

Abstract
Dual-color imaging of acridine orange (AO) and EGFP fused to a vesicular glutamate transporter or the vesicle-associated membrane proteins 2 or 3 has been used to visualize a supposedly well-defined subpopulation of glutamatergic astrocytic secretory vesicles undergoing regulated exocytosis. However, AO metachromasy results in the concomitant emission of green and red fluorescence from AO-stained tissue. Therefore, the question arises whether AO and EGFP fluorescence can be distinguished reliably. We used evanescent-field imaging with spectral fluorescence detection as well as fluorescence lifetime imaging microscopy to demonstrate that green fluorescent AO monomers inevitably coexist with red fluorescing AO dimers, at the level of single astroglial vesicles. The green monomer emission spectrally overlaps with that of EGFP and produces a false apparent colocalization on dual-color images. On fluorophore abundance maps calculated from spectrally resolved and unmixed single-vesicle spectral image stacks, EGFP is obscured by the strong green monomer fluorescence, precluding the detection of EGFP. Hence, extreme caution is required when deriving quantitative colocalization information from images of dim fluorescing EGFP-tagged organelles colabeled with bright and broadly emitting dyes like AO. We finally introduce FM4-64/EGFP dual-color imaging as a remedy for imaging a distinct population of astroglial fusion-competent secretory vesicles.
INTRODUCTION
Fluorescence colocalization imaging is a powerful method for exploring the targeting of molecules to intracellular compartments and to screen for their association and interaction. In such experiments, distinct fluorophores are attached to the molecular targets of interest and imaged into spectrally separated detection channels. The fluorescence intensity in each channel ideally is dominated by spatial and concentration information derived from one fluorophore only. In this case, the dual-color fluorescence intensity images then correspond to diffraction-limited fluorophore maps, which can be overlaid or displayed side-by-side and the amount of colocalization can be calculated using different estimators (1–3).
One recent application of dual-color fluorescence detection has been the identification of vesicular compartments and study of their dynamics and fusion in nonsecretory exocytoses, the primary function of which is the transfer of the organelle membrane and its embedded proteins to the cell surface rather than the release of the vesicular cargo (4), or for studying regulated secretion of vesicular compartments in nonspecialized secretory cells (5), like macrophages, fibroblasts (6) or astrocytes (7) (see, e.g., (8) for review). In contrast to presynaptic nerve terminals or neuroendocrine cells that harbor large numbers of secretory vesicles close to the plasma membrane in readiness for exocytosis, nonsecretory cells lack such obvious morphological and functional specializations. Any attempt to study the release of biologically active substances from these cells must therefore first pinpoint the right vesicular compartment among the numerous endosomes, lysosomes, caveolae, and transport carriers, which is typically done by overexpressing a suitable marker (9) and then specifically follow the fate of the identified organelle, typically by using a second fluorescent reporter of membrane fusion.
Acridine orange (AO) is a weak base and metachromatic, fluorescent cationic dye (10,11) and photosensitizer (12,13) that stains live (12,14) and fixed tissue (15,16) with variable hues of fluorescence. At the cellular level, AO relocation and color change has been widely used as a test for cell viability (17,18), and for studying pH gradients across vesicular (19–22), as well as lyso- (23–25) and endosomal (26,27) membranes.
Dilute AO solutions or AO molecules bound to polyanions and isolated from each other emit green orthochromic fluorescence. Concentrated solutions, aggregates of AO, or molecules bound close by to neighboring sites, have orange or red metachromatic fluorescence (12,28). Note that “metachromasy” is here operationally defined as the hypsochromic (shift in absorption to shorter wavelength) and hypochromic (decrease in intensity of emitted fluorescence) change in color exhibited by certain dyes in the presence of water under the following conditions: 1), increase in dye concentration; 2), temperature decrease; 3), salting out; and 4), interaction with substrates that favor water intercalation and/or proximity or stacking of dye monomers. Like thionine, methylene blue, toluidine blue, rhodamine 6G, and phronine G, AO derives its metachromatic properties from the progressive formation of dimers and higher aggregates with increasing concentration (10,11). Low temperature and ionic strength favor AO metachromasy (27,29,30). Due to these photochemical properties and because of its passive accumulation inside acidic vesicles as well as the pronounced and rapid fluorescence change associated with exocytosis, AO has been popular as a reporter of membrane fusion and vesicle release (20,31,32). The opening of the fusion pore equilibrates the acidic luminal pH, leading to the rapid dequenching of AO fluorescence, which is observed as a loss of metachromatic red fluorescence and a concomitant flash of orthochromatic green fluorescence, followed by the dissipation of AO monomers in the extracellular space. Although not easy to discern from the intracellular burst of AO-loaded vesicles, which is commonly observed at high incident powers (23,24,33), this characteristic spatiotemporal profile has generally been accepted as a hallmark of single-secretory granule exocytosis (34–36).
AO reports secretory-vesicle fusion but indiscriminately stains acidic lyso- and endosomal compartments as well. In many cell types, endosomes and lysosomes can undergo regulated exocytosis, too (see (37) for review). To avoid this ambiguity and to specifically study the release of an identified class of vesicular organelles, AO has recently been used in conjunction with EGFP fused to different secretory vesicle proteins in double-labeling experiments (7,38–40).
Inspired by these earlier dual-color studies, we attempted AO/EGFP detection in mouse cortical astrocytes expressing EGFP fusion proteins with the vesicle-associated membrane protein-2 (VAMP2, also termed synaptobrevin-2), VAMP3 (VAMP3, also called cellubrevin), or one of the vesicular glutamate transporters. Astrocytes, one type of macroglia, have recently become a subject of intense and renewed interest, because of their possible role as active partners in glutamatergic synaptic signaling (see (8) and references therein). We then asked to what extent single vesicles can be identified as EGFP-positive in AO-labeled astrocytes. We identified individual, near-membrane vesicles with evanescent-field microscopy (35,36) and used spectral single vesicle imaging and linear spectral unmixing (SILU, see (41) for a detailed description) to detect the presence of EGFP, as well as the monomeric and dimeric forms of AO. We here demonstrate that considerable heterogeneity exists among vesicular compartments concerning their AO loading and spectral emission profile, but that green fluorescent AO monomers inevitably coexist with red fluorescing AO dimers under all experimental conditions that have previously been used for AO/EGFP dual-color imaging (7,38–40). The green emission from AO monomers spectrally overlaps with that of EGFP and produces a significant amount of apparent co-localization, which is mistaken as a false-positive EGFP expression. Time- and space-correlated single-photon counting measurements corroborate our single-vesicle spectral data. We conclude that the EGFP/AO-fluorophore pair is inadequate for deriving quantitative information on secretory-vesicle identity. Finally, based on spectral and time-resolved dual-color imaging of single astroglial vesicles expressing VAMP3-EGFP we propose the red styrylpyridium dye FM4-64 as a robust and reliable alternative for imaging astroglial membrane fusion events of EGFP-labeled vesicles.
MATERIALS AND METHODS
Astrocyte preparation and EGFP expression
Pure astrocyte cultures were prepared from cortical hemispheres of OF1 mice at postnatal day P0-1 and ∼105 cells plated on 25-mm Marienfeld #1 borosilicate glass coverslips. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10 μg/ml streptomycin (42), 10 U/ml penicillin, and 5% fetal calf serum and were imaged during the first week after seeding, before reaching confluence. For imaging, we used astrocytes that grew isolated or in small clusters and were identified by their morphology as well as by the presence of spontaneous and evoked spreading calcium waves (data not shown). Immunocytochemical staining with antibodies against glial fibrillary acidic protein indicates that >90% of the cells in culture were positive for glial fibrillary acidic protein.
To highlight secretory vesicles on fluorescence images, we used lipofectamine 2000 (Invitrogen, Carlsbad, CA) to transfect each coverslip with 2 μg/ml of VAMP2 fused at its luminal C-terminus to EGFP, or its astrocytic analog VAMP3-EGFP, a gift by T. Galli, Paris, under the control of the ubiquitously active CMV (cytomegalovirus) promoter. On these constructs, the EGFP fluorophore is exposed to the low acidic intraluminal pH of ∼5.5 (see (41) for controls). Fusion protein expression was verified on the day after the transfection by comparing the fluorescence excitation and emission spectrum of transfected astrocytes with their nonlabeled (only autofluorescent) counterparts. Control transfections with cytoplasmically targeted EGFP (pEGFP-N1, Clontech, BD Biosciences, Franklin Lakes, NJ (41)) and immunocytochemical labeling with antibodies targeted against VAMP2 or VAMP3 ascertained the specific targeting of these constructs (data not shown). Cells were imaged 24 h after transfection.
