

Abstract
Voltage-gated sodium channels (Nav) are modulated by many bilayer mechanical amphiphiles, but whether, like other voltage-gated channels (Kv, HCN, Cav), they respond to physical bilayer deformations is unknown. We expressed human heart Nav1.5 pore α-subunit in oocytes (where, unlike αNav1.4, αNav1.5 exhibits normal kinetics) and measured small macroscopic currents in cell-attached patches. Pipette pressure was used to reversibly stretch the membrane for comparison of INa(t) before, during, and after stretch. At all voltages, and in a dose-dependent fashion, stretch accelerated the INa(t) time course. The sign of membrane curvature was not relevant. Typical stretch stimuli reversibly accelerated both activation and inactivation by ∼1.4-fold; normalization of peak INa(t) followed by temporal scaling (∼1.30- to 1.85-fold) resulted in full overlap of the stretch/no-stretch traces. Evidently the rate-limiting outward voltage sensor motion in the Nav1.5 activation path (as in Kv1) accelerated with stretch. Stretch-accelerated inactivation occurred even with activation saturated, so an independently stretch-modulated inactivation transition is also a possibility. Since Nav1.5 channel-stretch modulation was both reliable and reversible, and required stretch stimuli no more intense than what typically activates putative mechanotransducer channels (e.g., stretch-activated TRPC1-based currents), Nav channels join the ranks of putative mechanotransducers. It is noteworthy that at voltages near the activation threshold, moderate stretch increased the peak INa amplitude ∼1.5-fold. It will be important to determine whether stretch-modulated Nav current contributes to cardiac arrhythmias, to mechanosensory responses in interstitial cells of Cajal, to touch receptor responses, and to neuropathic (i.e., hypermechanosensitive) and/or normal pain reception.
INTRODUCTION
Voltage-gated sodium channels (Nav) trigger and propagate action potentials and modify rhythmic firing (1,2). The observation that mechanoreceptor current in touch-sensitive cells (Pacinian corpuscles) has a tetrodotoxin-dependent component (3,4) suggests that, in situ, membrane deformations can modulate Nav channel activity. Positive feedback amplification in excitable cells would ensure that even small mechanical modulations of Nav channel current could be physiologically important; for the same reason, minor kinetic dysmodulation of Nav channels tends to be pathologically significant (subtle kinetic features of Nav channel genetic polymorphs cause severe pathologies and kinetically unsubtle ones are typically lethal). With small deviations in Nav channel kinetics linked to cardiac arrhythmias, epilepsy, neuropathic pain, and so on (5,6), it is critical to determine whether Nav channel kinetics are modulated by membrane deformations. Other voltage-gated channels (VGCs) are modulated by stretch (7), but the mechanoresponsiveness of Nav channels has yet to be tested directly.
Moving voltage-sensor domains in VGCs make intimate and extensive contacts with bilayer molecules (8), and sensor motions are dependent on the presence of particular bilayer lipids (i.e., phospholipids) (9). The membrane channel for which the interaction between bilayer structure and protein structure is best understood is MscL, a bacterial mechanosensitive channel that acts as an osmotic valve. MscL's open/closed equilibria depend on the transbilayer pressure profile (also known as the lateral pressure profile) (10), which in turn depends on both chemical constituents and physical factors including bilayer tension. Whereas MscL is designed to gate in response to near-lytic bilayer stretch, a VGC is designed to gate with membrane depolarization. But the gating motions of VGCs entail multiple energetic negotiations with the bilayer, so it would seem almost unavoidable that bilayer deformations would also affect the conformational stability of VGCs. Our studies with Kv and HCN channels indicate that VGCs are indeed inextricably bilayer-mechanical proteins. In fact, unlike MscL, they are unable to ignore moderate elevations of bilayer tension (7). To establish whether the kinetics of Nav channels, like those of other VGCs, are modulated by membrane deformations, we have examined the responses to stretch of recombinant Nav1.5 expressed in oocytes, recording from patches before, during, and after stretch.
