

Abstract
The deep tissue penetration and submicron spatial resolution of multiphoton microscopy and the high detection efficiency and nanometer spectral resolution of a spectrograph were utilized to record spectral images of the intrinsic emission of mouse skin tissues. Autofluorescence from both cellular and extracellular structures, second-harmonic signal from collagen, and a narrowband emission related to Raman scattering of collagen were detected. Visualization of the spectral images by wavelength-to-RGB color image conversion allowed us to identify and discriminate tissue structures such as epidermal keratinocytes, lipid-rich corneocytes, intercellular structures, hair follicles, collagen, elastin, and dermal cells. Our results also showed morphological and spectral differences between excised tissue section, thick excised tissue, and in vivo tissue samples of mouse skin. Results on collagen excitation at different wavelengths suggested that the origin of the narrowband emission was collagen Raman peaks. Moreover, the oscillating spectral dependency of the collagen second-harmonic intensity was experimentally studied. Overall, spectral imaging provided a wealth of information not easily obtainable with present conventional multiphoton imaging systems.
INTRODUCTION
The intrinsic fluorescence of biological tissues originates from endogenous fluorophores that are primarily derived from the ultraviolet (UV)-emitting aromatic amino acids and vitamin derivatives. These include tryptophan, tyrosine, and phenylalanine (1,2), riboflavin, the nicotinamide ring of reduced pyridine nucleotides (NADH and NADPH (NAD(P)H)) derived from niacin, and the pyridoxine-based pyridolamine cross-links found in elastin and collagen (3). Autofluorescence is extensively used in biochemical research to study the structure, functions, and dynamics (4,5) of proteins, whereas intracellular autofluorescence is widely used to study cellular energy metabolism (6–8). In the latter, early methods of redox fluorometry involved one-photon (1P) excitation at near-UV and visible wavelengths of NAD(P)H and oxidized flavoproteins (FPs), respectively. However, in situ applications have been limited due to photobleaching, photodamage, and considerable light scattering and absorption in turbid tissue environments. These difficulties can be overcome by using multiphoton microscopy. Multiphoton microscopy (MPM) is a collective term used for various imaging microscopy techniques that include nonlinear excitation of a sample by using two or more photons. Commonly used modalities are two-photon (2P) excitation, three-photon (3P) excitation, second-harmonic generation (SHG), and third-harmonic generation (THG). Since its invention (9), 2P-excited fluorescence microscopy has been widely used in imaging biological specimens (10–13). Its advantages include intrinsic three-dimensional resolution, negligible out-of-focus photobleaching, reduced light scattering and photodamage, and improved fluorescence-collection efficiency (14,15). Particularly interesting is the application of 2P-excited fluorescence microscopy in autofluorescence imaging of cells and thick tissues (16–24). Several intrinsic fluorescent vitamin derivatives have 2P-excitation action cross sections that are maximal within the Ti:Sapphire (Ti:Sa) laser tuning range (700–1000 nm). Conversely, aromatic amino acids and indoleamine derivatives can be imaged by three-photon (3P) excitation (25,26) or by two-photon excitation using the frequency-shifted output of a Ti:Sa laser (27–29).
Although 2P-excited fluorescence microscopy is the primary imaging technique for thick tissue imaging, SHG can also be used to image certain types of biological samples. SHG imaging was one of the earliest forms of biological nonlinear microscopy, proposed and demonstrated decades ago (30). SHG occurs when an intense laser beam passes through and interacts with a highly polarizable material with noncentrosymmetric molecular organization. The generated second-harmonic signal is at twice the frequency (half the wavelength) of the light entering the material. A few biological materials are highly polarizable and assemble into large, ordered noncentrosymmetric structures. It has been demonstrated that strong SHG signals can be produced by collagen fibers as well as by dipolar protein structures such as microtubule arrays (31,32). Consequently, SHG has been used for high-resolution imaging with instrumentation similar to that of 2P-excited fluorescence microscopy (22,23,33,34). Similar to 2P excitation, SHG has a quadratic dependence on the laser light intensity, thus, it also has an intrinsic optical sectioning capability. In contrast, though, SHG does not involve an excited state, is energy conserving, and preserves the coherence of the laser light. Also, because SHG does not arise from absorption, photobleaching and phototoxicity are greatly reduced.
In addition to morphological imaging of tissue, spectroscopy provides complementary information that can be obtained through different spectroscopic techniques, such as absorption, autofluorescence, or Raman scattering. Tissue autofluorescence spectroscopy has the potential to identify endogenous fluorophores. The relative concentrations of these fluorophores are related to tissue physiological and pathological states. Although a number of studies have shown the potential of tissue autofluorescence spectroscopy in differentiating normal from diseased tissues (35), conflicting results were also reported (36,37). The main difficulty in tissue autofluorescence spectroscopy is the lack of structural and morphological information that can be related to the emission spectrum. In the last decade, studies have been made to correlate autofluorescence spectra with the morphology of the tissue by depth-resolved autofluorescence spectroscopic measurements and microscopic imaging (17,20,38). Spectral imaging is a promising technique that allows simultaneous recording of spectra and images and it has been applied to fluorescence microscopy (39,40) and FRET measurements (41,42). However, its application to tissue autofluorescence has been limited, primarily due to the difficulty in detecting the inherently weak intensity of autofluorescence and the low sensitivity of commercially available detectors in the visible-to-UV spectral region (16,17,43,44).
Although autofluorescence spectral imaging can potentially be applied to a variety of biological specimens, we focus our effort on the study of hairless mouse skin. Similar to the human skin, the mouse skin is divided into three layers: the epidermis, the dermis, and the subcutaneous tissue. The epidermis is the outermost portion of the skin and is composed of stratified squamous epithelium. The innermost layer of the epidermis consists of a single layer of cuboidal cells called basal cells. These cells differentiate and migrate toward the skin surface. The outer layer of the epidermis is called the stratum corneum, which is composed of flattened and dead cells. The basal cells of the epidermis divide, differentiate, and finally slough. As they migrate to the skin surface, the cells become more stratified and finally form the cornified layer of the stratum corneum. The dermis is composed of three types of fibers: collagen fibers, elastic fibers, and reticular fibers. The dermis also contains many specialized cells and structures such as fibroblasts and dendritic cells. The hair follicles, with their associated sebaceous and apocrine glands, are situated in the dermis.
In this study, we acquired and analyzed multiphoton-excited spectral images of thin excised mouse skin tissue sections, stacks of depth-resolved spectral images of thick excised tissue, and stacks of depth-resolved spectral images of in vivo mouse skin tissue samples. The thin tissue sections used here were slices perpendicular to the surface of excised mouse tissue. This sample enabled us to characterize the variation in emission spectrum of the excised tissue as a function of distance from the skin surface, without the effects of light scattering and absorption. In contrast, the emission spectra of the excised and in vivo mouse skin tissue as a function of focal depth from the skin surface is distorted by light scattering and absorption. We also investigated the biochemical differences between the thick excised and the in vivo skin tissue samples based on the acquired depth-resolved spectral images. A simple method of converting the spectral image data into RGB images enabled us to distinguish different structures at different depths within the skin-tissue specimens. Imaging depths based on the spectrum-integrated-intensity depth profile of the thick tissue samples were determined. A detailed analysis of the depth-resolved emission spectra including identification of endogenous fluorophores was carried out. Spectral measurements of pure samples of some of the main fluorophores in tissues were presented. Our results suggest that there is nonlinear Raman scattering of collagen in the visible wavelength region, which, to the best of our knowledge, has not been previously reported. Finally, the oscillating dependence of the backscattered SHG intensity with excitation wavelength was verified, and optimal wavelengths for second-harmonic generation were determined based on the fitting model.
Two-photon-excited fluorescence and second-harmonic generation
The theory of nonlinear optics is well studied, and a wide range of literature exists on the subject. The nonlinear polarization of a material can be generally expressed as
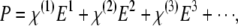 |
(1) |
where P is the induced polarization, 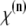 is the nth-order nonlinear susceptibility, and E is the electric field vector. The first term describes normal absorption and reflection of light; the second, second-harmonic generation (SHG), sum and difference frequency generation; and the third, light scattering, stimulated Raman processes, third harmonic generation, and both 2P and 3P excitation. Two-photon excitation is a resonant nonlinear process that involves the absorption of two photons of wavelengths λ1 and λ2, usually via a virtual state, to excite an electron to a real molecular vibronic state. Relaxation to the ground state results in emission of a photon (fluorescence). Practically, a single excitation source is used, such that λ1 = λ2 = λ, and the emitted photon has a wavelength greater than λ/2. In microscopy, the detected time-averaged 2P fluorescence is defined as (13)
is the nth-order nonlinear susceptibility, and E is the electric field vector. The first term describes normal absorption and reflection of light; the second, second-harmonic generation (SHG), sum and difference frequency generation; and the third, light scattering, stimulated Raman processes, third harmonic generation, and both 2P and 3P excitation. Two-photon excitation is a resonant nonlinear process that involves the absorption of two photons of wavelengths λ1 and λ2, usually via a virtual state, to excite an electron to a real molecular vibronic state. Relaxation to the ground state results in emission of a photon (fluorescence). Practically, a single excitation source is used, such that λ1 = λ2 = λ, and the emitted photon has a wavelength greater than λ/2. In microscopy, the detected time-averaged 2P fluorescence is defined as (13)
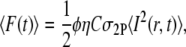 |
(2) |
where η, C, and 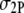 are the fluorescence quantum yield, concentration, and 2P action cross section of a fluorophore, respectively; φ is the instrumental fluorescence collection efficiency;
are the fluorescence quantum yield, concentration, and 2P action cross section of a fluorophore, respectively; φ is the instrumental fluorescence collection efficiency; 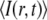 is the average spatial and temporal profile of the excitation laser pulses.
is the average spatial and temporal profile of the excitation laser pulses.