Acridine orange (AO) reference standards and single-organelle spectra
We prepared 20-mM aliquots of AO (C17H19N3, 3,6-bis(dimethylamino)acridine hydrochloride) from an analytical-grade spectroscopic standard (1%-AO solution, Sigma, St. Quentin, France) and further diluted these aliquots to the desired final concentration immediately before the experiments. For astrocyte labeling, the AO stock was diluted at least 4000-fold in extracellular saline containing, in mM: 140 NaCl, 5.5 KCl, 1.8 CaCl2, 1 MgCl2, 20 glucose, 10 HEPES, at pH 7.3. Final extracellular AO concentrations ([AO]o) ranged between 0.5 and 5 μM, as indicated. Astrocytes were incubated in AO-containing solution for 15 min at 37°C or 23°C, and thoroughly washed thereafter to remove excess dye. AO is a weak base with a pKa of 9.5–10 (27,28). Its neutral, lipophilic form can pass membranes whereas its protonated, hydrophilic one cannot. Incubation of cells with solution containing μM [AO]o leads to the diffusion of the neutral form through the plasma membrane into the cytoplasm and through the vesicular membranes into the vesicle lumen of organelles where AO is trapped and accumulated (22,30,43). Although some degree of variability in the labeling of individual organelles was observed (Fig. 1 A), 15-min loading was sufficient to attain mM intraorganelle [AO] as judged by the evolution of the single-organelle emission spectrum (see Results and Discussion). In the absence of stimulation, we observed no appreciable leakage of AO from vesicular compartments during the time-window of the experiment—confirmed by the negligible decrease in single-vesicle fluorescence corrected for photobleaching (data not shown). For [AO] ranging from 0.1 to 20 mM, the pH of in vitro spectroscopic standards is calculated by  which results in a pH between 9.5 and 10.7 in our reference spectroscopic standards. In control experiments, we verified that the shape of the AO spectrum was unchanged at vesicular pH (∼5.5) (data not shown), thereby confirming earlier observations (27).
which results in a pH between 9.5 and 10.7 in our reference spectroscopic standards. In control experiments, we verified that the shape of the AO spectrum was unchanged at vesicular pH (∼5.5) (data not shown), thereby confirming earlier observations (27).
FIGURE 1.

Principle of acridine orange (AO)/EGFP detection with dual-band fluorescence or single-secretory organelle spectral imaging and linear unmixing. (A) 488-nm excited total-internal reflection fluorescence (TIRF) image of an AO-loaded (5 μM) astrocyte after 15-min incubation at 37°C, viewed through a dual-view emission beam-splitter (535 ± 25-nm band-pass, 590 nm dichroic mirror, and 600 nm long-pass filter). Circles show spots that are visible in both the red and green detection channel. Scale bar is 5 μm. (B) Schematic microscope optical path for combined TIRF and epifluorescence. The beam angle and hence the penetration depth of the evanescent field is set by sliding the TIRF condenser (shaded) relative to the optical axis. A motorized filter wheel and dual-viewer device (boxed) permit sequential multispectral or (simultaneous) dual-color recordings, respectively. Obj, 1.45NA-×60 objective; DC1, DC3, 500DCLP dichroic mirrors; M1, (100%) mirror; and DC2, 590DCLP secondary dichroic mirror. (Inset) Transmission spectra of the band-pass filters used for spectral unmixing. (C) Epifluorescence spectral images of a VAMP2-EGFP expressing astroglial vesicle (top) and spectral background images, taken in a cell-free region (bottom). (D, top) Raw-data experimental organelle (solid) and background spectra (open circles) for the regions shown in red on panel C. Background-subtracted organelle spectrum before (middle) and after normalization (bottom).
Acquisition of spectral epifluorescence image stacks was interlaced with 458-nm evanescent-field excited fluorescence to identify near-membrane astroglial vesicular compartments (41) on a custom inverted microscope, fitted for combined epifluorescence and through-the-objective type total internal reflection fluorescence (TIRF, Fig. 1 B). This is due to the exponential intensity-dependence of the evanescent field; the single-vesicle fluorescence intensity picks up minor positional changes in z-direction, along the optical axis, which would impair spectral recordings. Regions of interest (ROIs) of diffraction-limited fluorescent spots or cytoplasmic or extracellular ROIs were identified on TIRF images and copied on the corresponding spectral epifluorescence image stack. Even though cultured astrocytes are particularly well suited for wide-field microscopy because their processes are very thin (<1 μm) (44) and single vesicles are clearly resolved in epifluorescence, the axial distance over which the microscope integrates fluorescence is much larger (∼2.2 μm) than the typical diameter of secretory vesicles (30–300 nm). As a consequence, spectral images contain fluorescence from planes above and below the focal plane in addition to the organelle under study (see below).
For evanescent-field excitation, we used an Omnichrome Ar+/Kr+ laser (Melles Griot, Carlsbad, CA) and HQ488/10 excitation clean-up (Chroma, Brattleboro, VT). Laser power was adjusted in function of [AO]o with neutral density filters so as to avoid photoinduced vesicle burst (23,24,33) (not shown). At 5 μM [AO]o, the measured power did not exceed 20 μW in the specimen plane, measured for a beam co-linear with the surface normal. The total illuminated near-membrane section (measured as the 1/e2-intensity decay) sampled by the evanescent-field was ∼200 nm.
Five epifluorescence emission spectral images were acquired sequentially upon 458-nm excitation with a TILL polychrome II (TILL Photonics, Gräfelfing, Germany). To acquire spectral image cubes, we used a 500DCLP dichroic mirror (Chroma) and discrete emission filters housed in a motorized and computer-controlled filter wheel (HQ510/20, HQ535/50, HQ560/40, HQ615/45, HQ670/40; see inset on Fig. 1 B). The resulting fluorescence images were further magnified (×2) to match the camera-pixel size to the calculated resolution and projected onto a GenIII-intensified PentaMax charge-coupled device (CCD) camera (Roper Scientific, Trenton, NJ). We acquired spectral image stacks with 1-s time resolution, which was sufficient to track and recenter moving organelles visually on consecutive frames, using Metamorph 7.0 software (Molecular Devices, Downingtown, PA).
We note that the displayed five-point spectra are a convolution of the fluorophore spectral emission profile and the instrument response and, depending on the exact choice of the center wavelengths and widths of emission bands, do not necessarily bear resemblance with the familiar (corrected) emission spectra known for these fluorophores. Instead, filters were chosen to give a unique signature to each fluorescent component. To be less sensitive to image noise and to better distinguish the green-emitting fluorophores, we slightly oversampled the yellow-green spectral region. For each [AO], we adjusted the gain of the intensified CCD camera so as to optimize the signal at 100-ms image acquisition time. Gain and exposure times were identical for all planes. For in vitro recordings, background spectra were acquired from water droplets using the same settings. For cellular spectral imaging, the spectral background was extracted from a cell-free region on the same coverslip. To verify that photobleaching did not deleteriously affect the sequentially collected spectral images (45), we collected EGFP, AO, and autofluorescence (AF) spectra in both ascending and descending order, confirming that the emission profiles were indistinguishable (not shown).
FM4-64 labeling and dual-color imaging
For Fig. 6, astrocytes were transfected with VAMP3-EGFP and were 24 h later incubated for 10 min in 6.7 μM FM4-64 dissolved in imaging buffer, without further stimulation. Cells were thoroughly rinsed thereafter during 30 min. Dual-band detection of EGFP and FM4-64 (46) was achieved with HQ535/50m and HQ675/50m band-pass filters and 590DCXR secondary dichroic. To detect the release of FM4-64 and dissipation of VAMP3-EGFP fluorescence, we measured the evolution with time of 488-nm excited fluorescence in a 1 μm2 ROI centered on the fluorescent spot, after subtraction of the local background that was measured in a concentric square (1.4-μm outer size). We also measured the correlation of FM4-64 destaining with the intracellular Ca2+ by incubating FM4-64 labeled astrocytes with Oregon Green-BAPTA-1-AM (2 μM, 40 min + 30 min wash, n = 10 cells). Slight mechanical stimulation was generated by a brief puff with control solution (imaging buffer) from a local perfusion pipette (Ø = 25 μm), placed at ∼50 μm from the target cell. Fluid speed was set at ∼0.6 ml/min.
