nicotinamide subsite of the NAD+-binding pocket of the enzyme, hydrogen bonding to the catalytic residue Glu988 [68] (Fig. 3). The degree of sensitization in cells is related to the potency of the PARP inhibition. Inhibitor studies in vitro have shown the IC50 for 4-AN and 3-aminobenzamide (3-AB) to be 0.18 and 33 μM, respectively [69] and studies in cells demonstrated that 4-AN is 1,000-fold more potent than 3-AB at inhibiting PARP activity [70].
Fig. 3.
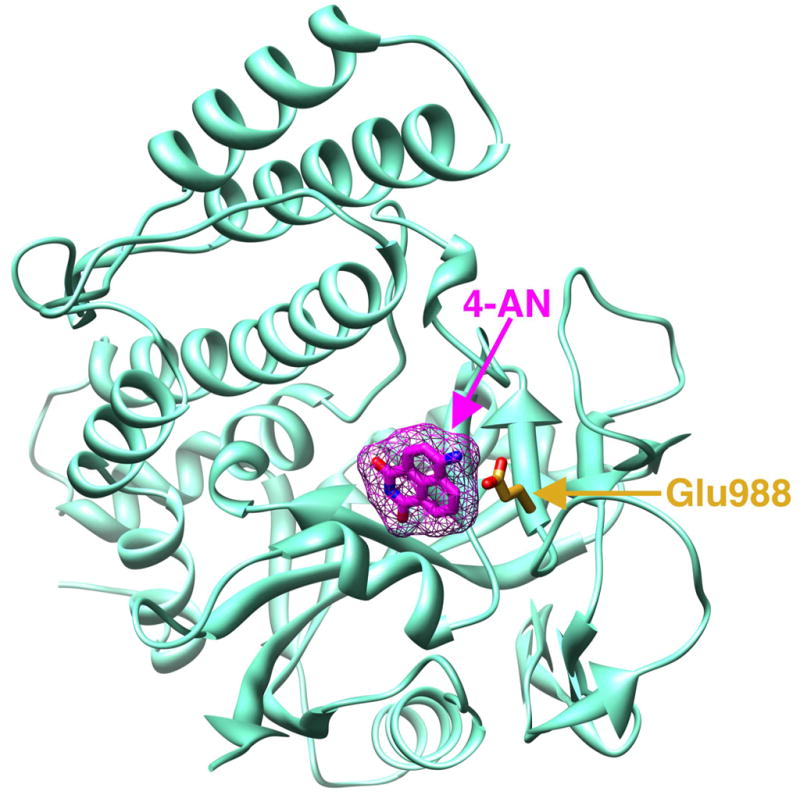
Crystal structure of the catalytic domain of PARP-1 in complex with the inhibitor 4-AN. Ribbon representation of the carboxyl-terminal catalytic domain of chicken PARP-1 complexed with 4-AN; accession code 2PAX [68]. The inhibitor (magenta) tightly binds to the nicotinamide portion of the NAD+ binding pocket and hydrogen bonds with the conserved active site residue Glu988 (orange). This figure was prepared in Chimera [116].
Both wild-type and pol β null cells are extremely sensitized to MMS by co-incubation with 4-AN [71] and the extent of sensitization is both time- and 4-AN dose-dependent [66]. Survival following exposure to MMS and utilizing the optimal 4-AN schedule (10 μM for 24 h) is shown in Figure 4. When comparing the IC90 for MMS, an ~ 40- and 80-fold sensitization is observed in pol β +/+ and −/− cells, respectively. Thus, under conditions where PARP activity is inhibited by 4-AN, the pol β null cells demonstrate almost twice the hypersensitivity to MMS than under control conditions (Fig. 4). The effect of 4-AN is compared with that obtained with the less potent inhibitors 3-AB and 1,5-dihydroxyisoquinoline (DIQ). For these two inhibitors, there was only a slightly enhanced sensitization in pol β −/− compared with pol β +/+ cells, therefore the sensitivity ratio of the two cell lines in the presence of the inhibitors remained more similar to that obtained in the absence of inhibitor (Fig. 4). Importantly, in the presence of all the inhibitors, the pol β null cell hypersensitivity to MMS is still observed. The results demonstrate that PARP activity is critical in protection of cells against MMS-induced DNA adducts both in the presence or absence of pol β-dependent repair pathways. Cells exposed to potent PARP inhibitors are more sensitive to MMS than are PARP-1 −/− cells [66]. In the absence of PARP-1, BER may be less efficient, hence the moderate MMS hypersensitivity. In the presence of an inhibitor, PARP-1 can still bind DNA and intermediates of BER, but, in the absence of poly(ADP-ribosyl)ation, binding of the protein to DNA, and disruption of BER, may be prolonged.
Fig. 4.
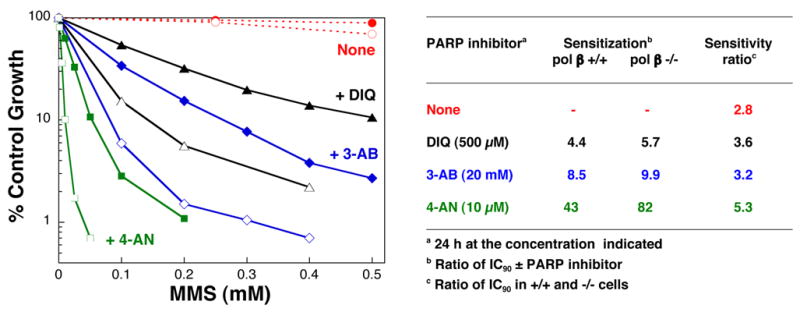
PARP inhibition results in sensitization to MMS. Shown is the sensitivity of pol β +/+ (closed symbols) and −/− cells (open symbols) to a 1 h exposure to MMS combined with a 24 h exposure to the PARP inhibitors 4-AN (squares), 3-AB (diamonds) and DIQ (triangles), as described [66]. The concentration of inhibitor used, its ability to potentiate MMS cytotoxicity in pol β +/+ and −/− cells, and the sensitivity ratio of pol β +/+ and −/− cells in the presence of the inhibitor are given in the table to the right. For all three inhibitors, the results with maximally effective concentrations are presented. The IC90 for MMS in pol β +/+ and −/− cells under the conditions used is 2.22 and 0.80 mM, respectively.