Second-harmonic generation, on the other hand, is of primarily electronic origin and is a nonresonant process. Two photons of the same energy “coalesce” to a virtual state within the specimen to generate a photon of exactly twice the energy of the incident photon. The nonlinear interaction between a system and an electric field that is responsible for second-harmonic generation is described as the second order of the nonlinear polarization, expressed in Eq. 1, and can be rewritten in Cartesian coordinate as (45)
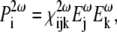 |
(3) |
where subscripts denote Cartesian components, superscripts indicate relevant frequencies, and 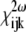 is a 3 × 3 × 3 third-rank tensor known as the second-order nonlinear optical susceptibility.
is a 3 × 3 × 3 third-rank tensor known as the second-order nonlinear optical susceptibility.
Assuming a representation of the form 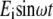 for the incident electric field, the resulting polarization P becomes, according to Eq. 1,
for the incident electric field, the resulting polarization P becomes, according to Eq. 1,
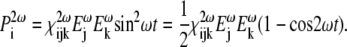 |
(4) |
The unit term gives rise to a direct-current polarization within the material and the term 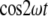 to a polarization wave that oscillates at twice the fundamental frequency ω and that acts as a source for the second-harmonic output field.
to a polarization wave that oscillates at twice the fundamental frequency ω and that acts as a source for the second-harmonic output field.
The elements of the tensor 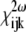 defined in Eq. 1 reflect the symmetry and nonlinear optical properties of the material. Due to the symmetry selection rules, the elements of the tensor
defined in Eq. 1 reflect the symmetry and nonlinear optical properties of the material. Due to the symmetry selection rules, the elements of the tensor 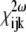 vanish for material with inversion symmetry. Hence, no SHG effect will be observed for an isotropic material or for centrosymmetric molecules. Together with this restriction, the specimen must be relatively transparent to the fundamental illumination wavelength and the generated harmonics.
vanish for material with inversion symmetry. Hence, no SHG effect will be observed for an isotropic material or for centrosymmetric molecules. Together with this restriction, the specimen must be relatively transparent to the fundamental illumination wavelength and the generated harmonics.
High localized instantaneous intensity is provided by a focused beam into the specimen. Using a plane-wave approximation, the second-harmonic intensity 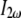 generated at the exit surface of a specimen of length ℓ can be expressed as (46)
generated at the exit surface of a specimen of length ℓ can be expressed as (46)
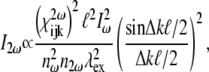 |
(5) |
where 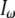 is the intensity of the fundamental wave;
is the intensity of the fundamental wave; 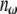 and
and 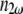 the refractive indices of the sample at fundamental and second-harmonic frequencies, respectively;
the refractive indices of the sample at fundamental and second-harmonic frequencies, respectively; 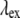 is the wavelength of the fundamental or excitation wave, and
is the wavelength of the fundamental or excitation wave, and 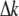 is the phase mismatch between the fundamental wave
is the phase mismatch between the fundamental wave 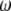 and second-harmonic wave
and second-harmonic wave 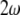 and has a relation to the relevant refractive indexes given by
and has a relation to the relevant refractive indexes given by
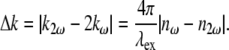 |
(6) |
From Eq. 5, it is seen that the second-harmonic power depends mainly on the square of the excitation intensity, the phase mismatch 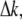 and the interaction length ℓ. The magnitude of the SHG wave can be resonance-enhanced when the energy of the second-harmonic signal overlaps with an electronic absorption band (47). Here, the total second-order response is a sum of the nonresonant and resonant contributions:
and the interaction length ℓ. The magnitude of the SHG wave can be resonance-enhanced when the energy of the second-harmonic signal overlaps with an electronic absorption band (47). Here, the total second-order response is a sum of the nonresonant and resonant contributions:
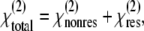 |
(7) |
where the resonant term arises from a sum-over-states expression. In some cases, the resonant contribution may dominate the detected SHG, resulting in an enhancement of one order of magnitude or more.
MATERIALS AND METHODS
Spectral imaging instrument
A diagram of the home-made experimental setup is shown in Fig. 1. The setup was optimized for high detection sensitivity over a broad wavelength range (340–650 nm). The excitation light source was a mode-locked titanium-sapphire (Ti:Sa) laser (Tsunami, Spectra-Physics, Sunnyvale, CA) pumped by a 5W neodymium-yttrium vanadate (Nd:YVO4) laser (Millennia, Spectra-Physics, Sunnyvale, CA). The laser was tunable from 700 nm to 1000 nm with typical pulse widths of 70- to 100-fs pulses at a repetition rate of 82 MHz, and typical output average powers of 0.6–1.0 W.
FIGURE 1.

Schematic diagram of the spectral imaging setup. The intrinsic emission of a sample is epidetected without an aperture. The signal is spectrally dispersed by two prisms, focused with an achromat, and detected with a back-illuminated CCD array.
The laser light was attenuated by a dual filter wheel (5254, New Focus, CA) before passing through a UV-VIS-IR achromat (Bernhard Halle Nachfl., Berlin, Germany). Beam scanning was accomplished by a galvanometer mirror scanner (040EF, LSK, Stallikon, Switzerland). In addition to the beam-scanning mechanism, the microscope was also equipped with an XYZ piezotranslation (sample) stage (Physik Instrumente, Karlsruhe/Palmbach, Germany). The laser light was focused by an objective lens on the sample, and the fluorescence emission was collected by the same objective lens. The results reported here were acquired in the inverted geometry using air- (20×/0.75) and oil-immersion (40×/1.30) objectives (160-mm tube length, Fluor, Nikon, Japan) with estimated lateral resolutions of 0.62 μm and 0.36 μm and axial resolutions of 1.4 μm and 0.68 μm, respectively. Both resolutions were calculated using an excitation wavelength of 764 nm.
The emission passed through a dichroic mirror and was filtered by a set of short-pass Schott optical glass filters (total thickness, 7 mm; BG-40, Schott, Mainz, Germany). These filters were fitted onto the entrance port of an optically isolated “black box”, which housed the spectrograph. The filtered light entering the black box was dispersed by two prisms and focused on the charge-coupled device (CCD) camera by another UV-VIS-IR achromat (diameter, 40 mm, focal length, 160 mm; Bernhard Halle Nachfl., Berlin, Germany). The spectrograph was equipped with a thermoelectrically cooled, back-illuminated CCD camera (16-bit, ST-133 controller, typical read noise 3 e− rms at 100-kHz digitization; Spec-10:2KBUV, Princeton Instruments, Trenton, NJ).
To provide us the ability to measure both the low-wavelength SHG and the weak tissue autofluorescence, the overall detection efficiency of the spectral imaging system was optimized in the wavelength range 330–600 nm. Moreover, the acquisition of spectral images is a tradeoff between the spectral resolution, signal/noise ratio (SNR), photobleaching of the sample, and image acquisition speed. The dispersion prisms and achromats that were used have transmissions of >90% from 300 nm to 2000 nm, whereas the overall transmission of short-wavelength pass filters and dichroic mirror is >50% from 400 nm to 600 nm. On the other hand, the quantum efficiency of the UV/AR-coated CCD is >50% from 300 nm to 700 nm. The use of optical elements with high transmission and a detector with high sensitivity allowed us to acquire wide-spectral-range (330–600 nm) 100-channel spectral images at an acceptable frame rate of <2 min/frame (224 × 224 pixels). The overall transmission of the system was estimated to be ∼30% at 500 nm and ∼5% at 375 nm. It should be emphasized here that optimization of SNR is further limited by the fact that fluorophores suffer from photobleaching with high excitation average intensities. In our experiments, the exposure time was limited to 2.1 ms per pixel and employed excitation powers of <5 mW. Photobleaching was not observed at this low power level. Data processing and visualization were carried out using a program (SpecView) written in IDL 6.0 (Research Systems, Boulder, CO).
Wavelength and instrument spectral response (flatfield) calibration of the instrument was carried out to ensure accurate spectral measurements. A white light source and a calibrated monochromator were used to calibrate the instrument wavelength and the calibration accuracy was better than 0.5 nm over the whole wavelength range. The flatfield correction was calculated by measuring the spectra of two standard fluorophores: tryptophan in water and quinine sulfate in perchloric acid (48).
Animal model
The Committee on Animal Research of the Erasmus University Rotterdam approved the experimental protocol. The skin of female inbred albino hairless mice (SKH1 HR, Charles River, Someren, The Netherlands) was used either excised or in vivo. The mice were fed on a diet free of chlorophyll (Hope Farms BV, Woerden, Netherlands) for a minimum of 2 weeks before starting the experiments to remove the autofluorescence emission from mouse skin centered at 675 nm attributed to pheophorbide-a (49). The skin of normal mice was freshly excised and was either left intact (thick excised tissue) for deep-tissue imaging or cut in 20-μm sections (thin excised tissue) perpendicular to the epidermal layer. For in vivo imaging the mice were placed on a home-made temperature-controlled microscope stage with a coverslip. During imaging, the mice were anaesthetized using Hypnorm (0.5 ml/kg, Janssen Pharmaceutica, Tilburg, The Netherlands) and diazepam (2.5 ml/kg) administered intraperitoneally. To prevent dehydration, the mouse received, intraperitoneally, 0.3 ml of 0.9% sterile NaCl solution.