FIGURE 6.

VAMP3-EGFP/FM4-64 dual-color imaging of astroglial secretory vesicle fusion. (A) (Left) Experimental single-organelle fluorescence emission spectrum of VAMP2-EGFP and FM4-64 double-labeled astroglial vesicles (n = 8, mean ± SD) are virtually free of emission cross-talk. Detection bands are indicated by rectangles. (Right) Dual-viewer 488-nm excited epifluorescence images of an EGFP/FM4-64 double-labeled astrocyte and their pseudo-color merge (bottom). (B) Raw-data fluorescence of the calcium indicator Oregon Green-BAPTA-1-AM (OGB-1, 2 μM, 40 min) in response to mechanical stimulation. Brief ejection of physiological saline from a local perfusion pipette generated recurrent and reversible calcium ([Ca2+]i) transients. (C) Concomitant loss of VAMP3-EGFP and FM4-64 fluorescence upon exocytosis of the doubly-labeled vesicle identified on panel A, arrow. Note the slower destaining kinetics for cellubrevin-EGFP relative to FM4-64. The value t = 0 represents the onset of fusion, defined as the first point where the FM4-64 signal dropped below the pre-destaining average intensity minus three times its standard deviation. The organelle shown fused with a lag of 10 s after mechanical stimulation.
Spectral unmixing and detection of fluorophore presence
For unmixing, we excised 3 × 3 μm ROIs containing an isolated fluorescent spot from spectral image stacks (top row on Fig. 1 C). Individual epifluorescence image planes were aligned to compensate movement or offset resulting from different filter thickness, and residual spectral background (bottom row) was subtracted. Linear unmixing and the detection of fluorophore presence were automated for the resulting ministacks (41) and took three steps:
For each organelle, the noise-reduced spectrum was obtained from the spectral pixel vectors
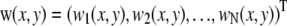 of the nine pixels (x,y) of a 575 × 575 nm quadratic ROI centered on the diffraction-limited organelle image (red box on Fig. 1 C). Outlier values resulting from CCD “hot” pixels or excess noise were removed. The remaining pixel spectral vectors were averaged to yield the organelle spectrum w(i) of the Airy-sized ROI i. Spectra were normalized to equal energy, by division with
of the nine pixels (x,y) of a 575 × 575 nm quadratic ROI centered on the diffraction-limited organelle image (red box on Fig. 1 C). Outlier values resulting from CCD “hot” pixels or excess noise were removed. The remaining pixel spectral vectors were averaged to yield the organelle spectrum w(i) of the Airy-sized ROI i. Spectra were normalized to equal energy, by division with 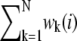 (Fig. 1 D).
(Fig. 1 D).- Mixed spectra were fitted with a linear weighted sum of a predefined spectral library containing m pure components, X = (x1,…,xm),
We used a least- squared-error method to unmix the organelle spectra and to obtain the normalized contributions (abundance coefficients) of EGFP, AO monomers (m.AO), and AO dimers (d.AO) forming a triple vector,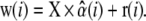
(1) 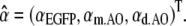 r(i) are the residues not explained by the linear mixing model. To determine the relative abundance of m spectral components, the number of acquired spectral images N needs to be equal to or larger than m. The spectral vector
r(i) are the residues not explained by the linear mixing model. To determine the relative abundance of m spectral components, the number of acquired spectral images N needs to be equal to or larger than m. The spectral vector 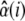 is obtained by minimizing the sum of squared residuals, rT · r, for each organelle i. In contrast to commercial spectral imaging and linear unmixing (SILU) approaches (reviewed, e.g., in (47)), we did not impose a strict sum-to-one condition,
is obtained by minimizing the sum of squared residuals, rT · r, for each organelle i. In contrast to commercial spectral imaging and linear unmixing (SILU) approaches (reviewed, e.g., in (47)), we did not impose a strict sum-to-one condition, 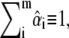 nor did we apply the commonly used nonnegativity constraint,
nor did we apply the commonly used nonnegativity constraint,  By relaxing these boundary conditions, we ascertain that the abundance-coefficient estimates are not biased, see (41,48) for a critical discussion. As a consequence, values of αi can assume apparently physically nonmeaningful values. However, this is not a problem if the calculated confidence interval of the fluorophore abundance estimate is larger than its negative amplitude (41).
By relaxing these boundary conditions, we ascertain that the abundance-coefficient estimates are not biased, see (41,48) for a critical discussion. As a consequence, values of αi can assume apparently physically nonmeaningful values. However, this is not a problem if the calculated confidence interval of the fluorophore abundance estimate is larger than its negative amplitude (41). - We finally verified that the unmixed signal was derived from the fluorescent spot under study, and not resulting from diffuse cytoplasmic fluorescence above and below focus. To this end, we derived fluorophore abundance maps that display the contribution of each dye (i.e., the product of fluorophore abundance and sum of spectral intensities) in each pixel (see, e.g., Fig. 5 B) and applied a center-versus-surround statistical test. For example, the contribution of EGFP to pixel (x,y) is found by denormalizing the EGFP abundance
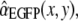
where wk (x, y) is the raw-data intensity of pixel (x, y) viewed in spectral band k. The sum runs over N = 5 detection bands. Next, we averaged IEGFP(x,y) over the nine pixels each of 16 surrounding 1 × 1 μm ROIs i so as to obtain the peripheral EGFP distribution (Ii)1≤i≤16 in the cytoplasm around the organelle as well as the central fluorescent spot itself. The center-versus-surround signal/background ratio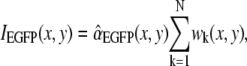
(2)
follows a Student law if the central organelle region is not an outlier relative to the 16 peripheral regions and hence can be compared to a threshold defined by a Student distribution. We reasoned that fluorophore presence was unambiguously detected on an organelle, if 1), the central ROI containing the organelle was diffraction-limited in size; 2), if the confidence interval of the fluorophore abundance estimate was not too big; and 3), if the central ROI was an outlier relative to the surround (Eq. 3). See Nadrigny et al. (41) for details.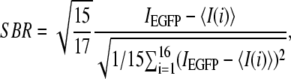
(3)
FIGURE 5.

AO obscures the EGFP signal on spectral images. (A) Examples of normalized mixed organelle spectra extracted from a VAMP2-EGFP expressing astrocyte before (top) and after (bottom) AO labeling (5 μM, 15 min, 23°). Note the variable degree of red hue. Reconstituted fluorophore abundance maps (left, and confidence interval of the SILU estimate, right, see Materials and Methods) before (B) and after AO loading (C). EGFP, but not acridine orange monomers (m.AO) or dimers (d.AO), were detected before AO labeling. After AO accumulation in the vesicle lumen, the EGFP signal is totally obscured by the bright m.AO and d.AO fluorescence. Units are numbers of pure spectra of EGFP, m.AO, and d.AO, respectively. Exposure times were 100 ms before and 10 ms after AO loading, respectively. Scale bar is 1 μm. The hole in the postloading EGFP image is due to the absence of a nonnegativity constraint on the abundance coefficients. (D) Log-intensity histogram of green fluorescence (TIRF 488 nm, HQ535/50m) of near-membrane organelles expressing VAMP2-EGFP (open bars), or labeled with AO during 15 min, with 5 μM [AO] at 23°C (gray) or 37°C (red). Graph pools data from 35 cells.
Dual-color colocalization analysis
Colocalization estimates calculated from unmixed single-vesicle fluorophore abundance maps were compared with conventional dual-color fluorescence detection. For dual-color fluorescence imaging, we used 488-nm TIRF/epifluorescence imaging. The spectral detection downstream of the 500DCLP dichroic was bypassed and fluorescence directed on a custom image beamsplitter with a 590DXCR secondary dichroic mirror (Fig. 1 B), allowing the side-by-side projection of a green (HQ535/50m) and red (HQ600LP) color channel (all from Chroma Technology, Rockingham, VT).
We measured colocalization either by image segmentation, proceeding in the same way as we did for the detection of dyes on unmixed images by calculating the SBR on each emission channel (Eq. 3) or by calculating Pearson's correlation coefficient (49),
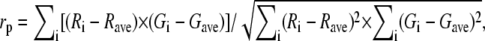 |
(4) |
where R and G are the intensities in the red and green channels, respectively, while the indices i and ave designate one pixel and the average among all pixels of the region of interest studied. The resulting colocalization estimates, along with the results obtained for the same organelles by spectral unmixing, are summarized in Table 3.