Extreme potentiation of the cytotoxicity of other monofunctional methylating agents, including SN1-type agents (e.g., MNU and TMZ) that produce significant O6-methyl- in addition to N-methyl-adducts, has also been documented (Table 1) [66,72]. In addition, dominant-negative inhibition of PARP-1 results in a marked decrease in cell survival following low doses of MNNG [73]. The extreme sensitization of pol β null cells to MNU by a PARP inhibitor [66] contrasts the data obtained in double knockout cells [32]. The results suggest that the absence of PARP-1 protein (PARP-1 −/− cells) and the inhibition of PARP activity (+ 4-AN) do not result in equivalent MNU hypersensitivity in a pol β −/− background. Interestingly, the degree of sensitization to Me-lex is considerably reduced when compared with MMS [74]. In contrast to 7-methylguanine which is cytotoxic only in the event of ineffective BER, the minor groove adduct, 3-methyadenine, is known to be a lethal replication blocking lesion in DNA [75]. Sensitization to hmdUrd is also seen in the presence of 4-AN [66] further substantiating our hypothesis that the hypersensitivity of the pol β null cells reflects accumulation of cytotoxic repair intermediates following removal of damaged bases from DNA. PARP-1 −/− cells, already demonstrating a hypersensitivity phenotype, are further sensitized to MMS and hmdUrd by 4-AN [66]. The data suggest a limited role for other proteins with PARP activity in addition to PARP-1 in protection against BER intermediate-induced cytotoxicity.
The results obtained with a combination of 4-AN and the oxidative stress agents (e.g., hydrogen peroxide) are very different. Wild-type, pol β null and PARP-1 −/− cells are slightly sensitized by 4-AN but this effect is extremely small compared with that seen with MMS [30,66]. Thus, there is a requirement for PARP activity in protection against hydrogen peroxide-induced cytotoxicity, but this role is not as important as in protection against MMS-induced cytotoxicity. The results emphasize that there are differences in repair of methylated and oxidative DNA lesions. For methylated DNA, repair is initiated by a monofunctional glycosylase resulting in formation of the 5′-dRP intermediate, whereas, for oxidized base lesions, the bifunctional glycosylases excise the damaged base without formation of the PARP-1 binding cytotoxic dRP intermediate. The minimal 4-AN-induced sensitization of cells to hydrogen peroxide is in contrast to the moderate hypersensitivity, similar to MMS, of PARP-1 −/−cells to the same agent [66]. These results are again consistent with the idea that the absence of PARP-1 protein (PARP-1 −/− cells) and the presence of inactivated PARP protein (PARP-1 wild-type + 4-AN) do not always result in an equivalent hypersensitivity phenotype.
4. Activation of cell signaling pathways as a result of inhibition of PARP activity
4.1. ATR and Chk1-dependent cell cycle arrest
As described above, PARP activity plays a major role in protection against the cytotoxicity induced by exposure of both pol β +/+ and −/− cells to MMS [66]. In wild-type fibroblasts treated with a lethal combination of MMS and 4-AN, a complete inhibition of DNA synthesis is apparent after 4 h and, by 24 h, all cells are arrested in S phase of the cell cycle [66] (Fig. 5A and B). Treatment with either MMS or 4-AN alone had no effect. Similar results were obtained in pol β −/− cells demonstrating the importance of PARP activity in prevention of cell cycle arrest in both the presence and absence of pol β-dependent repair pathways. In contrast to the results obtained in PARP-1-expressing cells, treatment of PARP-1 −/− fibroblasts with MMS + 4-AN did not result in inhibition of DNA synthesis at 4 h. Further, at later time points (longer than 16 h), the majority of the cells were arrested not in S phase, but in G2/M [66] (Fig. 5B). The results suggest there is a requirement for activity-inhibited PARP-1 protein in order to achieve both the DNA damage-induced perturbations in DNA synthesis and the S phase cell cycle arrest.
Fig. 5.

Cell cycle analysis of MMS + PARP inhibitor-treated mouse fibroblasts. (A) and (B) Shown is the proportion of cells in G0/G1 (black), S (red) and G2/M (blue) phases of the cell cycle. (A) Control untreated pol β +/+ cells are compared with cells treated with MMS (0.25 mM) + PARP inhibitor (4-AN; 10 μM) for the times indicated. (B) Pol β +/+, PARP-1 +/+ and PARP-1 −/− fibroblasts 24 h after exposure to MMS + 4-AN are compared with control untreated cells. (C) and (D) Wild-type cells were treated with MMS combined with 4-AN (10 μM continuously) in the presence (red) or absence (green) of caffeine (1 mM) as described [66]. Shown is the proportion of cells in (C) S phase (bromodeoxyuridine; BrdUrd positive) or (D) G2/M phase of the cell cycle compared with control untreated (blue) cells. Propidium iodide staining (a measure of DNA content) and BrdUrd incorporation (a measure of DNA synthesis) were analyzed by flow cytometry as described [66].
The S phase arrest observed following exposure of wild-type cells to a combination of MMS and 4-AN might be caused by checkpoint signaling in response to DNA damage. Alternatively, it may result from a passive mechanical block of DNA replication by inactive PARP protein irreversibly bound to damaged DNA; replication forks are known to be slowed or arrested by specific protein-DNA complexes [76] and to initiate checkpoint activation. Caffeine is known to inhibit the activity of both ataxia telangiectasia-mutated (ATM) and ATM and Rad3-related (ATR) upstream checkpoint kinases, and to override ATM- and ATR-dependent replication and DNA damage checkpoints [77,78]. Caffeine was found to have little effect on the early (at 4 h) MMS + 4-AN-induced inhibition of DNA synthesis in wild-type cells, but the synthesis block could not be maintained after 8 h (Fig. 5C). Further, by 16 - 24 h, addition of caffeine resulted in arrest of cells in G2/M rather than S phase (Fig. 5D). Since ATM activation is generally associated with double strand breaks resulting from irradiation damage, it is more likely that ATR is the signaling protein involved in the recognition of DNA damage associated with MMS treatment and inhibition of PARP activity. An ATR-mediated checkpoint regulates phosphorylation and activation of Chk1 [79], an essential effector kinase that plays a key role in both G2/M and S phase DNA damage and replication checkpoints [80,81]. Analysis of signaling pathways by western blotting of cell extracts revealed an activation of Chk1 following treatment with MMS and 4-AN. The appearance of phosphorylated Chk1 could be suppressed by caffeine [66]. Taken together, these data suggest that inhibition of PARP activity, and formation of activity inhibited PARP protein, result in sensitization to MMS through activation of a Chk1-and most likely an ATR-dependent S phase checkpoint.