Preparation of samples
Two-photon-excited fluorescence spectra were recorded for purified samples of major component compounds in skin tissues. Samples of β-nicotinamide adenine dinucleotide phosphate-reduced tetrasodium salt (β-NADPH), flavin adenine dinucleotide disodium salt dihydrate (FAD-Na2 2H20), keratin solution from human epidermis, type I insoluble Achilles tendon collagen, bovine neck ligament elastin, and synthetic melanin prepared by oxidation of tyrosine with hydrogen peroxide were purchased from Sigma-Aldrich Chemie (Steinheim, Germany). β-NADPH was dissolved in phosphate-buffered saline (PBS) buffer (pH 7.35) to a concentration of 100 nM. FAD was dissolved in a 50-mM tris buffer (pH 7.6) to a concentration of 94 μM. For further purification, the melanin was treated with 3% HCl at room temperature several times and collected by centrifugation. The black precipitate was first washed with water (seven times) and successively with acetone (five times) and ether (three times). The black, amorphous powder was dried at 50°C. A saturated solution of the purified melanin in water was made by sonification at 40°C for 30 min and collected after centrifugation. The spectral measurements were done at room temperature. The excitation wavelength was fixed at 764 nm. To avoid photobleaching of the specimens, the excitation power was limited to 5 mW, as measured at the sample stage.
We also measured the 2P-excitation action cross section of collagen solution obtained by dissolving acid-soluble collagen type I from human placenta (Sigma-Aldrich Chemie, Steinheim, Germany) in 0.04 M acetic acid solution. A solution of 14.4 μM fluorescein in water of pH 12.14 was used as a reference dye in the determination of the 2P action cross section according to the procedure in previous studies (15,50). The same experimental conditions were used for the 2P fluorescence spectral measurements of both the fluorescein and collagen solutions.
Calibration procedure
Accurate determination of 2P-excitation action cross sections is usually complicated by the temporal coherence in the excitation focal region. This can be avoided by use of a standard calibration sample of known 2P-excitation action cross sections and spectra, in our case 14.4 μM fluorescein in water of pH 12.14. For a given excitation wavelength, the degree of temporal coherence is the same for both the calibration sample and the fluorophore being studied, in our case collagen solution. The ratio of the experimentally measured collagen fluorescence signal is (50)
 |
(8) |
where 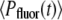 and
and 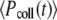 are the incident powers for the fluorescein and collagen solutions, respectively. The 2P-excitation action cross section of collagen solution becomes
are the incident powers for the fluorescein and collagen solutions, respectively. The 2P-excitation action cross section of collagen solution becomes
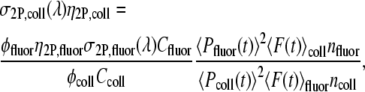 |
(9) |
where 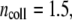
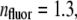
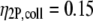 (51),
(51), 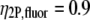 (50). The fluorescence detection efficiencies of our instrument were calculated for both fluorescein and collagen solutions, taking into account the measured spectral responses of the optics, and the CCD and fluorescence-emission spectra of the fluorophores.
(50). The fluorescence detection efficiencies of our instrument were calculated for both fluorescein and collagen solutions, taking into account the measured spectral responses of the optics, and the CCD and fluorescence-emission spectra of the fluorophores.
RGB color visualization of the spectral image
To visualize the three-dimensional (i.e., x and y dimensions and wavelength channel) images, they were transformed into 11-channel RGB images. A 16-bit, 100-wavelength channel spectral image can be represented as a set of image planes 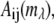 where i and j denote the x and y positions, respectively in the image (i, j = 1–224) and
where i and j denote the x and y positions, respectively in the image (i, j = 1–224) and 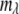 denotes the wavelength channel number (
denotes the wavelength channel number (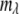 = 1–100). The transformation into an RGB image involved two main steps:
= 1–100). The transformation into an RGB image involved two main steps:
- Data reduction to 8-bit, 11-wavelength channel spectral image,
where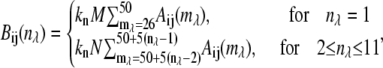
(10) 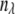 is the new wavelength channel dimension (
is the new wavelength channel dimension (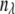 = 1–11),
= 1–11), 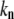 is the wavelength channel-dependent correction factor to compensate for the nonlinearity of the wavelength interval, and M and N are the normalizing factors for the data bit-size reduction. The optimal value for the ratio N/M was found to be 20:1 for images acquired at depths <40 μm and 10:1 at depths ≥40 μm. Here, the first 25 wavelength channels (330–360 nm) were not used, because they contain no information. The next 25 wavelength channels (
is the wavelength channel-dependent correction factor to compensate for the nonlinearity of the wavelength interval, and M and N are the normalizing factors for the data bit-size reduction. The optimal value for the ratio N/M was found to be 20:1 for images acquired at depths <40 μm and 10:1 at depths ≥40 μm. Here, the first 25 wavelength channels (330–360 nm) were not used, because they contain no information. The next 25 wavelength channels (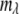 = 26–50, corresponding to 380 ± 20 nm) were summed to form channel 1 of the reduced spectral image, and each subsequent five channels were summed to form wavelength channels 2–11 (400–600 nm) of the reduced spectral image.
= 26–50, corresponding to 380 ± 20 nm) were summed to form channel 1 of the reduced spectral image, and each subsequent five channels were summed to form wavelength channels 2–11 (400–600 nm) of the reduced spectral image. - Conversion of the reduced spectral image into a 24-bit RGB image,
where R, G, and B are the red, green, and blue respective values in RGB color space,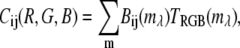
(11) 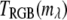 is the wavelength channel-dependent RGB value derived from approximations of the RGB value for visible wavelengths.
is the wavelength channel-dependent RGB value derived from approximations of the RGB value for visible wavelengths.
Analysis of spectral image data
The spectral image data were analyzed using three approaches: 1), depth-integrated spectral profiling; 2), spectrum-integrated depth profiling; and 3), spectrum-depth profiling. For the three approaches, the data were obtained by averaging the spectra of selected xy areas depicted in Figs. 2 A, 3 A, and 4 A (white dashed rectangles). The purpose of selecting an image area as an alternative to using the whole image is to simplify the analysis of the tissue structure by neglecting the highly fluorescing hair shaft or follicle. For the thin excised tissue, depth is defined by the x dimension of the image measured from the surface of the skin. The z dimension measured from the uppermost layer of the skin of the 3D spectral images of the thick excised tissue and the in vivo tissue samples defined the depth.
FIGURE 2.

(A) Spectral image of unstained mouse skin tissue section (thickness 20 μm; colors represent emission wavelength as indicated in the color bar). (B) Brightfield microscopic image of the subsequent H&E-stained tissue section. D, dermis; E, epidermis; hf, hair follicle; dc, dermal cells; sc, stratum corneum; bc, basal cells.
FIGURE 3.

Spectral imaging of thick excised mouse tissue sample at different depths below the skin surface: (A) 0 μm; (B) 5 μm; (C) 10 μm; (D) 20 μm; (E) 30 μm; (F) 40 μm; (G) 50 μm; (H) 60 μm; (I) 70 μm; (J) 80 μm; (K) 90 μm; and (L) 100 μm. The excitation wavelength is 764 nm and the objective is a 40×/1.30 oil immersion objective. Colors represent emission wavelength as indicated in the color bar. Excitation intensity: (A–E) 4 mW; (F–H) 8 mW; (I) 12 mW; (J) 20 mW; (K) 25 mW; and (L) 32 mW. Image intensity scaling factors: (A–F) 1.00; (G) 1.17; (H) 1.50; (I) 2.50; (J) 5.00; (K) 7.08; and (L) 6.25. All images are 224 × 224 pixels, 100 × 100 μm. c, corneocytes; sc, spinous cells; C, collagen fibers; bc, basal cells; hf, hair follicle.
FIGURE 4.

Spectral imaging of in vivo mouse skin tissue at different depths below the skin surface: (A) 0 μm; (B) 5 μm; (C) 10 μm; (D) 20 μm; (E) 30 μm; (F) 40 μm; (G) 50 μm; (H) 60 μm; (I) 70 μm; (J) 80 μm; (K) 90 μm; and (L) 100 μm below the surface of the sample. The excitation wavelength is 764 nm and the objective is a 40×/1.30 oil immersion objective. Colors represent emission wavelength as indicated in the color bar. Image intensity scaling factors: (A–I) 1.00; (J) 1.33; (K) 2.00; and (L) 2.67. All images are 224 × 224 pixels, 120 × 120 μm. c, corneocytes; hf, hair follicle; sc, spinous cells; is, intercellular structures; bc, basal cells; C, collagen fibers; E, elastin fibers; dc, dermal cells; sg, sebaceous glands.
RESULTS AND DISCUSSION
Identification of skin tissue structures from RGB spectral images
Fig. 2, A and B, shows the RGB spectral image of a thin excised tissue section and the brightfield microscopic image of a mouse skin section stained with hematoxylin and eosin (H&E), respectively. The H&E-stained tissue section was obtained subsequent to the unstained section used for spectral imaging. Although the morphology is not identical to the spectrally imaged section, the stained tissue section can be used as a reference in identifying tissue layers and structures. For instance, the epidermis is observed to have a bluish green color in the spectral image. Mouse skin epidermis consists of 1), the stratum corneum, the outermost layer of the skin; 2), the stratum spinosum, consisting of 2–3 cell layers; and 3), the basal layer, the farthest epidermal layer from the skin surface. Underlying the epidermis, the dermis is observed to have purple and cyan structures, as depicted in the spectral image. The dermis is mainly composed of structural proteins such as collagen and elastin fibers, which appear as white fibrous structures in the H&E-stained tissue section. A small population of dermal cells, such as fibroblasts and dendritic cells, are also known to be found in the dermis. These dermal cells appear as black elongated structures between the fiberlike structures in the stained section. Furthermore, a structure with strong green fluorescence, associated with a hair follicle, is observed.