TABLE 3.
SILU reveals the false classification of organelles by dual-color colocalization methods
| EGFP | AO | EGFP∩AO | |
|---|---|---|---|
| Threshold | 20 | 0 | 80 |
| Pearson | 40 | 0 | 60 |
| SILU | ND | 100 | ND |
Numbers give percentages of vesicular organelles that appear in the 535 ± 25-nm green channel and are classified EGFP, AO, or EGFP-AO, based on dual-color imaging and image segmentation (intensity threshold), calculation of Pearson's coefficient or spectral imaging and linear unmixing (SILU), respectively. Data pooled from 150 organelles in seven VAMP2-EGFP transfected cells subsequently loaded with 5 μM [AO] for 15 min at 23°C. ND, not detected.
Time-domain picosecond lifetime microscopy (FLIM) and data analysis
For picosecond (ps) time-resolved fluorescence excitation, the beam of a pulsed 440-nm diode laser running at 10 MHz (Picoquant, Berlin, Germany) was coupled to the epifluorescence path of a model No. TE2000 inverted microscope (Nikon, Tokyo, Japan) with a 455DC dichroic mirror (Omega, Brattleboro, VT). A 440/10 cleanup filter in the excitation path removed laser fluorescence. To minimize afterpulse, the laser operated at low power, resulting in a 200-ps (full width at half-maximum) instrument response function (black rectangles on Fig. 2 B). The same AO spectral standards as those used for steady-state fluorescence were measured, for low [AO], in a 200-μm cuvette (Hellma, Mullheim, Germany) to avoid potential surface contributions. High [AO] were measured using a quartz capillary or 10-μm cuvette (Hellma) to minimize reabsorption.
FIGURE 2.

Acridine orange (AO) metachromasy. (A) Concentration dependence of the five-point emission spectrum (510 ± 10, 535 ± 25, 560 ± 20, 615 ± 22.5, and 670 ± 20 nm) of aqueous AO solutions (gray traces, [AO] in mM).The spectral emission of EGFP expressed in the cytoplasm of cortical astrocytes is shown in green for comparison. The mixed AO spectrum depends on the relative abundance of green fluorescent m.AO and red fluorescent d.AO, which depends on [AO]. Measured AO spectra (gray) converged toward pure m.AO (yellow) or d.AO reference spectra (red) at low and high concentrations, respectively. (B) In vitro picosecond fluorescence lifetime imaging microscopy (FLIM) of AO corroborated the steady-state fluorescence measurements shown in panel A. The observed ps time-dependent fluorescence displayed a multiexponential decay with two principal components, τ1 = 1.85 ns and τ2 = 18 ns. IRF, instrument response function. Table 1 summarizes their relative amplitudes as a function of [AO]. Differences to 100% are due to minor components with τ3 = 5 and τ4 = 11 ns with a relative amplitude <7%.
Fluorescence was viewed through a 493ALP long-pass filter (Omega) and imaged on a space-correlated single-photon counting detector (quadrant-anode detector, EuroPhoton, Berlin, Germany). Briefly, replacing the disk anode of a standard multichannel-plate photomultiplier tube with a quadrant-anode, x,y spatial information is gained in addition to fluorescence lifetime. The incident photon produces a cone-shaped cloud of electrons at the output face of the MCP (50). The center of the cloud hits the quadrant anode at a position identical to that of the detected photon. The quadrant anode splits the detected charge in four portions, whose individual magnitudes depend on the xy coordinate of the incident photon. The measured charges are amplified by charge-sensitive shaping amplifiers and read-out by analog-digital circuitry and software for the calculation of the xy position (51). The temporal information is derived from an auxiliary pulse from a second MCP that is inverted, amplified, discriminated, and then used as a start pulse into a time-to-amplitude converter, together with a stop pulse from a photodiode.
Picosecond fluorescence decays obtained from pure AO solutions were analyzed by assuming that the measured decay of the fluorescence I(t) and the laser pulse are related to each other by a convolution integral,
 |
(5) |
where G(t) is the undistorted fluorescence. Fluorescence lifetime components τi (Fig. 2 B) were obtained by fitting a sum of exponential functions with the observed decay curves,
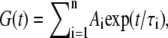 |
(6) |
where the preexponential factors Ai give the contribution of each lifetime component to the measured fluorescence.
RESULTS AND DISCUSSION
Acridine orange-labeled astroglial vesicular compartments display both metachromatic red and orthochromatic green fluorescence
Mouse cortical astrocytes incubated in saline containing 5 μM AO (15 min, 37°C) (7,40) simultaneously displayed a granular green and red fluorescence when viewed with a dual-viewer device housing a 590DXCR secondary dichroic separating the green (HQ535/50m) and red (HQ600LP) detection channels. This filter combination is similar to those used earlier for AO/EGFP dual-color imaging (7,38–40). Fig. 1 A shows a representative 488-nm evanescent-field excited dual-color fluorescence image pair. We use grayscale intensity images rather than pseudo-color to better appreciate the relative fluorescence intensities in both detection channels. Although no EGFP was present in the astrocyte, a rapid inspection by eye reveals an appreciable amount of fluorescent puncta that appear on both the red and green images (circles). Thus, with the filters that have typically been used for EGFP and AO detection, AO alone produces signals in both detection channels. Room temperature (23°C) instead of physiological temperature (37°C) did not appreciably change this observation (data not shown).
To what extent can the green AO signal be separated from EGFP fluorescence? We recently introduced a new live-cell spectral imaging and linear unmixing technique for detecting the presence and colocalization of spectrally overlapping fluorophores on single diffraction-limited point objects, based on their spectral signature across several detection channels (41). Here, we applied this technique to detect the presence of AO monomers (m.AO), AO dimers (d.AO), and EGFP on single vesicles in VAMP2-EGFP transfected and AO-labeled mouse cortical astrocytes.
For spectral imaging, the sample was first viewed in dual-color TIRF (see Fig. 1 B for a schematic layout of the microscope). Rapidly thereafter, we sequentially acquired five spectral images in discrete, partially overlapping detection bands (inset on Fig. 1 B). Regions that identified near-membrane organelles on TIRF images were transferred onto the corresponding spectral epifluorescence image stack to identify near-membrane organelles. We excised and aligned 3 × 3 μm regions encompassing isolated, diffraction-limited fluorescent spots to build spectral ministacks (Fig. 1 C) and extract single-organelle spectra from an Airy-sized region centered on the fluorescent spot (Fig. 1 D, top). Typically, some 10 ROIs were extracted from the TIRF image of the footprint of an astroglial process, viewed at a total magnification of ×120. The spectral background was removed (Fig. 1 D, middle) and the resulting spectrum divided by the sum of its component intensities to yield the normalized organelle spectrum w(i) (Fig. 1 D, bottom).
Pure fluorophore reference spectra of AO monomers and dimers
Due to bleedthrough and spectral overlap, each of the five spectral detection bands do not represent a single fluorescent species. However, each of the pure fluorophores produced a unique five-point spectrum (Fig. 2 A). Pure AO reference spectra were obtained in vitro by microspectrofluorimetry of a small AO droplet on the microscope. When gradually increasing [AO] from 0.1 mM to 20 mM, its peak emission shifted from green-yellow to orange-red (Fig. 2 A). TIRF and epifluorescence spectra were indistinguishable, indicating the absence of self-absorption at these concentrations (data not shown).
AO metachromasy is thought to result from the coexistence of green (orthochromatic) fluorescing AO monomers (m.AO) with red (metachromatically) fluorescing dimers (d.AO) (10). Thus, the experimental AO emission spectrum should reflect up to a factor ɛφ and an instrument constant reflecting the sampled detection bands, their relative abundance in the image region. The value ɛ denotes the molar extinction, and φ the fluorescence quantum yield of each species.
To prepare a pure solution of nonaggregated m.AO as a spectral reference (yellow trace on Fig. 2 A), we diluted 0.5 mM AO in 20% detergent (Triton-X100) following the protocol by Knof and co-workers (52). The measured spectra of dilute AO solutions with 0.1 mM [AO] were virtually indistinguishable from the m.AO reference spectrum. In line with earlier bulk spectroscopic observations (10,27) we found a convergence toward a stable red-shifted spectrum with increasing [AO] (Fig. 2 A, red trace), indicative of the preeminence of d.AO for [AO] ≥ 20 mM. Our results suggest that the polychromatic emission of single AO-labeled astroglial organelles (Fig. 1 A) could potentially be explained by the coexistence of m.AO with a green peak fluorescence and orange-red fluorescing d.AO at the level of single vesicles.