4.2. Chk1-dependent apoptosis
Inhibition of PARP activity with 4-AN renders cells extremely sensitive to killing by even very low doses of MMS (i.e., levels that produce no cytotoxicity in the presence of normal PARP activity). When a sub-lethal concentration of 4-AN is combined with a non-toxic concentration of MMS, there is greater than 99% cell growth inhibition (Fig. 6A). A DNA fragmentation assay revealed the DNA laddering pattern characteristic of apoptosis, indicating that both wild-type and pol β null cells treated with the low dose MMS + 4-AN combination die by an apoptotic pathway [71]. Using flow cytometry, it was found that greater than 70% of treated wild-type cells are apoptotic between 48 and 72 h, with the appearance of very few necrotic cells (Fig. 6B). Channeling of cell death into an apoptotic pathway, in the absence of significant cell death by necrosis, suggests there may be a signaling pathway that activates programmed cell death in response to the inhibition of PARP activity. However, addition of caffeine did not significantly affect the mechanism of cell death or the extent of cell growth inhibition. In contrast, the Chk1 inhibitor, UCN-01, was able to protect cells against MMS + 4-AN-mediated apoptosis resulting in an increased proportion of viable cells [71]. The decrease in apoptotic cell death as a result of UCN-01 exposure was linked with an abrogation of MMS + 4-AN-induced mitochondrial membrane depolarization, a phenotype associated with most models of apoptosis [82,83]. The results are consistent with a role for an ATR-and Chk1-dependent signaling pathway in the cellular response to PARP inhibition after DNA damage by MMS. Combination of the methylating triazene TMZ with the PARP inhibitor 3-AB results in an increased percentage of cells with hypodiploid DNA content, an indicator of cell death by apoptosis, relative to treatment with TMZ alone [84,85]. It is not known if Chk1-dependent signaling is involved under these conditions.
Fig. 6.
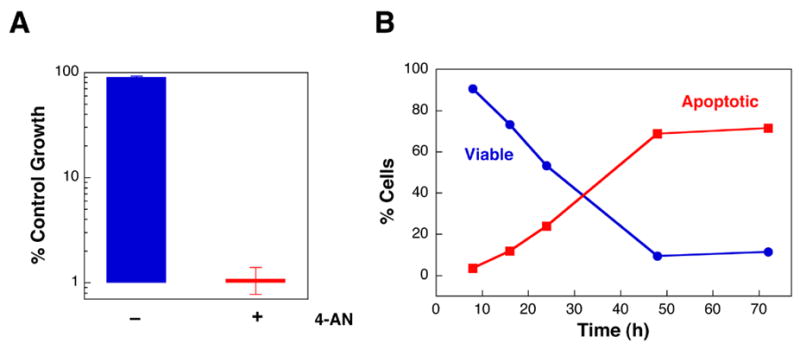
Apoptotic cell death following treatment of cells with MMS and PARP inhibitor. Wild-type mouse fibroblasts were exposed to a sub-lethal concentration of MMS (0.25 mM) in the presence or absence of a non-toxic level of PARP inhibitor (4-AN; 10 μM continuously). (A) The combination of MMS + 4-AN results in at least 99% cell growth inhibition [71]. (B) Time course of disappearance of intact viable cells and appearance of apoptotic cells following treatment with the MMS + 4-AN combination. The mode of cell death was determined by flow cytometry after staining with annexin-V and propidium iodide as described [71].
5. Influence of the BER inhibitor, methoxyamine, on methylation-induced cytotoxicity
As discussed above, the 5′-dRP group formed during monofunctional glycosylase-initiated single-nucleotide BER, and implicated in MMS-induced cytotoxicity, is removed by the dRP lyase activity of pol β. Methoxyamine (MX) is a chemical that can react with the aldehydic C1’ atom of the abasic site rendering it refractory to the β-elimination step of the dRP lyase mechanism [86,87]. In addition, the MX-adducted AP site is relatively resistant to enzymatic removal by APE [3]. Thus, MX effectively blocks single-nucleotide BER, however, repair may occur through alternate long-patch pathways [88]. Exposure to MX sensitizes pol β wild-type, but not pol β null cells, to the cytotoxic effects of MMS and MNU [3] (Fig. 7A). Further, MX is able to sensitize pol β null cells expressing either full-length or the 8-kDa domain of pol β. MX has no effect on UV-induced cytotoxicity [3]. The data are consistent with the idea that MX specifically blocks pol β-dependent dRP lyase activity and hence single-nucleotide BER. In other studies, MX significantly increased the number of TMZ-induced DNA single strand breaks in colon cancer cells and also potentiated the cytotoxic effects of TMZ in both MMR proficient and deficient cell lines [89,90].
Fig. 7.
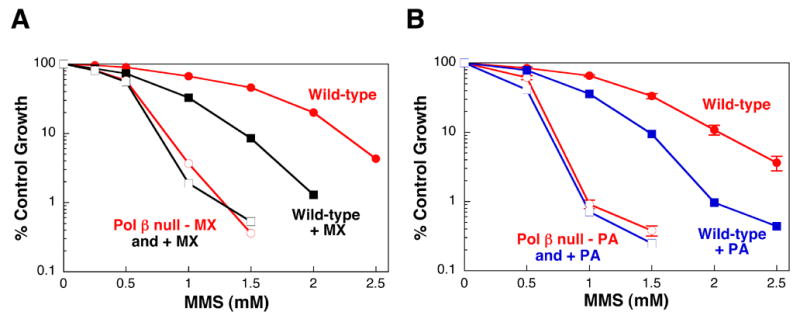
Effect of inhibition of BER on cellular sensitivity to MMS. Wild-type (closed symbols) and pol β null (open symbols) mouse fibroblasts were treated with a range of MMS concentrations for 1 h in the presence (squares) or absence (circles) of (A) MX or (B) PA. (A) Incubation with MX (30 mM) was for a total of 4 h as described [3]. (B) Cells were pretreated with PA (300 μM) for 7 h and the incubation was continued following MMS for a total of 24 h [92]. Cell sensitivity was determined by growth inhibition assays [3].
6. Small molecule inhibitors of pol β identified by NMR
The natural product, koetjapic acid, has been described as a pol β inhibitor [91]. The binding site for this compound was localized by NMR chemical shift mapping to the region of pol β 8-kDa domain that provides the dRP lyase activity of the enzyme [92]. The NMR binding study was extended to include structurally similar synthetic terpenoids and finally other synthetic compounds containing aromatic, or other hydrophobicity, in combination with two carboxylate groups. Dissociation constants for binding were determined for these compounds, and the binding sites on the pol β 8-kDa domain were mapped. Nine of these compounds with high binding affinities were further characterized for their ability to potentiate MMS-induced cytotoxicity. The working hypothesis was that, if the mechanism of sensitization is pol β specific, sensitization would be seen in wild-type, but not in pol β null cells, and the resulting sensitivity to MMS in the wild-type cells would mimic that observed in pol β null cells. Treatment of cells with the most active and specific compound identified, pamoic acid (PA), resulted in sizeable sensitization of wild-type cells to MMS with only a negligible effect in pol β null cells (Fig. 7B) [92]. In addition, PA was able to sensitize pol β null cells expressing either full-length or the 8-kDa domain of pol β. This is consistent with the observation that it is the dRP lyase activity of pol β found in the 8-kDa domain that is required to protect against MMS-induced cytotoxic repair intermediates. PA was also found to be inhibitory for activity in in vitro assays for the dRP lyase and DNA polymerase activities of pol β. The results suggest that binding of PA to the 8-kDa domain of pol β results in inhibition of both the major activities associated with the full length protein.