The inherent 3D resolution and penetration capability of the two-photon technique allows us to image deep into thick tissues. Figs. 3 and 4 show the RGB spectral images at different depths of thick excised tissue (Fig. 3) and in vivo tissue (Fig. 4) samples. Note that the image sizes of the thick excised tissue images are 100 μm × 100 μm, whereas the in vivo images are 120 μm × 120 μm. The surface of the skin in Fig. 3, A and B, and Fig. 4, A and B, shows green autofluorescence of the stratum corneum with the typical polygonal structures of corneocytes. At a depth of 10 μm, fluorescence of the spinous cells of the stratum spinosum is observed (Figs. 3 C and 4 C). Interestingly, the nuclei show much less fluorescence than the cellular cytoplasm. Furthermore, the colors and intensity between the two samples vary: the cellular cytoplasm of the in vivo sample is more blue (Fig. 4 C) than that of the thick excised tissue sample (Fig. 3 C). This color discrepancy reflects the difference in biochemistry between the living and dead cells, hence the difference in spectral emission. The spectral images of the in vivo tissue also reveal green intercellular structures (see, for example, Fig. 4 C), which is likely to be caused by lipids, as discussed in detail in a later section. The cells at a depth of 20 μm for both the thick excised tissue and in vivo sections (Figs. 3 D and 4 D) are smaller in size compared to the spinous cells from the overlying layer (Figs. 3 C and 4 C). Both the size and depth of these cells correspond to those of epidermal basal cells. Again, the basal cells of the in vivo sample are more intense and more blue than those of the excised tissue biopsy.
Tissue sections deeper than 20 μm reveal the second-harmonic signals of collagen, which appear as purple fiber structures indicative of the dermis. Within the dermis, other structures, including elastin fibers (cyan, thin fibers), dermal cells (cyan, grain-like clusters), hair follicles (bright green, round), and sebaceous glands (blue, deep-lying structures) (see Figs. 3, E–L, and 4, E–L) are positively identified by morphological comparison with known histology of skin. It is well known that the dermis is largely composed of the extracellular matrix molecules collagen and elastin. Additionally, small densities of cells are known to be found in the mouse dermis. A recent immunofluorescence study has identified dermal macrophages as the major population, ∼60% of nucleated cells in the mouse dermis (52). Small populations of dermal dendritic cells (DC), migrating Langerhans' cells (LC), and fibroblasts are also found in mouse dermis. Fibroblasts are responsible for the production of skin structural proteins such as collagen, elastin, and glucosaminoglycans (53). It is also well known that mouse dermis contains hair follicles, which in hairless mice are inactive. Normally associated with hair follicles, sebaceous glands are lipid-producing structures. These holocrine glands secrete lipids (sebum) that coat the hair, and in furry mammals these lipids have been hypothesized to play an important role in water repulsion and thermoregulation (54).
Comparison between thin excised, thick excised, and in vivo tissue emission spectra
Shown in Fig. 5 are typical spectra of thin excised tissue sections, thick excised tissue, and in vivo tissue samples. These spectra are integrated from the skin surface to a distance of 100 μm from the skin surface. The SHG peak was found to be ∼20-fold more intense than the spectral emission between 400 nm and 600 nm. Thus, to present the entire spectrum more clearly, the data from 400 nm to 600 nm were multiplied by a factor of 20.
FIGURE 5.

Integrated spectra of mouse skin tissue section (triangles), thick excised tissue (circles), and in vivo (squares) samples.
The observed tissue emission spectra are generally characterized by 1), a SHG peak at half the excitation wavelength (i.e., 382 nm); 2), a narrowband emission from 400 nm to 430 nm; and 3), a broadband emission from 430 nm to 600 nm. The SHG peak of the thin excised tissue section emission spectrum is noticeably sharper (full-width at half-maximum (FWHM), 5 nm) than the SHG peak of the thick excised tissue (FWHM, 10 nm) and the in vivo (FWHM, 14 nm) emission spectra. This difference in spectral widths can be explained by the degradation of spectral resolution of the spectral imaging instrument due to scattering of the detected light. This apparent limitation is a consequence of the lack of an aperture in the spectral imaging detection. It should be noted that an aperture would dramatically reduce the light-detection sensitivity for spectral imaging of layers deep inside the tissue.
The broadband emission peaks of the spectra differ significantly from each other: the thin excised tissue section emission peaks more to the blue (peak wavelength/FWHM, 483 nm/125 nm) than either the thick excised tissue (peak wavelength/FWHM, 515 nm/111 nm) or the in vivo (peak wavelength/FWHM, 503 nm/125 nm) tissue samples. The spectral variation between the thin excised tissue section and the thick excised tissue sample, which are biochemically similar, is largely due to scattering effects in thick tissues. Analysis of the data (Fig. 5) showed that the ratio between the thin excised and thick excised tissue spectra varied with wavelength as ∼λ−4 (data not shown). Evidently, thick tissue spectra are strongly altered by wavelength-dependent scattering processes. The difference in depth-integrated spectra of the thick excised tissue and in vivo tissue is ascribed to biochemical differences. In both thick tissue samples, it is reasonable to assume that the detected optical signals suffer from the same overall scattering behavior. The normalized difference spectrum between the thick excised tissue and in vivo tissue emission spectra is depicted in Fig. 6 (squares). Here, the difference spectrum was corrected for the wavelength-dependent scattering factor. The depth-integrated difference spectrum, representing ∼14% relative to the in vivo emission spectrum (integrated from 430 to 600 nm), is found to be similar to the 2P-excited fluorescence measurement on a pure sample of NADPH (Fig. 6, solid line). However, the difference between the spectra of the in vivo and thick excised tissue is much larger (∼41%) at a depth of 10 μm.
FIGURE 6.

Normalized difference spectrum between in vivo emission and thick excised tissue emission spectra (open squares) and normalized measured 2P-excited fluorescence spectrum of NADPH (solid line) using 764 nm 2P-excitation wavelength.
To assess the effect of scattering with optical depth, the SHG (382 nm) and autofluorescence (integrated from 425 nm to 600 nm) intensity profiles of the thin excised tissue section, thick excised tissue, and in vivo tissue samples measured relative to the surface of the skin, from 0 to 100 μm, are compared (Fig. 7, A and B). The data for the depth profile of the thin tissue section were obtained from a single spectral image recorded ∼5 μm below the surface of the specimen. At this imaging depth, scattering effects are negligible, and therefore this image could be used for a reference. The spectral images of the in vivo specimen and the biopsy were obtained from various depths. The data were normalized such that their respective spectrum-integrated autofluorescence intensities at 0 μm were equal in values. At distances >20 μm from the skin surface, the SHG intensity profile of the thin tissue section is approximately constant, whereas its autofluorescence intensity profile remains fairly constant over the whole range (Fig. 7, A and B). Here, the profiles are plotted against the distance from the skin surface, not the depth. Since these data were obtained from a single layer of the tissue section consisting of the epidermal and dermal cross-sections, these intensity profiles depict skin tissue SHG and autofluorescence intensity profiles without tissue scattering effects. In contrast, the intensity of the thick excised and in vivo tissue samples are attenuated with depth. For instance, the SHG intensity profiles of the thick samples show an exponential decay behavior at distances >40 μm from the skin surface. This intensity degradation is caused by tissue absorption and scattering. Previous studies have shown that tissue absorption is primarily due to hemoglobin, and for mouse tissues, its effect is minimal compared to tissue scattering. By fitting the intensity profiles with an exponential function (Figs. 7, A and B, solid lines), we calculated the imaging depth (1/e) values at the SHG wavelength (382 nm) for the in vivo and thick excised tissue samples as 20 μm and 25 μm, respectively. Similarly, the imaging depth values in the autofluorescence spectral range from 425 to 600 nm for the in vivo and thick excised tissue samples are estimated to be 34 μm and 45 μm, respectively. Additionally, the intensity profiles from two other in vivo depth spectral images (n = 4) are fitted (data not shown) resulting in average imaging depth values of 20 ± 3 μm for the SHG emission and 40 ± 6 μm for the autofluorescence emission.
FIGURE 7.

(A) SHG (382 nm) and (B) autofluorescence (integrated from 425 to 600 nm) intensity profiles of mouse skin thin excised tissue (triangles); thick excised tissue (circles); and in vivo tissue (squares) samples.
Biochemical interpretation of tissue intrinsic emission
The inherent optical sectioning capability of nonlinear excitation combined with sensitive spectroscopic acquisition provides a wealth of information on the depth-resolved intrinsic emission spectra of tissues. Shown in Fig. 8, A–C, are depth-resolved spectra of the fixed tissue section, excised tissue biopsy, and in vivo samples, respectively. The SHG peaks for the three cases are either not present or low in relative intensity at depths <20 μm. It is well known that collagen fibers, which are the main source of SHG, are abundant in the dermis but not present in the overlying epidermis. Thus, from the depth-resolved spectra, the epidermis of the mouse is found to be ∼20 μm in thickness, consistent with previous estimates based on the brightfield microscope image of an H&E-stained skin biopsy section (Fig. 2 B).
FIGURE 8.

Depth-resolved spectra of (A) thin excised tissue section, (B) thick excised tissue, and (C) in vivo samples of mouse skin. Colors represent normalized intensity as shown in the color bar. Representative spectra for the depths (D) 0 μm, (E) 10 μm, (F) 20 μm; (G) 30 μm, (H) 50 μm,and (I) 100 μm are also presented for the thin tissue section, thick excised tissue, and in vivo samples. Each color map or spectrum is divided into two spectral regions: 330–400 nm; and 400–600 nm; the longer wavelength region is multiplied by a factor of 20.