Picosecond fluorescence lifetime imaging microscopy (FLIM) corroborates SILU data
Mixed spectra reflect the relative concentration and brightness of EGFP, m.AO, and d.AO. In contrast, time-resolved single-photon counting histograms to measure the fluorescence lifetime are independent of fluorophore concentration. Fig. 2 B shows typical ps-time resolved AO fluorescence decay curves measured with a time- and space-correlated single-photon counting detector, at different values of [AO] and pH. Decays were well described by a sum of exponential functions (Eq. 6) with two principal lifetime components of τ1 = 1.85 ns and τ2 = 18 ns, corresponding to the short and long lifetimes of the m.AO and d.AO, respectively. In line with our spectroscopic measurements, the relative proportion of d.AO increased with [AO], but nonaggregated m.AO remained present at all studied [AO] between 0.1 and 10 mM. Table 1 summarizes the preexponential amplitudes Ai indicating the fractional presence of the different lifetime components. A small component (<7%) of unknown origin with τ3 at ∼11 ns was consistently observed and was mainly present at high mM [AO]. At the low concentration end, a faint fourth component of ∼5 ns could sometimes be detected (not shown). Further studies need to establish the origin of these components that may result from some impurity.
TABLE 1.
Multiexponential regression analysis of ps-time resolved fluorescence decay curves
| Ai (0.1 mM) | Ai (1 mM) | Ai (10 mM) | |
|---|---|---|---|
| τ1 = 1.85 ns (m.AO) | 99.3 | 97.6 | 67.5 |
| τ2 = 18 ns (d.AO) | 0 | 0.15 | 25.5 |
| τ3 = 11 ns | 0.7 | 2.25 | 7.0 |
Our FLIM data corroborates the spectral-intensity based observation that even at 10 mM [AO], green fluorescing m.AO coexists with red-fluorescing d.AO. The measured lifetimes for m.AO and d.AO are compatible with earlier studies that found 3–4 ns and 13–14 ns for m.AO and d.AO, respectively (52,53), based on a two-component regression analysis. Importantly, for equimolar AO solutions, we found no significant differences in FLIM-derived m.AO and d.AO abundances between pH 5 and pH 7 (Fig. 2 B), which reinforces the validity of the in vitro reference spectra for unmixing AO spectra of acidic organelles.
SILU reliably detects pure spectral components in the absence of autofluorescence
Before using the previously determined reference spectra to unmix single-vesicle spectral image ministacks, we next verified whether unmixing spectral image stacks from control samples containing only one fluorescent species correctly identified the pure spectral component present. In these control experiments, the algorithm is searching for all three predefined fluorophore spectra and should converge to the one fluorophore only. As SILU yields for each unmixed ROI a three-vector  the result should be the three unit vectors corresponding to pure fluorescent species spanning the fluorophore space. The scatter of the estimated
the result should be the three unit vectors corresponding to pure fluorescent species spanning the fluorophore space. The scatter of the estimated  around the theoretical values is expected to reveal the noise-sensitivity and trial-to-trial variability of our estimate under the chosen experimental conditions, and is indicative of our performance to separate close-by fluorophore spectra.
around the theoretical values is expected to reveal the noise-sensitivity and trial-to-trial variability of our estimate under the chosen experimental conditions, and is indicative of our performance to separate close-by fluorophore spectra.
For each fluorophore i, the abundance 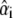 can vary between 0 (absence of dye i) and 1 (for a pure sample exclusively containing i). In the absence of noise, if all fluorophores present were known and if the background subtraction completely removed, any offset, mixed organelle vectors should lie in a plane for which
can vary between 0 (absence of dye i) and 1 (for a pure sample exclusively containing i). In the absence of noise, if all fluorophores present were known and if the background subtraction completely removed, any offset, mixed organelle vectors should lie in a plane for which 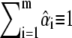 (gray, inset of Fig. 3 A). We deliberately relaxed the sum-to-unity and positivity constraints so that the unbiased estimates of fluorophore abundance (see Materials and Methods and (41,48)) can lie outside this plane (41). For better clarity and graphic representation, we plot on Fig. 3 the projection of the
(gray, inset of Fig. 3 A). We deliberately relaxed the sum-to-unity and positivity constraints so that the unbiased estimates of fluorophore abundance (see Materials and Methods and (41,48)) can lie outside this plane (41). For better clarity and graphic representation, we plot on Fig. 3 the projection of the  onto the two-dimensional plane. Thus, in the absence of noise, pure spectral components are expected to populate the corners of this parametric triangular plot.
onto the two-dimensional plane. Thus, in the absence of noise, pure spectral components are expected to populate the corners of this parametric triangular plot.
FIGURE 3.

Astroglial autofluorescence interferes with the detection of monomeric AO (m.AO). (A) Spectral imaging of pure control samples and subsequent linear unmixing with reference spectra of m.AO, dimeric AO (d.AO), and EGFP correctly detects pure fluorescent species. (B) Astroglial autofluorescence (AF) interferes with m.AO detection (C), due to the virtually identical spectral profile of the AF and AO emission.
For each control condition, SILU correctly recognized the origin of 25 ministacks extracted, for AO from pure control droplets containing either 0.1 (green dots) or 20 mM AO (red dots), or from cytoplasmic regions on images of astrocytes transfected with cytoplasmically expressed EGFP (from pEGFP-N1) construct (blue dots, Fig. 3 A). In some astrocytes, a high density of autofluorescent granular inclusions with a broad yellow-green fluorescence emission profile was observed that interfered with EGFP detection (41). When we tried to include autofluorescence in the unmixing, Fig. 3 B, we observed that SILU falsely classified as m.AO-containing unlabeled astrocytes that displayed high levels of autofluorescence (see the large scatter of gray spots around the expected corner position on Fig. 3 B). The deficiency to correctly classify AF and m.AO is understood by realizing the very close spectral proximity between astroglial autofluorescence and a mixture of (0.8 ± 0.13) × m.AO + (0.15 ± 0.12) × d.AO (at 0.5 mM [AO]) (Fig. 3 C, open symbols) that closely mimics the measured AF spectrum (gray trace, average of n = 25 organelles). Therefore, we subsequently restricted our analysis to astrocytes that displayed low autofluorescence, as judged by the absence of a detectable signal in the 670 ± 20-nm band before AO labeling.
Single-vesicle spectroscopy reveals a large variability among AO-labeled organelles
As the EGFP emission displays a broad spectral overlap with m.AO fluorescence, even small amounts of m.AO are expected to contaminate the green detection channel and to be mistaken for EGFP expression on dual-band recordings (Fig. 2 A). The question arises whether experimental conditions can be found under which red d.AO fluorescence dominates and green fluorescing m.AO is negligible, so that the green detection band would be free for EGFP detection. To detect the presence of m.AO and d.AO at the single-organelle level, we systematically varied the conditions of AO loading and used SILU followed by a center-versus-surround statistical test to unequivocally attribute the detected signal to the organelle (see Fig. 2 legend and Materials and Methods for details).
Fig. 4, A and B, show representative mixed organelle spectra extracted from astrocytes labeled with AO (5 μM, 15 min at 23°C and 37°C, respectively (7,39)), whereas Fig. 4 C represent unmixing results that encompass the range of obtained results. For a clearer graphical representation, only a small selection of organelle spectra (Fig. 4, A and B) and unmixing results (Fig. 4 C) are shown. The full data set from 150 unmixed astroglial vesicles (n = 12 cells) is given in Table 2.
FIGURE 4.

Single vesicle spectral unmixing of astrocytes after AO loading. Examples of normalized emission spectra of astroglial vesicular organelles, after loading with 5 μM AO, 15 min at 23°C (A), and at 37°C (B), respectively. Although extracted from an Airy-sized region of interest centered on the organelle, these spectra may contain signals from the organelles from above and below focus. To unambiguously detect the presence of monomeric (m.AO) or dimeric AO (d.AO), we used a statistical test that compares the organelle spectrum with its (peripheral) background. Green and red color-code for organelles on which m.AO or d.AO only were detected; yellow indicates the detection of both, i.e., colocalization on the same organelle. (C) Detection of m.AO and d.AO in single organelles. Each barbell represents one organelle. Their extremities give the result of the detection test for the presence of m.AO and d.AO, respectively. The dashed line indicates the threshold level corresponding to 95% certainty for detecting fluorophore presence in the studied spot. Green barbells symbolize organelles that exclusively contain m.AO; red, only d.AO; and yellow stands for organelles in which both the presence of m.AO and d.AO was detected (see Table 2). Error bars are confidence intervals defined for 70% certainty.