7. Potential role of BER modulators in chemotherapy
Since PARP-1 facilitates BER, and inhibition of its activity sensitizes cells to certain DNA damaging agents (Table 1), it is an attractive target for improving anticancer chemotherapy. There is considerable interest in developing more potent nontoxic PARP inhibitors with the possibility for use in a clinical setting. As described above, PARP inhibition results in cellular sensitization to the chemotherapeutic methylating agent TMZ, particularly in MMR-deficient cells, just as it does to the model methylating agents MMS and MNU [66,90,93–96]. Further, preclinical studies have demonstrated that novel PARP inhibitors can enhance the antitumor activity of TMZ against mouse tumors and human colon and glioma xenograft models [72,97–99]. TMZ is effective in the treatment of astrocytoma and glioblastoma since it is able to cross the blood-brain barrier [100,101], and combination regimens have been developed. A phase 1 trial of a PARP inhibitor in combination with oral TMZ has recently been completed [102].
Data with MMS in mouse fibroblasts suggests that equivalent cell kill can be obtained using a high concentration of a methylating agent alone, or a low concentration combined with a PARP inhibitor [71]. After exposure of cells to high concentrations of MMS, PARP-1 will become extremely activated. In an effort to resynthesize its substrate NAD+, cellular ATP levels also become depleted, and cell death is by necrosis [71]. With the MMS and PARP inhibitor combination, cellular ATP levels are conserved and cells can die by ATP-dependent apoptosis [71]. In cancer chemotherapy, combination with a PARP inhibitor may be advantageous, since a considerably lower dose of chemotherapeutic agent can be utilized and, in addition, it is more likely that cell death will be by apoptosis [71]. Apoptosis is preferred in vivo since apoptotic cells undergo phagocytosis by macrophages, thus disappearing without causing inflammation. In contrast, necrotic cell death induces an inflammatory response when cells swell, lose the integrity of their plasma membrane and release their cellular contents. Since apoptotic cell death following treatment with MMS and 4-AN involves activation of Chk1-dependent signaling, it is interesting to consider modulation of DNA damage-induced cell signaling as a target for potentiating chemotherapy.
It has been demonstrated that MX can sensitize pol β-expressing cell lines to methylating agents. The data with TMZ combined with MX clearly confirm that BER has a protective effect against this agent in tumor cell lines [89,90]. Follow up studies demonstrated a MX-induced enhancement of TMZ in human colon cancer xenografts [103]. It is notable that MX potentiated the antitumor effects of TMZ in both MMR-proficient and -deficient tumors. The authors conclude that BER may be a useful pharmacological target through which tumor cells can be sensitized to alkylating therapeutic agents. A phase I clinical trial in humans has been initiated [102].
In recent years, there has been considerable effort towards isolating and identifying natural product inhibitors of pol β. Active agents, as ascertained by inhibition of pol β DNA synthesis activity in in vitro assays, have been discovered in extracts from bacteria, marine organisms, fungi and higher plants [91,104–109]. Many of the “pol β inhibitory” compounds (glycoglycerolipids, sulfolipids, triterpenoids, phenalenone derivatives) have been found to inhibit other polymerases as well (e.g., DNA polymerases α, γ and λ, and HIV-1 reverse transcriptase) [110–115]. The primary aim given for these studies is the possibility of inhibiting repair of DNA adducts formed after treatment with DNA-damaging anticancer agents, and thus potentiating chemotherapeutic treatments. As noted above, a synthetic small molecule inhibitor, PA, was found to bind to the 8-kDa domain of pol β resulting in inhibition of both activities associated with this protein [92]. Further, most likely as a result of this inhibitory activity in cells, PA can sensitize cells with pol β-dependent repair activity to MMS [92]. Importantly, these results illustrate that NMR mapping techniques can be an effective in the selection of small molecule enzyme inhibitors including those with potential for use in a clinical setting.
8. Final words
The results summarized here illustrate that we have gained considerable insight into the importance of pol β, PARP-1 and XRCC1 in the survival of mouse fibroblasts following specific types of genotoxic stress. When these proteins are functionally eliminated either by targeted gene deletion or the use of selective inhibitors, cells become hypersensitive to agents, such as the DNA methylator MMS, that are known to induce BER. This is taken as an indication of BER roles for pol β, PARP-1 and XRCC1, and such an assignment is entirely consistent with biochemical studies of these proteins in purified form. It is important to consider that activity-inhibited proteins may still retain their ability to interact with damaged DNA and BER partner proteins in the cell, and therefore maintain a role in BER coordination. For PARP-1, deletion of the protein and inhibition of its activity do not always result in an equivalent hypersensitivity phenotype.
The genetic and inhibitor studies available to date have not given a clear picture of whether pol β, PARP-1, and XRCC1 are additive or epistatic in protection against monofunctional methylating agent-induced cytotoxicity. Pol β gene deletion and inhibitor-induced PARP inactivation, the two BER deficiency hypersensitivity phenotypes summarized in detail in Table 1, are additive since PARP inhibitors sensitize both pol β +/+ and −/− cells to specific agents producing damage repaired by BER. Published studies with PARP-1 −/− pol β −/−cells are so far limited, but suggest that the hypersensitivity phenotypes as a result of deletion of both genes is not additive. Double gene knockouts of XRCC1 and PARP-1, or XRCC1 and pol β have not been reported. To our knowledge, the effect of PARP, BER or pol β inhibitors in XRCC1 −/−, or the effect of BER or pol β inhibitors in PARP-1 −/− mouse fibroblasts has not been explored. There is a need for further studies to investigate these questions. In addition, it should be interesting in the future to more thoroughly examine the linkages between signaling for cell death/DNA damage checkpoint activation, and elimination of the individual steps of BER.