The broadband emission spectra within the epidermis show variation with depth. In general, the epidermal emission near the skin surface (i.e., stratum corneum) peaks more to longer wavelengths than in the basal layer. This apparent blueshifting of the emission peaks with increasing depth, depicted as black dotted lines in Fig. 8, A–C, cannot be due to scattering. Scattering results in progressive redshift of spectra with increasing depth, because a shorter wavelength of light has a higher scattering probability. Thus, the observed variations in spectra are most likely due to biochemical differences between the layers of the epidermis. As previously discussed, the cells of the epidermis originate from the basal layer and move into the upper layers of the skin as they undergo differentiation. The differentiation process includes the accumulation of keratin, a major contributor to cellular autofluorescence in the epidermis, and certain other proteins, and the accumulation of lipids. Our measurement on keratin fluorescence shows a peak at ∼465 nm using 764 nm 2P-excitation light (Fig. 9, circles), consistent with previous measurements (55). Another major source of cellular fluorescence is reduced NAD(P)H, which has a characteristic blue fluorescence (19,20,24,56,57). Fig. 9 (triangles) shows the measured fluorescence of NADPH. Since nicotinamide adenine dinucleotide is only fluorescent when reduced, it is expected that due to oxidation the cells of thick excised tissue will show less NADPH fluorescence than in the in vivo tissue. Finally, oxidized flavin adenine dinucleotide (FAD), also known to be found in epidermal cells, emits green fluorescence (24,44,57,58), peaking at ∼520 nm (Fig. 9, squares).
FIGURE 9.

Normalized measured spectral emission of pure samples of keratin (circles), NADPH (upright triangles), FAD (squares), collagen fiber (diamonds), elastin (inverted triangles), and melanin (left triangles) using 764 nm excitation wavelength.
In the late stage of differentiation near the surface of the skin, small organelles called lamellar granules, where the accumulated lipids are packaged, migrate to the apical end of the keratinocyte. The main lipids associated with lamellar granules are phospholipids, glycolipids, and cholesterol. Phospholipids are known to emit green fluorescence at ∼540 nm (56), whereas cholesterol emits at ∼370 nm (59). These lipids are converted to ceramides, fatty acids, cholesterol, and cholesterol esters that comprise the intercellular spaces of the stratum corneum (as observed in Fig. 4, A–C). The ordered structure of ceramides allows the formation of bilayers with a closely packed interior, thus dramatically reducing the permeability of the outer skin to water. The high concentration of lipids in the stratum corneum may explain the long-wavelength emission at the surface of the skin (Fig. 8 D).
The SHG peak dominates the spectra of the dermis at depths >20 μm. For the tissue section, the SHG peak is constant in spectral width and intensity between 50 μm and 100 μm (Fig. 8 A). In contrast, the SHG peaks for the biopsy and in vivo share similar characteristics: 1), spectral broadening with depth; and 2), rapid attenuation at depths >50 μm (Fig. 8, B and C). These are consistent with the increasing effect of tissue scattering with increasing depth. Similar to the SHG peaks, the narrowband emission peaks are also limited to the dermis. However, differences in relative intensities between the three samples are observed. For instance, at depths of 20–50 μm, the narrowband emission peak of the in vivo sample is more prominent than in the other two samples (Fig. 8, F–H). The spectral shape of the broadband emission (from 400 nm to 600 nm) remains constant from 30 μm to 100 μm, although the intensity varies with depth for the thick excised tissue and in vivo samples. Between the tissue types, the broadband emission peaks and spectral widths (FWHM) of the three tissue types in the dermis vary significantly. For instance, the broadband emission peaks and spectral widths for the tissue section, biopsy, and in vivo samples at 50 μm are 485 nm and 122 nm, 509 nm and 122 nm, and 497 nm and 149 nm, respectively. Compared to the tissue section and the biopsy broadband emission, the in vivo emission is notably broader, indicating that there are other fluorophores, such as reduced NAD(P)H in the dermal dendritic cells, present in the dermis for this type of tissue. Here, the measured depth-resolved spectra did not vary significantly between different specimens of the same tissue type.
To gain a better knowledge of the fluorophores responsible for the emission in the dermis, the spectral emission of pure samples of collagen and elastin were measured (Fig. 9). Whereas similarities of the collagen emission to that measured in the dermis are observed, including an intense SHG and a narrowband emission peak, it is also characterized by a broadband emission tail. Elastin, on the other hand, has a broadband emission that peaks at ∼495 nm and a width of 95 nm. Taking into account the broad nature of the measured emission spectra in the dermis, fluorophores other than collagen and elastin are expected to be present in the dermis.
Emission microspectroscopy of skin tissue components
Another advantage of spectral imaging is the capability to measure intrinsic emission spectra of the identified tissue structures directly from the acquired images. Fig. 10 shows normalized measured intrinsic emission spectra of epidermal and dermal cells (Fig. 4, C and F), hair follicles (Fig. 4 C), dermal collagen, and elastin (Fig. 4 E) obtained from in vivo mouse skin tissue spectral images. It is evident in Fig. 10 that discrimination between different tissue structures based on the emission spectra alone is very difficult because of the spectral overlap. Combining the spectrum with the structural pattern (such as shape and size) results in reliable discrimination of the tissue components.
FIGURE 10.

Normalized measured emission spectra of epidermal cells (upright triangles), dermal cells (left triangles), hair follicles (squares), dermal collagen (diamonds), and elastin (inverted triangles) obtained from the in vivo mouse skin tissue spectral images using 764 nm excitation light.
Although there are considerable overlaps between the spectra of the tissue components, the peak wavelengths and spectral shapes show significant variations. Significantly similar to the emission of pure type I collagen (Fig. 9, diamonds), the dermal collagen emission spectrum (Fig. 10, diamonds) shows a sharp SHG peak, a narrowband emission peak, and a broadband emission tail. This further confirms that the fibrillar collagen in the mouse dermis consists mostly of type I collagen. The emission of the epidermal (spinous) cells (Fig. 10, triangles) is described by a spectrum similar to NADPH (Fig. 9, upright triangles) and keratin (Fig. 9, circles) fluorescence. At present, it is unclear which of these two fluorophores dominate epidermal cell autofluorescence. The dermal elastin emission spectrum (Fig. 10, inverted triangles) is characteristically similar to the emission spectrum of a pure sample of elastin (Fig. 9, inverted triangles). Interestingly, the emission spectrum of dermal cells (Fig. 10, left triangles) is significantly similar to that of elastin (Fig. 10, inverted triangles). This may mean that the observed dermal cells are dominantly fibroblasts. The emission of hair follicles (Fig. 10, squares) shows a broader spectrum and higher peak wavelength compared to the other tissue components. Hair follicles are mainly composed of keratin and melanin. Melanin, which peaks at ∼550 nm (Fig. 9, left triangles), is responsible for the intense greenish-yellow autofluorescence of mouse hair follicles. Studies have shown that melanin has an unusual absorption spectrum that exhibits a monotonous decreasing absorption from the UV, over the visible range, up to the near infrared (60). This causes reabsorption of the emission, especially in optically thick samples, such as the hair follicle. In addition, melanin fluorescence has strong environment sensitivity; for instance, it exhibits a broader emission spectrum and is shifted to longer wavelengths in solutions. These observations may explain the differences between the spectra of pure melanin and the hair follicle.
Origin of the collagen emission
To assess the origin of the narrowband emission, which we reported in a previous study (16), emission spectra of type I collagen fibers at different excitation wavelengths were recorded (Fig. 11). The narrowband emission is found to consist of two narrow peaks that shift with excitation wavelength. This confirms our initial assumption that the narrowband emission is not due to fluorescence excitation. It should also be noted here that a separate experiment was carried out to show that the observed peaks are not artifacts (i.e., stray SHG light). In this experiment we placed an aperture (diameter, 75 μm) at the back focal plane of the 160-mm-tube length objective and used pure collagen fiber as the specimen.
FIGURE 11.

Spectral emission of collagen fibers using different excitation wavelengths. (Inset) Spectra are shown in wavenumbers relative to the corresponding SHG emission peak.
Plotting the collagen emission spectra in wavenumbers (Fig. 11, inset) reveals that the peak shifts with respect to the corresponding SHG peaks are constant within experimental error. This is indicative of Raman scattering, and the average values of the relative wavenumber shifts are 1750 ± 110 cm−1 and 2250 ± 160 cm−1 at a sampling step of ∼137 cm−1. Additionally, anti-Stokes Raman peaks are observed at −1700 ± 140 cm−1 and −1000 ± 30 cm−1 (see Fig. 11, inset). Due to the broadband excitation source, further characterization of the narrowband emission to resolve distinct Raman peaks was not carried out. Previous Raman spectroscopic studies have shown that the most prominent collagen bands are the amide stretches (amide III, C-N-H stretch, 1244 and 1270 cm−1; amide I, C-C-N stretch, 1662 cm−1) and the CH2 band (1447 cm−1) (61). Changes in the collagen secondary structure are most notably manifested as changes in the amide I band contour (1650–1680 cm−1) (62–64). Moreover, the amide I envelope for collagen with nonreducible and reducible cross-links contains vibrational bands near 1660 and 1690 cm−1, respectively (65). Interestingly, we also observed two broad emission peaks at ∼380 nm and ∼440 nm (data not shown) from our fitting using multiple Gaussian curves (width 550 cm−1 for all Raman peaks). We attribute these two bands to collagen fluorescence. Collagen-linked fluorescence includes two bands with excitation/emission maxima around 335:385 nm and 370:440 nm, corresponding to pepsin-digestible (pentosidine) and collagenase-digestible cross-links, respectively (66–68). The 380-nm collagen fluorescence emission is most likely to be 3P-excited, whereas the 440-nm collagen fluorescence is 2P-excited.