TABLE 2.
Percentage of vesicles containing green fluorescent AO monomers (m.AO) only, red fluorescent AO dimers (d.AO) only, or both, respectively, as a function of [AO]o
| [AO]o (μM) | m.AO | d.AO | m.AO ∩ d.AO |
|---|---|---|---|
| 0.5 | 38 | 4 | 58 |
| 2 | 28 | 4 | 68 |
| 5 | 6 | 44 | 50 |
| 5 (37°C) | 7 | 61 | 32 |
T = 23°C unless indicated. Data from 150 spectral images of isolated near-membrane organelles in 12 cells.
Confirming our earlier impression gained from dual-band recordings, SILU revealed a large heterogeneity among the spectra of AO-labeled organelles (Fig. 4, A and B). Although increasing [AO]o from 0.5 to 5 μM increased the fraction of d.AO-positive organelles at the expense of only m.AO-containing organelles, one important result of Fig. 4 C is that even after 15-min incubation with 5 μM [AO] at room-temperature, an appreciable number of astroglial vesicular compartments still contains green fluorescing m.AO (see Table 2). In addition, despite a large variability among measured organelle spectra within a single astrocyte, we can conclude from the comparison of these spectra with pure AO spectra at different [AO] that AO accumulated at mM levels inside near-membrane vesicular compartments. Incubation at physiological temperature favored AO accumulation and shifted the organelle spectrum to the red, witnessing the effect of increasing AO aggregation with raising [AO] (see example spectra shown in Fig. 4 B). Nevertheless, even at 37°C (5 μM [AO]o, 15 min (7)), SILU still detected green fluorescing m.AO in 39% of the AO-labeled organelles, versus 56% after incubation at 23°C (39). Virtually all organelles (96%) contained m.AO at lower [AO]o (2 μM (40)). Perhaps longer incubation times and higher [AO]o could be a cure? Unfortunately, those conditions that favor d.AO formation and produce a majority of red-fluorescing organelles make these particularly vulnerable to photoinduced burst (23,24,33). Under our experimental conditions, 25 μM in the specimen plane were the upper limit for avoiding AO-mediated photodamage (data not shown).
Our single-organelle spectral data from cortical astrocytes incubated in low μM concentrations of AO and imaged at low illumination intensity indicate that green m.AO fluorescence inevitably coexists with red-emitting d.AO under all experimental conditions previously reported to detect EGFP and AO labeling. Thus, AO/EGFP double labeling is expected to produce erroneous results when dual-color fluorescence is used to obtain quantitative colocalization information.
AO totally obscures EGFP expression, leading to a false classification of vesicles
To quantify the amount of astroglial vesicles that are falsely classified as EGFP positive on dual-color images in the presence of green m.AO fluorescence, astrocytes were transfected with a VAMP2-EGFP construct and imaged with both dual-band and spectral detection. Subsequent AO labeling under observation on the microscope stage following the protocol of Crippa and co-workers (39) (5 μM [AO]o, 15 min, 23°C) guaranteed that EGFP-expressing cells were viewed.
Fig. 5 A displays typical spectra recorded before AO loading of single astroglial vesicles that expressed VAMP2-EGFP. Although image noise produces some variability among organelles, the EGFP spectral signature is readily recognized (see Fig. 2 A) and SILU reliably detected EGFP expression. Fig. 5 B displays an example of the calculated fluorophore abundance maps along with the confidence interval of the SILU estimate, demonstrating the presence of EGFP on the organelle (top row) as well as the complete absence of AO, both in its monomeric green (Fig. 5 B, middle row) and dimeric red forms (bottom).
Addition of AO to the extracellular fluid caused the organelle spectra to shift to the red. Again, spectra varied widely among organelles, indicating their different capacity to take up and accumulate AO (Fig. 5 A, bottom panel). However, to our surprise, when subsequently unmixing the ministacks, SILU systematically detected m.AO but failed to detect EGFP on green fluorescing organelles (Fig. 5 C and Table 3). The deficiency to detect EGFP after AO loading was not due to the absence of VAMP2-EGFP expression because unmixing before AO loading accurately detected EGFP (Fig. 5 B, top row), nor was it due to the incapacity of SILU to distinguish EGFP and m.AO spectra (see Fig. 3). Rather, the contribution of EGFP to the green part of the mixed organelle spectrum was so small that the spectra of EGFP positive AO-charged vesicles were virtually indistinguishable from spectra of organelles that contained AO only. Thus, on spectral images, the EGFP fluorescence only adds a minor correction to the dominant AO spectrum. This observation is understood when considering the single-vesicle intensity histogram on Fig. 5 D. On average, the green fluorescence (HQ535/50m) of organelles in AO-labeled astrocytes was ∼10-fold brighter compared to their EGFP-containing counterparts, before AO loading. Corroborating and extending our results from Fig. 4, green fluorescence from AO-labeled organelles was stronger at 23°C than at physiological temperature, consistent with the expected lower abundance of m.AO at 37°C (red histogram bars) compared to room-temperature incubation (black bars). In conclusion, on spectral recordings, the bright green emission of AO monomers totally obscures the faint EGFP signal.
Pooling data from 150 organelles in seven astrocytes, Table 3 summarizes these observations and relates them to the interpretation of the same data set that would have resulted when analyzing the same data set by calculating colocalization from threshold- or Pearson-coefficient-type dual-color fluorescence. Although SILU failed in detecting EGFP after AO labeling, linear unmixing unraveled information that is obscured on dual-band recordings, namely that the green fluorescence detected on every single astroglial vesicle was due to m.AO and not EGFP. Instead, depending on the method used, classical colocalization analysis would have erroneously suggested that 20–40% of the organelles contained EGFP labeling only.
In summary, the green m.AO fluorescence not only contaminates the green color channel on dual-band recordings but also precludes the spectrally resolved detection of EGFP at the single-vesicle level. Because the faint EGFP fluorescence only imprints a small modulation onto the bright m.AO signal and since m.AO fluorescence extends from 510 to 570 nm, it is unlikely that different detection bands or a finer spectral sampling could change this negative result.
Imaging secretory vesicle fusion of an identified subpopulation of astroglial vesicles
What then would be a suitable experimental strategy that permits the tagging, tracking, and monitoring of exocytosis of a well-defined subpopulation of astroglial vesicular compartments? To benefit from the large array of EGFP-fusion constructs available, this approach should be 1), compatible with EGFP detection; 2), permit data acquisition with high-temporal resolution so as to follow the fine detail of individual-vesicle dynamics; and 3), thus not require changing filters between acquisitions.
FM4-64 is a red-emitting activity-dependent marker of endocytosed secretory vesicles with its emission spectrum sufficiently removed from that of EGFP to permit single-wavelength excitation imaging. In Fig. 6 A, we display the five-point spectra of EGFP and FM4-64. FM4-64 belongs to the same styryl pyridinium family of dyes as FM1-43 and is virtually not fluorescent in water, but increases its fluorescence quantum yield by more than two orders of magnitude upon insertion in lipid membranes. Its peak excitation (558 nm) and emission (734 nm) are both red-shifted compared to FM1-43 due to three double bonds linking the positively charged head and lipophilic tail group. From a spectroscopic standpoint, both EGFP and FM4-64 can be simultaneously and efficiently excited (with 99% and 93% of their peak absorption, respectively) at 488 nm, leading to an almost balanced (43 vs. 57% cross-excitation) excitation for FM4-64 and EGFP when using our TILL polychromatic narrowband light source (3). Single-color excitation offers the possibility of simultaneously projecting, side-by-side, the green and red images on the same imaging detector (bands on the left panel and image panels on the right of Fig. 6 A).