We previously tested stretch on Nav1.4 channel α-subunits, choosing that isoform for its anomalously slow gating (in oocytes), which, we reasoned, would facilitate accurate kinetic measurements (11). Instead, we uncovered a dramatic irreversible stretch effect that side-tracked the issue of reversible stretch effects: for αNav1.4, stretch transforms the anomalous slow-mode into fast-mode gating (a phenomenon also reported by another group (12)). What underlies this is unknown (among the many possibilities are irreversible changes in channel-protein interactions like G-proteins and/or cytoskeleton, and stretch-induced disorganization of channel-caveolin and/or channel-lipid microdomain interactions).
Here, we revisit Nav channel mechanosensitivity using the human heart Nav1.5 α-subunit, an isoform whose gating in oocytes is like that in mammalian cells. Nav1.5 is the major Nav isoform in mammalian cardiomyocytes (13). It triggers and propagates action potentials and contributes to sinoatrial node pacemaking (14). Late Nav1.5 current features in neonatal cardiomyocytes (15) and postinfarction pacemaker myocytes (16) suggest a role for Nav1.5 in plasticity. Cardiomyocyte Nav1.5 channels occur in caveolae (17,18), plasma membrane structures whose bilayer mechanical specializations include cholesterol enrichment and high radius of curvature. Nav 1.5 is found in DRG (19), limbic (20), and certain central (21) and autonomic neurons (22). It is expressed in some skeletal muscle myopathies (23). Jejeunal circular smooth-muscle fibers express Nav1.5 (24,25), as do interstitial cells of Cajal (26), the multifunctional gut cells (27) that mediate mechanosensitive responses in the stomach (28); perhaps related, the epidemiological genomics of sudden infant death reveal an association between a Nav1.5 cardiac channelopathy (long QT syndrome) and gastrointestinal symptoms (29). In subpopulations of human T-lymphocytes, Nav1.5 is needed for invasive activity (30).
Kinetically speaking, evidence suggesting that bilayer mechanics matter to Nav1.5 includes the fact that the inhibition potency of fatty acids on cardiac INa correlates with membrane fluidity (31). In Nav1.4, diverse amphiphilic molecules whose shapes alter the perichannel bilayer modulate inactivation, with bilayer elasticity (not bilayer curvature) being the important bilayer mechanical factor (32,33). Nav channels are bilayer-embedded proteins with multiple conformations. If those conformations have structurally different bilayer-channel interfaces, local restructuring of the bilayer accompanies each conformation change. The relevant lateral interface energetics can be summarized by the lateral pressure profile (34–36). In brief, at each bilayer leaflet's lipid-water interface, a large surface tension pulls outward on any membrane-embedded protein, whereas nearer the bilayer midplane, lateral forces are compressive, tending to compact the protein in that region. The task of the peripherally disposed voltage sensor array in a VGC is to use energy stored in the electric field to generate lateral forces that can pull open the hinged gating bundle of the central pore domain (37). In this light, it would be surprising if perturbors of the bilayer's lateral pressure profile, be they physical or chemical, failed to modulate the gating of VGCs.
Physical factors that contribute to bilayer mechanics include temperature (elevated temperatures thin the bilayer (38)) and pressure (hyperbaric pressure thickens and stiffens the bilayer (39)), as well as the factor varied here, extension (or “stretch”). Membrane stretch thins and softens the bilayer; in doing so, it simultaneously increases the (“interfacial”) surface tension at each leaflet and reduces the extent of midbilayer compression (35).
Kv, Cav, and HCN channels all exhibit reversible gating changes with stretch (40–42). We postulate (43) that their stretch-modulated gating arises not through changes in the intensity of the electric field but by stretch alteration of the lateral pressure profile at the channel-bilayer interface. This view is inferred from Shaker Kv channels, in which stretch accelerates the independent voltage-dependent transitions without changing the quantity of gating charge moved (44). In Shaker ILT, the concerted pre-pore opening transition decelerates with stretch, but again, without changing the amount of charge moved (45).