Our results suggest that nonlinear optical processes are involved in the generation of the Raman signals, mainly because they are found to be Stokes-shifted relative to the second-harmonic rather than to the excitation frequency. In addition to second-harmonic generation, other high-order nonlinear optical processes have been reported in collagen. Its high nonlinear optical response is due to its noncentrosymmetric structure and high molecular hyperpolarizability, just 10-fold less than crystalline quartz (34,69,70). The precise nonlinear process that effectively generated the observed Raman peaks is difficult to identify from these results. It is possible that one or more interacting nonlinear processes, such as 1), (two-photon) hyper-Raman scattering, 2), second-harmonic-induced (stimulated) Raman scattering, and 3), (stimulated-) Raman-scattering-induced second-harmonic generation, may have contributed to the observed Raman band. Although these high-order nonlinear processes are expected to produce inherently weak signals, resonance of the multiphoton excitation wavelength with a multiphoton absorption band of collagen may enhance the signal. In fact, our measurements on the 2P-excitation action cross section of collagen (see Fig. 12, solid line) support the hypothesis of resonance enhancement of the related nonlinear optical process or processes. Although collagen Raman bands have been well characterized using UV and near-infrared excitation, to the best of our knowledge, no published work has reported on nonlinear Raman scattering of collagen.
FIGURE 12.

Excitation wavelength dependence of measured SHG intensity of collagen fibers (open squares) and the sine-squared function fit (solid line). The measured 2P-excitation action cross section of 4 mg/ml type I collagen solution is also shown (dashed line). 1 GM = 10−50 (cm4 s)/photon.
Nonlinear (hyper-) Raman spectroscopy combined with an imaging modality such as nonlinear microscopy may be used to obtain direct chemical information about the structures in the skin in vivo. For instance, the Raman bands that correspond to the amide I envelope for collagen with cross-links may reveal information on the structure, organization, and integrity of collagen fibers. As such, it could be a tool for (early) detection of collagen denaturation or proteolysis, which have been shown to accompany cancer growth in tissues (71,72).
Oscillating spectral dependence of collagen second-harmonic intensity and observation of two-photon resonance effects
To determine the spectral dependency of the backscattered second-harmonic intensity and optimal excitation wavelength for its generation, type I collagen fibers were excited with various wavelengths from 715 to 880 nm. Our experimental data, shown in Fig. 12 (squares), depict a peak at ∼840 nm and have an oscillatory curve. A previous experimental study also suggested an oscillating dependence of the second-harmonic intensity with the excitation wavelength, but this was based on only four data points (22). Rewriting Eq. 5 using the dependence of the phase mismatch, 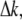 with the wavelength of the fundamental wave (as defined in Eq. 6), the dependence of the second-harmonic intensity with the excitation wavelength is derived as
with the wavelength of the fundamental wave (as defined in Eq. 6), the dependence of the second-harmonic intensity with the excitation wavelength is derived as
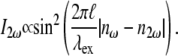 |
(12) |
In Eq. 5, the second-harmonic intensity has its maxima at
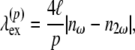 |
(13) |
where p is an odd integer. The result from data fitting using a single-parameter function of the form 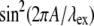 is shown in Fig. 12 (line), where A = 1.883 μm. Using
is shown in Fig. 12 (line), where A = 1.883 μm. Using 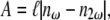 the wavelengths at which the SHG has its maxima for collagen, given by
the wavelengths at which the SHG has its maxima for collagen, given by 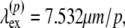 are
are 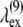 = 837 nm, and
= 837 nm, and 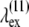 = 685 nm. Despite being generally in good agreement with the sine-squared function, the data points between 740 and 770 nm deviate considerably from the fitting model. This deviation is likely to be caused by resonance enhancement of SHG due to its overlap with the measured 2P-excitation band of collagen depicted in Fig. 12 (dashed line).
= 685 nm. Despite being generally in good agreement with the sine-squared function, the data points between 740 and 770 nm deviate considerably from the fitting model. This deviation is likely to be caused by resonance enhancement of SHG due to its overlap with the measured 2P-excitation band of collagen depicted in Fig. 12 (dashed line).
The results presented here imply an optimal excitation wavelength of ∼837 nm for second-harmonic imaging of type I collagen in skin tissues. Moreover, the results suggest additional maxima in the SHG efficiency at 685 nm and 1076 nm. The use of low wavelengths such as 685 nm for nonlinear excitation has its advantages in 2P and 3P autofluorescence imaging of biological specimens that are not too optically thick (in the order of tens of microns). In general, aromatic amino acids and vitamin derivatives have high 2P- and 3P-excitation cross sections at low excitation wavelengths. Conversely, the use of long wavelengths such as 1076 nm has the advantage of deeper penetration of the excitation light in thick biological samples. In this case, excitation by 3P excitation also results in autofluorescence emission, but with higher nonlinear excitation intensity requirement. Another important matter to consider for deep tissue imaging is the effect of optical scattering that strongly attenuates both the excitation and the emitted SHG. Thus, simultaneous imaging of autofluorescence and SHG effectively demands a sensitive imaging system, particularly in the UV region below 380 nm. Equally important to take into account are the limitations introduced by imaging systems that include excitation-wavelength tuning range, spectral sensitivity of the detector, and spectral transmission of optical elements such as dichroic mirrors, glass filters, objective lenses, and (scanning) mirrors.
SUMMARY AND CONCLUSIONS
Intrinsic emission spectral imaging has distinct advantages over imaging specimens stained with multiple fluorescent dyes. First, the specimen needs no staining preparation. Preparation procedures such as sectioning, fixation, and staining are typically necessary to resolve fine structures within thick tissues. However, cytotoxic and phototoxic effects inevitably accompany specimen staining. Photobleaching of specimens produce toxic free radicals and limit the study of tissue and cellular dynamics for a long time. This work demonstrates that tissue intrinsic emission spectral imaging is capable of resolving fine structures within thick tissues. Similar to previous multiphoton studies on tissues (20,22,23,33), we were able to observe cellular structures in the epidermis and the extracellular matrices in the dermis. Our work, however, is unique in its use of pixel-by-pixel emission spectra to enhance the contrast between biochemically different structures. This distinct feature of the imaging method enabled us to observe and discriminate dermal cells from collagen or elastin. To the best of our knowledge, observation of dermal cells such as fibroblasts and dendritic cells within thick skin tissues have not been reported in previous intrinsic emission multiphoton studies.
Other techniques in fluorescent label-free microscopy that are capable of discriminating cellular and extracellular structures, as well as endogenous fluorophores, include fluorescence lifetime imaging microscopy, coherent anti-Stokes Raman microscopy, and infrared and Raman imaging microscopy. One common issue with imaging label-free specimens is the tradeoff between the amount of information obtained and the speed of acquisition. For instance, fluorescence lifetime imaging microscopy yields limited information on the concentration of fluorophores and gives high uncertainties in discriminating between different fluorophores (73,74). On the other hand, infrared and Raman microscopy offer high accuracy in the determination of spatially resolved concentration and molecular structure information but suffer from long acquisition times (∼1 h) (75,76). This work demonstrates the potential of spectral imaging in providing the ability to discriminate multiple tissue components. However, it is important to realize that we were able to discriminate not between fluorophores, but between tissue components containing these fluorophores. The overlapping spectra limit the ability to discriminate between tissue components due to the broad emission of the tissue intrinsic fluorophores. This drawback was nevertheless circumvented by taking into account the structural pattern of the tissue components. Through the combination of spectral signature and structural pattern (i.e., morphological shape and size), we were able to identify a number of tissue components.
The results on the imaging depth of the multiphoton system were found to be consistent with the results of previous studies on multiphoton-excited fluorescence in turbid media and biological samples with values ranging from 25 to 70 μm (77–79). Although the imaging depth of the multiphoton microscope described here is in the order of a few tens of microns, acquisition of quality images in sections deeper than the imaging depth can be achieved, as was evident from the results (e.g., see Fig. 4 J). The spatial resolution, which is the key determining factor for the quality of an image, is determined by the point-spread function (PSF) of the excitation beam. It has been previously shown that the lateral PSF is minimally influenced by depth, even in turbid media, provided that the refractive index of the microscope objective is matched with that of the specimen (78). However, the increase in excitation and emission scattering with increasing depth limits the number of emission photons available for collection. This decreases the SNR and, consequently, degrades the acquired image. As mentioned earlier, the lack of an aperture also contributes to the degradation of the spectral image. Despite the fact that the excitation lateral PSF remains fairly unaffected by excitation scattering, the projected beam pattern of the emission on to the CCD is significantly influenced by emission scattering. Aside from observable spectral broadening of the detected signal, evident in the measured SHG spectra at deep tissue sections (e.g., see Fig. 8 G), emission scattering also degrades the color (spectral) contrast of the acquired images. Although the lateral PSF is largely unaffected by depth, refractive-index mismatch between the immersion fluid and the sample significantly broadens the axial PSF of the excitation beam inside a turbid medium (78). This axial PSF broadening results in deterioration of the resolution, increase in out-of-focus fluorescence signal, and decrease in SNR.
Our experimental results have shown that the optimal excitation wavelength for second-harmonic generation in collagen fibrils is ∼837 nm. Excitation wavelengths <800 nm are suitable for simultaneous imaging of 2P autofluorescence and SHG, considering that the optimal 2P-excitation bands of the endogenous fluorophores in biological tissues are in the range 700–800 nm (23) and that the signal of SHG is more intense relative to the autofluorescence (∼20-fold at 764 nm). However, the sensitivity of the imaging system between 350 and 400 nm will play a crucial role in the detection of second-harmonic signal when exciting below 800 nm. Although we account for the observed discrepancy between the experimental data and the model with 2P-resonance effects, a better explanation is still needed. The fundamental principles underlying the generation of backscattered second-harmonic signal in collagen fibers are not yet fully understood. Experimental studies have shown that the ratio between forward and backward SHG in type I collagen fibers is ∼10 (31,34). It has also been suggested that the observed backscattered SHG from collagen fibrils can be explained by the fact that the second harmonic radiates from a shell of a collagen fibril rather than from its bulk (34).