Acknowledgments
We thank our colleagues in the DNA Repair and Nucleic Acid Enzymology group for their contributions. In particular we thank Dr. William A. Beard for critical reading of the manuscript and help with figure preparation and Jennifer Myers for editorial assistance. This research was supported by the Intramural Research Program of the NIH, and NIEHS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson SH. Mammalian base excision repair and DNA polymerase β. Mutat Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 2.Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- 3.Horton JK, Prasad R, Hou E, Wilson SH. Protection against methylation-induced cytotoxicity by DNA polymerase β-dependent long patch base excision repair. J Biol Chem. 2000;275:2211–2218. doi: 10.1074/jbc.275.3.2211. [DOI] [PubMed] [Google Scholar]
- 4.Beard WA, Wilson SH. Purification and domain-mapping of mammalian DNA polymerase β. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 5.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 6.Lindahl T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog Nucleic Acid Res Mol Biol. 1979;22:135–192. doi: 10.1016/s0079-6603(08)60800-4. [DOI] [PubMed] [Google Scholar]
- 7.Wallace S. AP endonucleases and DNA glycosylases that recognize oxidative DNA damage. Environ Mol Mutagen. 1988;12:431–477. doi: 10.1002/em.2860120411. [DOI] [PubMed] [Google Scholar]
- 8.Perrino FW, Loeb LA. Animal cell DNA polymerases in DNA repair. Mutat Res. 1990;236:289–300. doi: 10.1016/0921-8777(90)90012-t. [DOI] [PubMed] [Google Scholar]
- 9.Randahl H, Elliott GC, Linn S. DNA-repair reactions by purified HeLa DNA polymerases and exonucleases. J Biol Chem. 1988;263:12228–12234. [PubMed] [Google Scholar]
- 10.Mosbaugh DW, Linn S. Excision repair and DNA synthesis with a combination of HeLa DNA polymerase β and DNase V. J Biol Chem. 1983;258:108–118. [PubMed] [Google Scholar]
- 11.Singhal RK, Wilson SH. Short gap-filling synthesis by DNA polymerase β is processive. J Biol Chem. 1993;268:15906–15911. [PubMed] [Google Scholar]
- 12.Wiebauer K, Jiricny J. Mismatch-specific thymine DNA glycosylase and DNA polymerase β mediate the correction of G.T mispairs in nuclear extracts from human cells. Proc Natl Acad Sci USA. 1990;87:5842–5845. doi: 10.1073/pnas.87.15.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dianov G, Price A, Lindahl T. Generation of single-nucleotide repair patches following excision of uracil residues from DNA. Mol Cell Biol. 1992;12:1605–1612. doi: 10.1128/mcb.12.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singhal RK, Prasad R, Wilson SH. DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testes nuclear extract. J Biol Chem. 1995;270:949–957. doi: 10.1074/jbc.270.2.949. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava DK, Vande Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 16.Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hübscher U, Dogliotti E. Mammalian base excision repair by DNA polymerases δ and ε. Oncogene. 1998;17:835–843. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- 17.Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- 18.Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. DNA polymerase λ mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J Biol Chem. 2005;280:18469–18475. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- 19.Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase β mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase β and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- 21.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 22.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 23.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 24.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair. 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 25.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 26.Newlands ES, Stevens MFG, Wedge SR, Wheelhouse RT, Brock C. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev. 1997;23:35–61. doi: 10.1016/s0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- 27.D’Atri S, Tentori L, Lacal PM, Graziani G, Pagani E, Benincasa E, Zambruno G, Bonmassar E, Jiricny J. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol Pharmacol. 1998;54:334–341. doi: 10.1124/mol.54.2.334. [DOI] [PubMed] [Google Scholar]
- 28.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 29.Boorstein RJ, Chiu LN, Teebor GW. A mammalian cell line deficient in activity of the DNA repair enzyme 5-hydroxymethyluracil-DNA glycosylase is resistant to the toxic effects of the thymidine analog 5-hydroxymethyl-2′-deoxyuridine. Mol Cell Biol. 1992;12:5536–5540. doi: 10.1128/mcb.12.12.5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton JK, Baker A, Vande Berg BJ, Sobol RW, Wilson SH. Involvement of DNA polymerase β in protection against the cytotoxicity of oxidative damage. DNA Repair. 2002;1:317–333. doi: 10.1016/s1568-7864(02)00008-3. [DOI] [PubMed] [Google Scholar]
- 31.Fortini P, Pascucci B, Belisario F, Dogliotti E. DNA polymerase β is required for efficient DNA strand break repair induced by methyl methanesulfonate but not by hydrogen peroxide. Nucleic Acids Res. 2000;28:3040–3046. doi: 10.1093/nar/28.16.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dantzer F, de la Rubia G, Ménissier-de Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 33.Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J Biol Chem. 1999;274:15230–15236. doi: 10.1074/jbc.274.21.15230. [DOI] [PubMed] [Google Scholar]
- 34.Raaphorst GP, Ng CE, Yang DP. Comparison of response to radiation, hyperthermia and cisplatin in parental and polymerase β knockout cells. Int J Hyperthermia. 2002;18:33–39. doi: 10.1080/02656730110072352. [DOI] [PubMed] [Google Scholar]
- 35.Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 36.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell Biol. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 37.Bennett RAO, Wilson DM, III, Wong D, Demple B. Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc Natl Acad Sci USA. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. EMBO J. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrigan JA, Opresko PL, von Kobbe C, Kedar PS, Prasad R, Wilson SH, Bohr VA. The Werner syndrome protein stimulates DNA polymerase β strand displacement synthesis via its helicase activity. J Biol Chem. 2003;278:22686–22695. doi: 10.1074/jbc.M213103200. [DOI] [PubMed] [Google Scholar]