Fig. 6 A (right panel) illustrates that 10-min incubation of cortical astrocytes with low μM [FM4-64], followed by 30-min wash were sufficient to produce a highly contrasted punctuate pattern of FM-positive spots that partially overlapped with EGFP-VAMP3 (cellubrevin), an astroglial analog of VAMP2 (7,54). To test whether VAMP3-EGFP/FM4-64-positive organelles could be released in response to near-membranous calcium elevations ([Ca2+]i), we mechanically stimulated astrocytes with a brief pulse application of extracellular saline from a perfusion pipette positioned in close proximity (Fig. 6 B). Over direct mechanical stimulation, this approach has the advantage of avoiding the risk of rupturing the plasma membrane (data not shown). Whereas no spontaneous destaining was observed under resting conditions, mechanical stimulation evoked brief [Ca2+]i transients that accompanied the secretory response carried by FM4-64/VAMP-3 positive organelles. In stark contrast to neuronal and neuroendocrine exocytosis, we often observed a considerable (∼s) delay between stimulation and fusion (not shown). Fig. 6 C shows the evolution with time of the concomitant loss of fluorescence in the FM4-64 and EGFP channel, lending further support to the colocalization of the two fluorophores at the single-secretory vesicle level (top). Plotting the evolution with time of the FM4-64 and VAMP3-EGFP fluorescence of the spot marked with an arrow on Fig. 6 A, reveals the different kinetics of loss of FM and VAMP3 from the vesicle (bottom): while FM4-64 is lost within <1 s, probably by rapid diffusion in the plasma membrane (D ∼ 1.2 μm2/s (55)), EGFP slowly dissipates with a τ of 2.8 s.
In conclusion, we demonstrate that, unlike AO/EGFP double labeling, the FM4-64/EGFP fluorophore pair permits simultaneous single-wavelength excitation imaging of both the identity and exocytosis of single astroglial EGFP-labeled astroglial vesicles. Due to its complicated photochemistry and interaction with cellular constituents, AO is a particularly problematic candidate for multicolor fluorescence imaging—both for dual-band and spectral detection. The example of AO/EGFP double labeling serves also as a powerful illustration that caution is required when deriving quantitative estimates from images of dim fluorescing EGFP-tagged organelles colabeled with strongly fluorescent and broadly emitting dyes. Even though spectral imaging and linear unmixing unambiguously identified m.AO as the source of green fluorescence in AO/VAMP2-EGFP double-labeled astrocytes, it is not a remedy for separating the overlapping EGFP/m.AO fluorescence. Their very different fluorophore brightness (the product of the molar extinction and quantum yield) and concentration preclude the detection of EGFP expression. Thus, proper controls must ascertain in each case individually the validity of the used multicolor imaging approach.
Finally, although our results do not exclude the possibility of the presence of a glutamatergic subpopulation of near-membrane astroglial vesicles competent for Ca2+-regulated exocytosis, they lead to a reconsideration of earlier experiments that suggested a high abundance of glutamatergic vesicles based on AO/EGFP dual-color fluorescence.
Acknowledgments
We thank W. Stühmer for the generous gift of the intensified CCD camera, E. Audinat, S. Charpak, M. Ducros, and T. Xu for comments on earlier versions of the manuscript. We acknowledge the help of I. Krämer (Karl-Thomas Bibliothek, Max-Planck Institute, Göttingen, Germany) in providing access to the older AO literature. A. Saab helped with the preparation of some figures.
This study was financed by grants from the Ministère National de la Recherche et de la Technologie, Action concertée incitative: dynamique et réactivité des assemblages biologiques (MRT, ACI DRAB, to M.O. and C.G.), the European Union (No. STRP NMP4-CT-2004-013880), a Franco-Chinese research collaboration grant (No. PRA B05-04: “Imagerie des circuits endocytiques mis en jeu lors de la secrétion astrogliale et neuroendocrine”), and a joint Franco-German Institut de la Santé et de la Recherche Médicale/Max-Planck Society AMIGO grant. F.N. and D.L. were recipients of a Ministère National de la Recherche et de la Technologie pre- and postdoctoral fellowship, respectively.
Editor: David W. Piston.
References
- 1.Garcia Peñarrubia, P., X. Férez Ruiz, and J. Galvez. 2005. Quantitative analysis of the factors that affect the determination of colocalization coefficients in dual-color confocal images. IEEE Trans. Image Proc. 14:1–8. [DOI] [PubMed] [Google Scholar]
- 2.Comeau, J. W. D., S. Costantino, and P. W. Wiseman. 2006. A guide to accurate fluorescence microscopy colocalization measurements. Biophys. J. 91:4611–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oheim, M., and D. Li. 2007. Quantitative colocalization imaging: concepts, measurements and pitfalls. In Imaging Cellular and Molecular Biological Function. F. Frischknecht, J. B. Pawley, and S. Shorte, editors. Springer, Heidelberg, Berlin.
- 4.Chieregatti, E., and J. Meldolesi. 2005. Regulated exocytosis: new organelles for non-secretory purposes. Nat. Rev. Mol. Cell Biol. 6:181–187. [DOI] [PubMed] [Google Scholar]
- 5.Borgonovo, B., E. Cocucci, G. Racchetti, P. Podini, A. Bachi, and J. Meldolesi. 2002. Regulated exocytosis: a novel, widely expressed system. Nat. Cell Biol. 4:955–963. [DOI] [PubMed] [Google Scholar]
- 6.Rao, S. K., C. Huynh, V. Proux-Gillardeaux, T. Galli, and N. W. Andrews. 2004. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 279:20471–20479. [DOI] [PubMed] [Google Scholar]
- 7.Bezzi, P., V. Gundersen, J. L. Galbete, G. Seifert, C. Steinhauser, E. Pilati, and A. Volterra. 2004. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat. Neurosci. 7:613–620. [DOI] [PubMed] [Google Scholar]
- 8.Montana, V., E. B. Malarkey, C. Verderio, M. Matteoli, and V. Parpura. 2006. Vesicular transmitter release from astrocytes. Glia. 54:700–715. [DOI] [PubMed] [Google Scholar]
- 9.Takamori, S. H., M. K. Stenius, E. A. Lemke, M. Gronborg, D. Riedel, H. Urlaub, S. Schenk, B. Brügger, P. Ringler, S. A. Müller, B. Rammner, F. Gräter, J. S. Hub, B. L. De Groot, G. Mieskes, Y. Moriyama, J. Klingauf, H. H. Grubmüller, and F. J. R. Wieland. 2006. Molecular anatomy of a trafficking organelle. Cell. 127:831–846. [DOI] [PubMed] [Google Scholar]
- 10.Zanker, V. 1952. On the detection of defined reversible binding (“reversible polymers”) of acridine orange by absorption flourescence measurements in agueous solution. Z. Phys. Chemie. 199:225–258. [Google Scholar]
- 11.Mataga, N. 1957. Note on the polymerization of dyes in solution. Bull. Chem. Soc. Jpn. 30:375–379. [Google Scholar]
- 12.Wolf, M. K., and S. B. Aronson. 1961. Growth, fluorescence and metachromasy of cells cultured in the presence of acridine orange. J. Histochem. Cytochem. 9:22–29. [DOI] [PubMed] [Google Scholar]
- 13.Delic, J., J. Coppey, H. Magdelenat, and M. Coppey-Moisan. 1991. Impossibility of acridine orange intercalation in nuclear DNA of the living cell. Exp. Cell Res. 194:147–153. [DOI] [PubMed] [Google Scholar]
- 14.Robbins, E., and P. I. Marcus. 1963. Dynamics of acridine orange-cell interactions. I. Interrelationships of acridine orange particles and cytoplasmic reddening. J. Cell Biol. 18:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstrong, J. A. 1956. Histochemical differentiation of nucleic acids by means of induced fluorescence. Exp. Cell Res. 11:640–643. [DOI] [PubMed] [Google Scholar]
- 16.von Bertalanffy, L. 1963. Acridine orange fluorescence in cell physiology, cytochemistry and medicine. Protoplasma. 57:51–83. [Google Scholar]
- 17.Strugger, S. 1940. Fluorescence microscopy study of the uptake and storage of acridine orange by live and dead plant cells. Jen. Zschr. Naturwiss. 73:98–230. [Google Scholar]