Spectrally resolved multiphoton 3D imaging microscopy offers a simple yet versatile method for imaging unstained biological tissues. It provides a wealth of information that surpasses what conventional multiphoton microscopy or autofluorescence spectroscopy offers. We have demonstrated the potential of spectral imaging for 1), producing high-quality depth images by taking advantage of the spectral information to visualize tissues in RGB color; 2), identifying and discriminating tissue cellular and extracellular structures, even in deep layers of the skin; and 3), identification of tissue fluorophores by correlating the emission spectra with morphological information. In the future, the observed collagen Raman peaks could potentially play a vital role in tissue diagnostics by probing the collagen denaturation state in diseased tissues.
Acknowledgments
The authors thank Dr. George W. H. Wurpel for illuminating discussions and help with this manuscript.
This work is part of the research program of the Stichting voor Fundamenteel Onderzoek der Materie (FOM, financially supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO)).
Editor: Alberto Diaspro.
References
- 1.Weber, G. 1960. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J. 75:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teale, F. W., and G. Weber. 1957. Ultraviolet fluorescence of the aromatic amino acids. Biochem. J. 65:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deyl, Z., K. Macek, M. Adam, and O. Vancikova. 1980. Studies on the chemical nature of elastin fluorescence. Biochim. Biophys. Acta. 625:248–254. [DOI] [PubMed] [Google Scholar]
- 4.Demchenko, A. P. 1986. Fluorescence analysis of protein dynamics. Essays Biochem. 22:120–157. [PubMed] [Google Scholar]
- 5.Lakowicz, J. R. 1983. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- 6.Masters, B. R. 1984. Noninvasive corneal redox fluorometry. Curr. Top. Eye Res. 4:139–200. [PubMed] [Google Scholar]
- 7.Chance, B., and B. Thorell. 1959. Localization and kinetics of reduced pyridine nucleotide in living cells by microfluorometry. J. Biol. Chem. 234:3044–3050. [PubMed] [Google Scholar]
- 8.Chance, B., P. Cohen, F. Jobsis, and B. Schoener. 1962. Intracellular oxidation-reduction states in vivo. Science. 137:499–508. [DOI] [PubMed] [Google Scholar]
- 9.Denk, W., J. H. Strickler, and W. W. Webb. 1990. Two-photon laser scanning fluorescence microscopy. Science. 248:73–76. [DOI] [PubMed] [Google Scholar]
- 10.Gerritsen, H. C., and C. J. De Grauw. 1999. Imaging of optically thick specimen using two-photon excitation microscopy. Microsc. Res. Tech. 47:206–209. [DOI] [PubMed] [Google Scholar]
- 11.Denk, W., K. R. Delaney, A. Gelperin, D. Kleinfeld, B. W. Strowbridge, D. W. Tank, and R. Yuste. 1994. Anatomical and functional imaging of neurons using 2-photon laser scanning microscopy. J. Neurosci. Methods. 54:151–162. [DOI] [PubMed] [Google Scholar]
- 12.Svoboda, K., W. Denk, D. Kleinfeld, and D. W. Tank. 1997. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 385:161–165. [DOI] [PubMed] [Google Scholar]
- 13.Xu, C., and W. W. Webb. 1997. Nonlinear and two-photon induced fluorescence. In Topics in Fluorescence Spectroscopy. J. R. Lakowicz, editor. Plenum Publishing, New York.
- 14.Denk, W., and K. Svoboda. 1997. Photon upmanship: why multiphoton imaging is more than a gimmick. Neuron. 18:351–357. [DOI] [PubMed] [Google Scholar]
- 15.Xu, C., W. Zipfel, J. B. Shear, R. M. Williams, and W. W. Webb. 1996. Multiphoton fluorescence excitation: New spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. USA. 93:10763–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palero, J. A., H. S. de Bruijn, A. van der Ploeg-van den Heuvel, H. J. C. M. Sterenborg, and H. C. Gerritsen. 2006. In vivo nonlinear spectral imaging in mouse skin. Opt. Express. 14:4395–4402. [DOI] [PubMed] [Google Scholar]
- 17.Laiho, L. H., S. Pelet, T. M. Hancewicz, P. D. Kaplan, and P. T. So. 2005. Two-photon 3-D mapping of ex vivo human skin endogenous fluorescence species based on fluorescence emission spectra. J. Biomed. Opt. 10:024016. [DOI] [PubMed] [Google Scholar]
- 18.Wu, Y. C., and J. N. Y. Qu. 2005. Two-photon autofluorescence spectroscopy and second-harmonic generation of epithelial tissue. Opt. Lett. 30:3045–3047. [DOI] [PubMed] [Google Scholar]
- 19.Piston, D. W., B. R. Masters, and W. W. Webb. 1995. Three-dimensionally resolved NAD(P)H cellular metabolic redox imaging of the in situ cornea with two-photon excitation laser scanning microscopy. J. Microsc. 178:20–27. [DOI] [PubMed] [Google Scholar]
- 20.Masters, B. R., P. T. C. So, and E. Gratton. 1997. Multiphoton excitation fluorescence microscopy and spectroscopy of in vivo human skin. Biophys. J. 72:2405–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masters, B. R., and P. T. C. So. 2001. Confocal microscopy and multi-photon excitation microscopy of human skin in vivo. Opt. Express. 8:2–10. [DOI] [PubMed] [Google Scholar]
- 22.Zoumi, A., A. Yeh, and B. J. Tromberg. 2002. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc. Natl. Acad. Sci. USA. 99:11014–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zipfel, W. R., R. M. Williams, R. Christie, A. Yu Nikitin, B. T. Hyman, and W. W. Webb. 2003. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc. Natl. Acad. Sci. USA. 100:7075–7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang, S., A. A. Heikal, and W. W. Webb. 2002. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 82:2811–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams, R. M., J. B. Shear, W. R. Zipfel, S. Maiti, and W. W. Webb. 1999. Mucosal mast cell secretion processes imaged using three-photon microscopy of 5-hydroxytryptamine autofluorescence. Biophys. J. 76:1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiti, S., J. B. Shear, R. M. Williams, W. R. Zipfel, and W. W. Webb. 1997. Measuring serotonin distribution in live cells with three-photon excitation. Science. 275:530–532. [DOI] [PubMed] [Google Scholar]
- 27.Palero, J. A., V. O. Boer, J. C. Vijverberg, H. C. Gerritsen, and H. J. C. M. Sterenborg. 2005. Short-wavelength two-photon excitation fluorescence microscopy of tryptophan with a photonic crystal fiber based light source. Opt. Express. 13:5363–5368. [DOI] [PubMed] [Google Scholar]
- 28.Lippitz, M., W. Erker, H. Decker, K. E. van Holde, and T. Basche. 2002. Two-photon excitation microscopy of tryptophan-containing proteins. Proc. Natl. Acad. Sci. USA. 99:2772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kao, F. J. 2004. The use of optical parametric oscillator for harmonic generation and two-photon UV fluorescence microscopy. Microsc. Res. Tech. 63:175–181. [DOI] [PubMed] [Google Scholar]
- 30.Sheppard, C., and R. Kompfner. 1978. Resonant scanning optical microscope. Appl. Opt. 17:2879–2882. [DOI] [PubMed] [Google Scholar]
- 31.Cox, G., E. Kable, A. Jones, I. Fraser, F. Manconi, and M. D. Gorrell. 2003. 3-dimensional imaging of collagen using second harmonic generation. J. Struct. Biol. 141:53–62. [DOI] [PubMed] [Google Scholar]
- 32.Theodossiou, T., G. S. Rapti, V. Hovhannisyan, E. Georgiou, K. Politopoulos, and D. Yova. 2002. Thermally induced irreversible conformational changes in collagen probed by optical second harmonic generation and laser-induced fluorescence. Lasers Med. Sci. 17:34–41. [DOI] [PubMed] [Google Scholar]
- 33.Campagnola, P. J., A. C. Millard, M. Terasaki, P. E. Hoppe, C. J. Malone, and W. A. Mohler. 2002. Three-dimensional high-resolution second-harmonic generation imaging of endogenous structural proteins in biological tissues. Biophys. J. 81:493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams, R. M., W. R. Zipfel, and W. W. Webb. 2005. Interpreting second-harmonic generation images of collagen I fibrils. Biophys. J. 88:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lohmann, W., M. Nilles, and R. H. Bodeker. 1991. In situ differentiation between nevi and malignant melanomas by fluorescence measurements. Naturwissenschaften. 78:456–457. [DOI] [PubMed] [Google Scholar]
- 36.Sterenborg, H. J. C. M., M. Motamedi, R. F. Wagner, M. Duvic, S. Thomsen, and S. L. Jacques. 1994. In vivo fluorescence spectroscopy and imaging of human skin tumors. Lasers Med. Sci. 9:191–201. [DOI] [PubMed] [Google Scholar]
- 37.Sterenborg, H. J. C. M., S. Thomsen, S. L. Jacques, M. Duvic, M. Motamedi, and R. F. Wagner Jr. 1995. In vivo fluorescence spectroscopy and imaging of human skin tumors. Dermatol. Surg. 21:821–822. [DOI] [PubMed] [Google Scholar]
- 38.Wu, Y. C., P. Xi, J. N. Y. Qu, T. H. Cheung, and M. Y. Yu. 2005. Depth-resolved fluorescence spectroscopy of normal and dysplastic cervical tissue. Opt. Express. 13:382–388. [DOI] [PubMed] [Google Scholar]
- 39.Ulrich, V., P. Fischer, I. Riemann, and K. Konigt. 2004. Compact multiphoton/single photon laser scanning microscope for spectral imaging and fluorescence lifetime imaging. Scanning. 26:217–225. [DOI] [PubMed] [Google Scholar]
- 40.Haraguchi, T., T. Shimi, T. Koujin, N. Hashiguchi, and Y. Hiraoka. 2002. Spectral imaging fluorescence microscopy. Genes Cells. 7:881–887. [DOI] [PubMed] [Google Scholar]
- 41.Kahn, E., A. Vejux, D. Dumas, T. Montange, F. Frouin, V. Robert, J. M. Riedinger, J. F. Stoltz, P. Gambert, A. Todd-Pokropek, and G. Lizard. 2004. FRET multiphoton spectral imaging microscopy of 7-ketocholesterol and Nile Red in U937 monocytic cells loaded with 7-ketocholesterol. Anal. Quant. Cytol. Histol. 26:304–313. [PubMed] [Google Scholar]
- 42.Zimmermann, T., J. Rietdorf, A. Girod, V. Georget, and R. Pepperkok. 2002. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett. 531:245–249. [DOI] [PubMed] [Google Scholar]
- 43.Chorvat, D., Jr., J. Kirchnerova, M. Cagalinec, J. Smolka, A. Mateasik, and A. Chorvatova. 2005. Spectral unmixing of flavin autofluorescence components in cardiac myocytes. Biophys. J. 89:L55–L57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rocheleau, J. V., W. S. Head, and D. W. Piston. 2004. Quantitative NAD(P)H/flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response. J. Biol. Chem. 279:31780–31787. [DOI] [PubMed] [Google Scholar]
- 45.Yariv, A. 1967. Quantum Electronics. John Wiley & Sons.