- 40.Mendez F, Kozin E, Bases R. Heat shock protein 70 stimulation of the deoxyribonucleic acid base excision repair enzyme polymerase β. Cell Stress Chaperones. 2003;8:153–161. doi: 10.1379/1466-1268(2003)008<0153:hspsot>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toueille M, El-Andaloussi N, Frouin I, Freire R, Funk D, Shevelev I, Friedrich-Heineken E, Villani G, Hottiger MO, Hübscher U. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase β and increases its DNA substrate utilisation efficiency: implications for DNA repair. Nucleic Acids Res. 2004;32:3316–3324. doi: 10.1093/nar/gkh652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marintchev A, Robertson A, Dimitriadis EK, Prasad R, Wilson SH, Mullen GP. Domain specific interaction in the XRCC1-DNA polymerase β complex. Nucleic Acids Res. 2000;28:2049–2059. doi: 10.1093/nar/28.10.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gryk MR, Marintchev A, Maciejewski MW, Robertson A, Wilson SH, Mullen GP. Mapping of the interaction interface of DNA polymerase β with XRCC1. Structure. 2002;10:1709–1720. doi: 10.1016/s0969-2126(02)00908-5. [DOI] [PubMed] [Google Scholar]
- 44.Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH. Specific interaction of DNA polymerase β and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J Biol Chem. 1996;271:16000–16007. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- 45.Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J Biol Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- 46.Narayan S, Jaiswal AS, Balusu R. Tumor suppressor APC blocks DNA polymerase β-dependent strand displacement synthesis during long patch but not short patch base excision repair and increases sensitivity to methylmethane sulfonate. J Biol Chem. 2005;280:6942–6949. doi: 10.1074/jbc.M409200200. [DOI] [PubMed] [Google Scholar]
- 47.Niimi N, Sugo N, Aratani Y, Koyama H. Genetic interaction between DNA polymerase β and DNA-PKcs in embryogenesis and neurogenesis. Cell Death Differ. 2005;12:184–191. doi: 10.1038/sj.cdd.4401543. [DOI] [PubMed] [Google Scholar]
- 48.Friedberg EC, Meira LB. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair. 2006;5:189–209. doi: 10.1016/j.dnarep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, Cleaver JE, Pedersen RA. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 50.Masson M, Niedergang C, Schreiber V, Muller S, Ménissier-de Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor RM, Thistlethwaite A, Caldecott KW. Central role for the XRCC1 BRCT I domain in mammalian DNA single-strand break repair. Mol Cell Biol. 2002;22:2556–2563. doi: 10.1128/MCB.22.8.2556-2563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor RM, Wickstead B, Cronin S, Caldecott KW. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr Biol. 1998;8:877–880. doi: 10.1016/s0960-9822(07)00350-8. [DOI] [PubMed] [Google Scholar]
- 53.Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 55.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. 2003:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 56.Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 57.Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 58.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 59.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283:593–596. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- 60.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 61.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okano S, Lan L, Caldecott KW, Mori T, Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol. 2003;23:3974–3981. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trucco C, Oliver FJ, de Murcia G, Ménissier-de Murcia J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998;26:2644–2649. doi: 10.1093/nar/26.11.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Ménissier-de Murcia J, de Murcia G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999;81:69–75. doi: 10.1016/s0300-9084(99)80040-6. [DOI] [PubMed] [Google Scholar]
- 65.Fernet M, Ponette V, Deniaud-Alexandre E, Ménissier-de Murcia J, De Murcia G, Giocanti N, Megnin-Chanet F, Favaudon V. Poly(ADP-ribose) polymerase, a major determinant of early cell response to ionizing radiation. Int J Radiat Biol. 2000;76:1621–1629. doi: 10.1080/09553000050201118. [DOI] [PubMed] [Google Scholar]
- 66.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest following DNA methylating agent exposure. J Biol Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 67.Molinete M, Vermeulen W, Burkle A, Ménissier-de Murcia J, Kupper JH, Hoeijmakers JH, de Murcia G. Overproduction of the poly(ADP-ribose) polymerase DNA-binding domain blocks alkylation-induced DNA repair synthesis in mammalian cells. EMBO J. 1993;12:2109–2117. doi: 10.1002/j.1460-2075.1993.tb05859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruf A, de Murcia G, Schulz GE. Inhibitor and NAD+ binding to poly(ADP-ribose) polymerase as derived from crystal structures and homology modeling. Biochemistry. 1998;37:3893–3900. doi: 10.1021/bi972383s. [DOI] [PubMed] [Google Scholar]
- 69.Banasik M, Komura H, Shimoyama M, Ueda K. Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J Biol Chem. 1992;267:1569–1575. [PubMed] [Google Scholar]
- 70.Schlicker A, Peschke P, Burkle A, Hahn EW, Kim JH. 4-Amino-1,8-naphthalimide: a novel inhibitor of poly(ADP-ribose) polymerase and radiation sensitizer. Int J Radiat Biol. 1999;75:91–100. doi: 10.1080/095530099140843. [DOI] [PubMed] [Google Scholar]
- 71.Horton JK, Stefanick DF, Wilson SH. Involvement of poly(ADP-ribose) polymerase activity in regulating Chk1-dependent apoptotic cell death. DNA Repair. 2005;4:1111–1120. doi: 10.1016/j.dnarep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 72.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, Durkacz BW, Hostomsky Z, Kumpf RA, Kyle S, Li J, Maegley K, Newell DR, Notarianni E, Stratford IJ, Skalitzky D, Thomas HD, Wang LZ, Webber SE, Williams KJ, Curtin NJ. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 73.Schreiber V, Hunting D, Trucco C, Gowans B, Grunwald D, de Murcia G, Ménisier-de Murcia J. A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc Natl Acad Sci USA. 1995;92:4753–4757. doi: 10.1073/pnas.92.11.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tentori L, Portarena I, Vernole P, Gold B, Graziani G. Apoptotic and genotoxic effects of a methyl sulfonate ester that selectively generates N3-methyladenine and poly(ADP-ribose) polymerase inhibitors in normal peripheral blood lymphocytes. Cancer Chemother Pharmacol. 2002;49:217–224. doi: 10.1007/s00280-001-0409-z. [DOI] [PubMed] [Google Scholar]