- 18.Foglieni, C., C. Meoni, and A. M. Davalli. 2001. Fluorescent dyes for cell viability: an application on prefixed conditions. Histochem. Cell Biol. 115:223–229. [DOI] [PubMed] [Google Scholar]
- 19.Lee, H. C., and J. G. Forte. 1980. A novel method for measurement of intravesicular pH using fluorescent probes. Biophys. Biochim. Acta. 601:152–166. [DOI] [PubMed] [Google Scholar]
- 20.Zoccarato, F., L. Cavallini, and A. Alexandre. 1999. The pH-sensitive dye acridine orange as a tool to monitor exocytosis/endocytosis in synaptosomes. J. Neurochem. 72:625–633. [DOI] [PubMed] [Google Scholar]
- 21.Melnik, V. I., L. S. Bikbulatova, N. V. Gulyaeva, and A. S. Bazyan. 2001. Synaptic vesicle acidification and exocytosis studied with acridine orange fluorescence in rat brain synaptosomes. Neurochem. Res. 26:549–554. [DOI] [PubMed] [Google Scholar]
- 22.Camacho, M., J. D. Machado, M. S. Motesinos, M. Criado, and R. Borges. 2006. Intragranular pH rapidly modulated exocytosis in adrenal chromaffin cells. J. Neurochem. 96:324–334. [DOI] [PubMed] [Google Scholar]
- 23.Olsson, G. M., A. Brunmark, and U. T. Brunk. 1989. Acridine orange-mediated photodamage of microsomal and lysosomal fractions. Virchows Arch. B Cell Pathol. 56:247–257. [DOI] [PubMed] [Google Scholar]
- 24.Zdolsek, J. M., G. M. Olsson, and U. T. Brunk. 1990. Photooxidative damage to lysosomes of cultured macrophages by acridine orange. Photochem. Photobiol. 51:67–76. [DOI] [PubMed] [Google Scholar]
- 25.Choi, S. H., D. H. Choi, J. J. Lee, M. S. Park, and B. C. Chun. 2002. Imidazoline drugs stabilize lysosomes and inhibit oxidative cytotoxicity in astrocytes. Free Radic. Biol. Med. 32:394–405. [DOI] [PubMed] [Google Scholar]
- 26.Moriyama, Y., T. Takano, and S. Ohkuma. 1982. Acridine orange as a fluorescent probe for the lysosomal proton pump. J. Biochem. (Tokyo). 92:1333–1336. [DOI] [PubMed] [Google Scholar]
- 27.Palmgren, M. G. 1991. Acridine orange as a probe for measuring pH gradients across membranes: mechanism and limitations. Anal. Biochem. 192:316–321. [DOI] [PubMed] [Google Scholar]
- 28.Moulik, S. P., S. Ghosh, and A. R. Das. 1976. Environment-induced spectral behavior of acridine orange dye. Ind. J. Chem. 14:302–305. [Google Scholar]
- 29.Bergeron, J. A., and M. Singer. 1958. Metachromasy: an experimental and theoretical reevaluation. J. Biophys. Biochem. Cytol. 4:433–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clerc, S., and Y. Barenholz. 1998. A quantitative model for using acridine orange as a transmembrane pH gradient probe. Anal. Biochem. 259:104–111. [DOI] [PubMed] [Google Scholar]
- 31.De Lisle, R. C., and J. A. Williams. 1987. Zymogen granule acidity is not required for stimulated pancreatic protein secretion. Am. J. Physiol. 6:711–719. [DOI] [PubMed] [Google Scholar]
- 32.Olsson, G. M., K. Rohberg, and I. Rundquist. 1990. The use of acridine orange cytofluorometry in the study of macrophage lysosomal exocytosis. Anal. Cell. Pathol. 2:179–188. [PubMed] [Google Scholar]
- 33.Zdolsek, J. M. 1993. Acridine orange-mediated photodamage to cultured cells. APMIS. 101:127–132. [DOI] [PubMed] [Google Scholar]
- 34.Avery, J., D. J. Ellis, T. Lang, P. Holroyd, D. Riedel, R. M. Henderson, J. M. Edwardson, and R. Jahn. 2000. A cell-free system for regulated exocytosis in PC12 cells. J. Cell Biol. 148:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steyer, J. A., H. Horstmann, and W. Almers. 1997. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 388:474–478. [DOI] [PubMed] [Google Scholar]
- 36.Oheim, M., D. Loerke, W. Stühmer, and R. H. Chow. 1998. The last few milliseconds in the life of a secretory granule. Docking, dynamics and fusion visualized by total internal reflection fluorescence microscopy (TIRFM). Eur. Biophys. J. 27:83–98. [DOI] [PubMed] [Google Scholar]
- 37.Holt, O. J., F. Gallo, and G. M. Griffiths. 2006. Regulating secretory lysosomes. J. Biochem. (Tokyo). 140:7–12. [DOI] [PubMed] [Google Scholar]
- 38.Tsuboi, T., C. Zhao, S. Terakawa, and G. A. Rutter. 2000. Simultaneous evanescent wave imaging of insulin vesicle membrane and cargo during a single exocytotic event. Curr. Biol. 10:1307–1310. [DOI] [PubMed] [Google Scholar]
- 39.Crippa, D., U. Schenk, M. Francolini, P. Rosa, C. Verderio, M. Zonta, T. Pozzan, M. Matteoli, and G. Carmignoto. 2006. Synaptobrevin2-expressing vesicles in rat astrocytes: insights into molecular characterization, dynamics and exocytosis. J. Physiol. (Lond.). 570:567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domercq, M., L. Brambilla, E. Pilati, J. Marchaland, A. Volterra, and P. Bezzi. 2006. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by TNFα and prostaglandins. J. Biol. Chem. 281:30684–30696. [DOI] [PubMed] [Google Scholar]
- 41.Nadrigny, F., I. Rivals, P. Hirrlinger, A. Koulakoff, L. Personnaz, M. Vernet, M. Allioux, M. Chaumeil, N. Ropert, C. Giaume, F. Kirchhoff, and M. Oheim. 2006. Detecting fluorescent protein expression and co-localization on single secretory vesicles with linear spectral unmixing. Eur. Biophys. J. 35:533–547. [DOI] [PubMed] [Google Scholar]
- 42.Robert, F., and T. K. Hevor. 2006. Abnormal organelles in cultured astrocytes are largely enhanced by streptomycin and intensely by gentamicin. Neuroscience. 144:191–197. [DOI] [PubMed] [Google Scholar]
- 43.Busch, G. L., H. Wiesinger, E. Gulbins, H. J. Wagner, B. Hamprecht, and F. Lang. 1996. Effect of astroglial swelling on pH of acidic intracellular compartments. Biophys. Biochim. Acta. 1285:212–218. [DOI] [PubMed] [Google Scholar]
- 44.Golovina, V. A. 2005. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J. Physiol. 564:737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neher, R., and E. Neher. 2004. Optimizing imaging parameters for the separation of multiple labels in a fluorescence image. J. Microsc. 213:46–62. [DOI] [PubMed] [Google Scholar]
- 46.Sharp, M. D., and K. Pogliano. 1999. An in vivo membrane fusion assay implicates SpoIIIE in the final stages of engulfment during Bacillus subtilis sporulation. Proc. Natl. Acad. Sci. USA. 96:14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris, A. T. 2006. Spectral mapping tools from the earth sciences applied to spectral microscopy data. Cytometry A. 69A:872–879. [DOI] [PubMed] [Google Scholar]
- 48.Shen, G., and L. M. Davis. Time-resolved spectral imaging at low signal levels. Biophysical Society Annual Meeting, February 18–22, 2006, Salt Lake City, UT. Abstracts, 1414-Pos.
- 49.Manders, E. M., F. J. Verbeek, and J. A. Aten. 1993. Measurement of co-localization of objects in dual-color confocal images. J. Microsc. 169:375–382. [DOI] [PubMed] [Google Scholar]
- 50.Lampton, M., and R. F. Malina. 1976. Quadrant anode image sensor. Rev. Sci. Instr. 47:1360–1362. [Google Scholar]
- 51.Kemnitz, K., L. Pfeifer, R. Paul, and M. Coppey-Moisan. 1997. Novel detectors for fluorescence lifetime imaging on the picosecond time scale. J. Fluor. 7:93–98. [Google Scholar]
- 52.Knof, J., F.-J. Theiss, and J. Weber. 1978. Influence of aggregation on the fluorescence decay of organic laser dyes. Z. Naturforsch. 33a:98–103. [Google Scholar]
- 53.Ban, T., K. Kasatani, M. Kawasaki, and H. Sato. 1983. Fluorescence decay of the acridine orange-sodium dodecyl sulfate system: formation of dye-riche induced micelles in the premicellar region. Photochem. Photobiol. 37:131–139. [Google Scholar]
- 54.Parpura, V., Y. Fang, T. Basarsky, R. Jahn, and P. G. Haydon. 1995. Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 377:489–492. [DOI] [PubMed] [Google Scholar]
- 55.Zenisek, D., J. A. Steyer, M. E. Feldman, and W. Almers. 2002. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron. 35:1085–1097. [DOI] [PubMed] [Google Scholar]