- 46.Bloembergen, N. 1965. Nonlinear Optics. Benjamin, New York.
- 47.Heinz, T. F., C. K. Chen, D. Ricard, and Y. R. Shen. 1982. Spectroscopy of molecular monolayers by resonant second-harmonic generation. Phys. Rev. Lett. 48:478–481. [Google Scholar]
- 48.Gardecki, J. A., and M. Maroncelli. 1998. Set of secondary emission standards for calibration of the spectral responsivity in emission spectroscopy. Appl. Spectrosc. 52:1179–1189. [Google Scholar]
- 49.Weagle, G., P. E. Paterson, J. Kennedy, and R. Pottier. 1988. The nature of the chromophore responsible for naturally occurring fluorescence in mouse skin. J. Photochem. Photobiol. B. 2:313–320. [DOI] [PubMed] [Google Scholar]
- 50.Albota, M. A., C. Xu, and W. W. Webb. 1998. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. Appl. Opt. 37:7352–7356. [DOI] [PubMed] [Google Scholar]
- 51.Chandra, M., K. Vishwanath, G. D. Fichter, E. Liao, S. J. Hollister, and M. A. Mycek. 2006. Quantitative molecular sensing in biological tissues: an approach to non-invasive optical characterization. Opt. Express. 14:6157–6171. [DOI] [PubMed] [Google Scholar]
- 52.Dupasquier, M., P. Stoitzner, A. van Oudenaren, N. Romani, and P. J. Leenen. 2004. Macrophages and dendritic cells constitute a major subpopulation of cells in the mouse dermis. J. Invest. Dermatol. 123:876–879. [DOI] [PubMed] [Google Scholar]
- 53.So, P. T. C., H. Kim, and I. E. Kochevar. 1998. Two-photon deep tissue ex vivo imaging of mouse dermal and subcutaneous structures. Opt. Express. 3:339–350. [DOI] [PubMed] [Google Scholar]
- 54.Thody, A. J., and S. Shuster. 1989. Control and function of sebaceous glands. Physiol. Rev. 69:383–416. [DOI] [PubMed] [Google Scholar]
- 55.Pena, A. M., M. Strupler, T. Boulesteix, and M. C. Schanne-Klein. 2005. Spectroscopic analysis of keratin endogenous signal for skin multiphoton microscopy. Opt. Express. 13:6268–6274. [DOI] [PubMed] [Google Scholar]
- 56.Ramanujam, N. 2000. Fluorescence spectroscopy in vivo. In Encyclopedia of Analytical Chemistry. R. A. Meyers, editor. J. Wiley & Sons, Chichester. 20–56.
- 57.Banerjee, B., B. Miedema, and H. R. Chandrasekhar. 1998. Emission spectra of colonic tissue and endogenous fluorophores. Am. J. Med. Sci. 316:220–226. [DOI] [PubMed] [Google Scholar]
- 58.Sokolov, K., J. Galvan, A. Myakov, A. Lacy, R. Lotan, and R. Richards-Kortum. 2002. Realistic three-dimensional epithelial tissue phantoms for biomedical optics. J. Biomed. Opt. 7:148–156. [DOI] [PubMed] [Google Scholar]
- 59.Maarek, J. M., L. Marcu, W. J. Snyder, and W. S. Grundfest. 2000. Time-resolved fluorescence spectra of arterial fluorescent compounds: reconstruction with the Laguerre expansion technique. Photochem. Photobiol. 71:178–187. [DOI] [PubMed] [Google Scholar]
- 60.Teuchner, K., J. Ehlert, W. Freyer, D. Leupold, P. Altmeyer, M. Stücker, and K. Hoffmann. 2000. Fluorescence studies of melanin by stepwise two-photon femtosecond laser excitation. J. Fluoresc. 10:275–281. [Google Scholar]
- 61.Frushour, B. G., and J. L. Koenig. 1975. Raman scattering of collagen, gelatin, and elastin. Biopolymers. 14:379–391. [DOI] [PubMed] [Google Scholar]
- 62.Morris, M. D., W. F. Finney, R. M. Rajachar, and D. H. Kohn. 2004. Bone tissue ultrastructural response to elastic deformation probed by Raman spectroscopy. Faraday Discuss. 126:159–168 (discussion 169–183). [DOI] [PubMed] [Google Scholar]
- 63.Lazarev, Y. A., B. A. Grishkovsky, and T. B. Khromova. 1985. Amide I band of IR spectrum and structure of collagen and related polypeptides. Biopolymers. 24:1449–1478. [DOI] [PubMed] [Google Scholar]
- 64.Tarnowski, C. P., S. Stewart, K. Holder, L. Campbell-Clark, R. J. Thoma, A. K. Adams, and M. A. Moore. 2003. Effects of treatment protocols and subcutaneous implantation on bovine pericardium: a Raman spectroscopy study. J. Biomed. Opt. 8:179–184. [DOI] [PubMed] [Google Scholar]
- 65.Paschalis, E. P., K. Verdelis, S. B. Doty, A. L. Boskey, R. Mendelsohn, and M. Yamauchi. 2001. Spectroscopic characterization of collagen cross-links in bone. J. Bone Miner. Res. 16:1821–1828. [DOI] [PubMed] [Google Scholar]
- 66.Gillies, R., G. Zonios, R. R. Anderson, and N. Kollias. 2000. Fluorescence excitation spectroscopy provides information about human skin in vivo. J. Invest. Dermatol. 115:704–707. [DOI] [PubMed] [Google Scholar]
- 67.Kollias, N., R. Gillies, M. Moran, I. E. Kochevar, and R. R. Anderson. 1998. Endogenous skin fluorescence includes bands that may serve as quantitative markers of aging and photoaging. J. Invest. Dermatol. 111:776–780. [DOI] [PubMed] [Google Scholar]
- 68.Na, R., I.-M. Stender, M. Henriksen, and H. C. Wulf. 2001. Autofluorescence of human skin is age-related after correction for skin pigmentation and redness. J. Invest. Dermatol. 116:536–540. [DOI] [PubMed] [Google Scholar]
- 69.Roth, S., and I. Freund. 1979. Second harmonic generation in collagen. J. Chem. Phys. 70:1637–1643. [Google Scholar]
- 70.Fine, S., and W. P. Hansen. 1971. Optical second harmonic generation in biological systems. Appl. Opt. 10:2350–2353. [DOI] [PubMed] [Google Scholar]
- 71.Hotary, K. B., E. D. Allen, P. C. Brooks, N. S. Datta, M. W. Long, and S. J. Weiss. 2003. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 114:33–45. [DOI] [PubMed] [Google Scholar]
- 72.Coussens, L. M., B. Fingleton, and L. M. Matrisian. 2002. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 295:2387–2392. [DOI] [PubMed] [Google Scholar]
- 73.Agronskaia, A. V., L. Tertoolen, and H. C. Gerritsen. 2004. Fast fluorescence lifetime imaging of calcium in living cells. J. Biomed. Opt. 9:1230–1237. [DOI] [PubMed] [Google Scholar]
- 74.Lakowicz, J. R., H. Szmacinski, K. Nowaczyk, K. W. Berndt, and M. Johnson. 1992. Fluorescence lifetime imaging. Anal. Biochem. 202:316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao, C., D. J. Moore, M. E. Rerek, C. R. Flach, and R. Mendelsohn. 2005. Feasibility of tracking phospholipid permeation into skin using infrared and Raman microscopic imaging. J. Invest. Dermatol. 124:622–632. [DOI] [PubMed] [Google Scholar]
- 76.Salzer, R., G. Steiner, H. H. Mantsch, J. Mansfield, and E. N. Lewis. 2000. Infrared and Raman imaging of biological and biomimetic samples. Fresenius J. Anal. Chem. 366:712–716. [DOI] [PubMed] [Google Scholar]
- 77.Dunn, A. K., V. P. Wallace, M. Coleno, M. W. Berns, and B. J. Tromberg. 2000. Influence of optical properties on two-photon fluorescence imaging in turbid samples. Appl. Opt. 39:1194–1201. [DOI] [PubMed] [Google Scholar]
- 78.de Grauw, G. J., J. M. Vroom, H. T. M. van der Voort, and H. C. Gerritsen. 1999. Imaging properties in two-photon excitation microscopy and effects of refractive-index mismatch in thick specimens. Appl. Opt. 38:5995–6003. [DOI] [PubMed] [Google Scholar]
- 79.Oheim, M., E. Beaurepaire, E. Chaigneau, J. Mertz, and S. Charpak. 2001. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J. Neurosci. Methods. 111:29–37. [DOI] [PubMed] [Google Scholar]