- 75.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 76.Hyrien O. Mechanisms and consequences of replication fork arrest. Biochimie. 2000;82:5–17. doi: 10.1016/s0300-9084(00)00344-8. [DOI] [PubMed] [Google Scholar]
- 77.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 78.Cortez D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem. 2003;278:37139–37145. doi: 10.1074/jbc.M307088200. [DOI] [PubMed] [Google Scholar]
- 79.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–14459. [PMC free article] [PubMed] [Google Scholar]
- 81.Chen Y, Sanchez Y. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair. 2004;3:1025–1032. doi: 10.1016/j.dnarep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 82.Castedo M, Ferri K, Roumier T, Metivier D, Zamzami N, Kroemer G. Quantitation of mitochondrial alterations associated with apoptosis. J Immunol Methods. 2002;265:39–47. doi: 10.1016/s0022-1759(02)00069-8. [DOI] [PubMed] [Google Scholar]
- 83.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis. 2003:115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- 84.Tentori L, Turriziani M, Franco D, Serafino A, Levati L, Roy R, Bonmassar E, Graziani G. Treatment with temozolomide and poly(ADP-ribose) polymerase inhibitors induces early apoptosis and increases base excision repair gene transcripts in leukemic cells resistant to triazene compounds. Leukemia. 1999;13:901–909. doi: 10.1038/sj.leu.2401423. [DOI] [PubMed] [Google Scholar]
- 85.Tentori L, Portarena I, Bonmassar E, Graziani G. Combined effects of adenovirus-mediated wild-type p53 transduction, temozolomide and poly (ADP-ribose) polymerase inhibitor in mismatch repair deficient and non-proliferating tumor cells. Cell Death Differ. 2001;8:457–469. doi: 10.1038/sj.cdd.4400832. [DOI] [PubMed] [Google Scholar]
- 86.Talpaert-Borle M, Liuzzi M. Reaction of apurinic/apyrimidinic sites with [14C]methoxyamine. A method for the quantitative assay of AP sites in DNA. Biochim Biophys Acta. 1983;740:410–416. doi: 10.1016/0167-4781(83)90089-1. [DOI] [PubMed] [Google Scholar]
- 87.Liuzzi M, Talpaert-Borle M. A new approach to the study of the base-excision repair pathway using methoxyamine. J Biol Chem. 1985;260:5252–5258. [PubMed] [Google Scholar]
- 88.Frosina G, Fortini P, Rossi O, Carrozzino F, Abbondandolo A, Dogliotti E. Repair of abasic sites by mammalian cell extracts. Biochem J. 1994;304:699–705. doi: 10.1042/bj3040699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taverna P, Liu L, Hwang HS, Hanson AJ, Kinsella TJ, Gerson SL. Methoxyamine potentiates DNA single strand breaks and double strand breaks induced by temozolomide in colon cancer cells. Mutat Res. 2001;485:269–281. doi: 10.1016/s0921-8777(01)00076-3. [DOI] [PubMed] [Google Scholar]
- 90.Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL. Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clin Cancer Res. 1999;5:2908–2917. [PubMed] [Google Scholar]
- 91.Sun DA, Starck SR, Locke EP, Hecht SM. DNA polymerase β inhibitors from Sandoricum koetjape. J Nat Prod. 1999;62:1110–1113. doi: 10.1021/np990104r. [DOI] [PubMed] [Google Scholar]
- 92.Hu HY, Horton JK, Gryk MR, Prasad R, Naron JM, Sun DA, Hecht SM, Wilson SH, Mullen GP. Identification of small molecule synthetic inhibitors of DNA polymerase β by NMR chemical shift mapping. J Biol Chem. 2004;279:39736–39744. doi: 10.1074/jbc.M402842200. [DOI] [PubMed] [Google Scholar]
- 93.Boulton S, Pemberton LC, Porteous JK, Curtin NJ, Griffin RJ, Golding BT, Durkacz BW. Potentiation of temozolomide-induced cytotoxicity: a comparative study of the biological effects of poly(ADP-ribose) polymerase inhibitors. Br J Cancer. 1995;72:849–856. doi: 10.1038/bjc.1995.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, Durkacz BW, Hostomsky Z, Newell DR. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res. 2000;6:2860–2867. [PubMed] [Google Scholar]
- 95.Tentori L, Portarena I, Torino F, Scerrati M, Navarra P, Graziani G. Poly(ADP-ribose) polymerase inhibitor increases growth inhibition and reduces G(2)/M cell accumulation induced by temozolomide in malignant glioma cells. Glia. 2002;40:44–54. doi: 10.1002/glia.10113. [DOI] [PubMed] [Google Scholar]
- 96.Calabrese CR, Batey MA, Thomas HD, Durkacz BW, Wang LZ, Kyle S, Skalitzky D, Li J, Zhang C, Boritzki T, Maegley K, Calvert AH, Hostomsky Z, Newell DR, Curtin NJ. Identification of potent nontoxic poly(ADP-Ribose) polymerase-1 inhibitors: chemopotentiation and pharmacological studies. Clin Cancer Res. 2003;9:2711–2718. [PubMed] [Google Scholar]
- 97.Tentori L, Leonetti C, Scarsella M, d’Amati G, Portarena I, Zupi G, Bonmassar E, Graziani G. Combined treatment with temozolomide and poly(ADP-ribose) polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site. Blood. 2002;99:2241–2244. doi: 10.1182/blood.v99.6.2241. [DOI] [PubMed] [Google Scholar]
- 98.Tentori L, Leonetti C, Scarsella M, D’Amati G, Vergati M, Portarena I, Xu W, Kalish V, Zupi G, Zhang J, Graziani G. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003;9:5370–5379. [PubMed] [Google Scholar]
- 99.Cheng CL, Johnson SP, Keir ST, Quinn JA, Ali-Osman F, Szabo C, Li H, Salzman AL, Dolan ME, Modrich P, Bigner DD, Friedman HS. Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol Cancer Ther. 2005;4:1364–1368. doi: 10.1158/1535-7163.MCT-05-0128. [DOI] [PubMed] [Google Scholar]
- 100.Yung WK, Prados MD, Yaya-Tur R, Rosenfeld SS, Brada M, Friedman HS, Albright R, Olson J, Chang SM, O’Neill AM, Friedman AH, Bruner J, Yue N, Dugan M, Zaknoen S, Levin VA. Multicenter phase II trial of temozolomide in patients with anaplastic astrocytoma or anaplastic oligoastrocytoma at first relapse. Temodal Brain Tumor Group. J Clin Oncol. 1999;17:2762–2771. doi: 10.1200/JCO.1999.17.9.2762. [DOI] [PubMed] [Google Scholar]
- 101.Yung WK, Albright RE, Olson J, Fredericks R, Fink K, Prados MD, Brada M, Spence A, Hohl RJ, Shapiro W, Glantz M, Greenberg H, Selker RG, Vick NA, Rampling R, Friedman H, Phillips P, Bruner J, Yue N, Osoba D, Zaknoen S, Levin VA. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83:588–593. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Madhusudan S, Middleton MR. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat Rev. 2005;31:603–617. doi: 10.1016/j.ctrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 103.Liu L, Nakatsuru Y, Gerson SL. Base excision repair as a therapeutic target in colon cancer. Clin Cancer Res. 2002;8:2985–2991. [PubMed] [Google Scholar]
- 104.Chen J, Zhang YH, Wang LK, Sucheck SJ, Snow AM, Hecht SM. Inhibitors of DNA polymerase β from Schoepfia californica. J Chem Soc Chem Commun. 1998:2769–2770. [Google Scholar]
- 105.Deng JZ, Sun DA, Starck SR, Hecht SM, Cerny RL, Engen JR. Chrysochlamic acid, a new diterpenoid-substituted quinol from Chrysochlamys ulei that inhibits DNA polymerase β. J Chem Soc Perkin Trans. 1999;1:1147–1149. [Google Scholar]
- 106.Mizushina Y, Takahashi N, Ogawa A, Tsurugaya K, Koshino H, Takemura M, Yoshida S, Matsukage A, Sugawara F, Sakaguchi K. The cyanogenic glucoside, prunasin (D-mandelonitrile-β-D-glucoside), is a novel inhibitor of DNA polymerase β. J Biochem (Tokyo) 1999;126:430–436. doi: 10.1093/oxfordjournals.jbchem.a022468. [DOI] [PubMed] [Google Scholar]
- 107.Deng JZ, Starck SR, Hecht SM. bis-5-Alkylresorcinols from Panopsis rubescens that inhibit DNA polymerase β. J Nat Prod. 1999;62:477–480. doi: 10.1021/np980522g. [DOI] [PubMed] [Google Scholar]
- 108.Deng JZ, Starck SR, Hecht SM. Pentacyclic triterpenoids from Freziera sp. that inhibit DNA polymerase β. Bioorg Med Chem. 2000;8:247–250. doi: 10.1016/s0968-0896(99)00276-x. [DOI] [PubMed] [Google Scholar]
- 109.Deng JZ, Starck SR, Sun DA, Sabat M, Hecht SM. A new 7,8-euphadien-type triterpenoid from Brackenridgea nitida and Bleasdalea bleasdalei that inhibits DNA polymerase β. J Nat Prod. 2000;63:1356–1360. doi: 10.1021/np000129m. [DOI] [PubMed] [Google Scholar]
- 110.Ogawa A, Murate T, Izuta S, Takemura M, Furuta K, Kobayashi J, Kamikawa T, Nimura Y, Yoshida S. Sulfated glycoglycerolipid from archaebacterium inhibits eukaryotic DNA polymerase α, β and retroviral reverse transcriptase and affects methyl methanesulfonate cytotoxicity. Int J Cancer. 1998;76:512–518. doi: 10.1002/(sici)1097-0215(19980518)76:4<512::aid-ijc12>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 111.Mizushina Y, Watanabe I, Ohta K, Takemura M, Sahara H, Takahashi N, Gasa S, Sugawara F, Matsukage A, Yoshida S, Sakaguchi K. Studies on inhibitors of mammalian DNA polymerase α and β: sulfolipids from a pteridophyte, Athyrium niponicum. Biochem Pharmacol. 1998;55:537–541. doi: 10.1016/s0006-2952(97)00536-4. [DOI] [PubMed] [Google Scholar]
- 112.Tanaka N, Kitamura A, Mizushina Y, Sugawara F, Sakaguchi K. Fomitellic acids, triterpenoid inhibitors of eukaryotic DNA polymerases from a basidiomycete, Fomitella fraxinea. J Nat Prod. 1998;61:193–197. doi: 10.1021/np970127a. [DOI] [PubMed] [Google Scholar]
- 113.Mizushina Y, Kamisuki S, Kasai N, Shimazaki N, Takemura M, Asahara H, Linn S, Yoshida S, Matsukage A, Koiwai O, Sugawara F, Yoshida H, Sakaguchi K. A plant phytotoxin, solanapyrone A, is an inhibitor of DNA polymerase β and λ. J Biol Chem. 2002;277:630–638. doi: 10.1074/jbc.M105144200. [DOI] [PubMed] [Google Scholar]
- 114.Perpelescu M, Kobayashi J, Furuta M, Ito Y, Izuta S, Takemura M, Suzuki M, Yoshida S. Novel phenalenone derivatives from a marine-derived fungus exhibit distinct inhibition spectra against eukaryotic DNA polymerases. Biochemistry. 2002;41:7610–7616. doi: 10.1021/bi020115a. [DOI] [PubMed] [Google Scholar]
- 115.Kasai N, Mizushina Y, Sugawara F, Sakaguchi K. Three-dimensional structural model analysis of the binding site of an inhibitor, nervonic acid, of both DNA polymerase β and HIV-1 reverse transcriptase. J Biochem (Tokyo) 2002;132:819–828. doi: 10.1093/oxfordjournals.jbchem.a003292. [DOI] [PubMed] [Google Scholar]
- 116.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 117.Ochs K, Sobol RW, Wilson SH, Kaina B. Cells deficient in DNA polymerase β are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res. 1999;59:1544–1551. [PubMed] [Google Scholar]
- 118.Boorstein RJ, Levy DD, Teebor GW. Toxicity of 3-aminobenzamide to Chinese hamster cells containing 5-hydroxymethyluracil in their DNA. Cancer Res. 1987;47:4372–4377. [PubMed] [Google Scholar]
- 119.Cantoni O, Cattabeni F, Stocchi V, Meyn RE, Cerutti P, Murray D. Hydrogen peroxide insult in cultured mammalian cells: relationships between DNA single-strand breakage, poly(ADP-ribose) metabolism and cell killing. Biochim Biophys Acta. 1989;1014:1–7. doi: 10.1016/0167-4889(89)90234-6. [DOI] [PubMed] [Google Scholar]
- 120.Huet J, Laval F. Potentiation of cell killing by inhibitors of poly(adenosine diphosphate-ribose) synthesis in bleomycin-treated Chinese hamster ovary cells. Cancer Res. 1985;45:987–991. [PubMed] [Google Scholar]
- 121.Bowman KJ, White A, Golding BT, Griffin RJ, Curtin NJ. Potentiation of anti-cancer agent cytotoxicity by the potent poly(ADP-ribose) polymerase inhibitors NU1025 and NU1064. Br J Cancer. 1998;78:1269–1277. doi: 10.1038/bjc.1998.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Suto MJ, Turner WR, Arundel-Suto CM, Werbel LM, Sebolt-Leopold JS. Dihydroisoquinolinones: the design and synthesis of a new series of potent inhibitors of poly(ADP-ribose) polymerase. Anticancer Drug Des. 1991;6:107–117. [PubMed] [Google Scholar]
- 123.Chalmers A, Johnston P, Woodcock M, Joiner M, Marples B. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int J Radiat Oncol Biol Phys. 2004;58:410–419. doi: 10.1016/j.ijrobp.2003.09.053. [DOI] [PubMed] [Google Scholar]
- 124.Sebolt-Leopold JS, Scavone SV. Enhancement of alkylating agent activity in vitro by PD 128763, a potent poly(ADP-ribose) synthetase inhibitor. Int J Radiat Oncol Biol Phys. 1992;22:619–621. doi: 10.1016/0360-3016(92)90889-p. [DOI] [PubMed] [Google Scholar]
- 125.Wedge SR, Porteous JK, Newlands ES. 3-aminobenzamide and/or O6-benzylguanine evaluated as an adjuvant to temozolomide or BCNU treatment in cell lines of variable mismatch repair status and O6-alkylguanine-DNA alkyltransferase activity. Br J Cancer. 1996;74:1030–1036. doi: 10.1038/bjc.1996.485. [DOI] [PMC free article] [PubMed] [Google Scholar]