We demonstrate here that Nav1.5 channel gating is inherently sensitive to membrane stretch: in a fully reversible manner, stretch caused acceleration of the rate-limiting voltage-dependent step leading to activation of ionic current. Inactivation also accelerated. Native Nav1.5 channels behaving in this way would provide a phasic positive feedback component to mechanotransduction. Judgments about the plausibility of such a scenario in any particular tissue or cell type will require further investigation, but it is noteworthy that the mechanical stimuli used here were no different from those that activate putative mechanotransducer (TRPC1-based) channels (46).
METHODS
Oocyte preparation and cRNA injection
Xenopus laevis oocytes were injected with 20–40 ng cRNA as described previously (42). The human heart Nav1.5 plasmid (pSP64T-hH1, kindly provided by Al George (47)) was linearized with XbaI and used to produce capped cRNA by in vitro transcription using the SP6 Ambion Message Machine (Austin TX).
Solutions
High-K bath solution contained (in mM) 89 KCl, 0.4 CaCl2, 5 HEPES, 0.8 MgCl2 (pH 7.5 with KOH). In the recording pipette solution the KCl was substituted with 89 mM NaCl (pH adjusted with NaOH), and 40 μM GdCl3 was added to block endogenous stretch-activated cation channels. As described previously (42), gadolinium was sometimes fully effective, but sometimes stretch-activated cation channel activity made records unusable, despite our addition of degassed acidified stock solution (100 mM GdCl3) to the degassed pipette solution immediately before recording (7).
Electrophysiology
Manually devitellinated oocytes were transferred to an inverted microscope rig. Recording pipettes, prepared as previously (45), had resistances of 2–4 MΩ; more details are given below (Resolving the currents). Suction of −10 to −15 mm Hg was usually sufficient to cause seals to form. Macroscopic currents measured from cell-attached patches (Axopatch 200B, Axon Instruments, Foster City, CA) at RT (20–22°C) were filtered at 5 kHz (Axopatch 200B low-pass filter) and analyzed using WinASCD (Guy Droogmans) and Origin (Microcal, Northampton, MA).
We used N51A (Garner Glass, Claremont, CA) borosilicate for pipettes and a soda-glass-coated polishing filament. Low-noise record requires the borosilicate, but polarization-induced patch creep may occur unless fire-polishing is done using a soda-glass-coated filament (7,48). We did not visually monitor patches, but our anecdotal experience (from brief unintentional use of a soda-glass-free polishing filament) confirmed that pipettes polished with a naked filament yielded highly unstable patches.
Experimental voltage protocols were controlled via pClamp 6. Vhold was −110 mV and P/N linear subtraction (N = 8) with hyperpolarizing steps was used, with suction applied during the P/N steps for the “stretch” runs. The magnitude of the residual capacitative transients did not change with stretch (for example, note the relaxation on return to holding voltage in the expanded traces of Fig. 3).
FIGURE 3.

Reversible effects of stretch in a single trace B/D/A train. (A) A no-stretch B/D/A (to show the reproducibility of voltage-elicited currents) for the same patch as B. (B) An experimental B/D/A (D = −30 mmHg, n = 1) whose first two elements are expanded below. Vhold provided −110 mV of driving force for the endogenous stretch-activated cation channels, so the “clean” stretch trace shows that gadolinium had effectively inhibited these channels. Scale bars: vertical, 20 pA; horizontal, 10 ms for trains, 1.5 ms for expanded sections.
Since gadolinium ions right-shift the g(V), it was necessary to locate the foot of the g(V) for each patch. Suction during seal formation dilutes and precipitates pipette-tip gadolinium to an unknown extent (bath solution bicarbonate is inevitable and precipitates gadolinium).
Membranes were stretched via negative pipette pressure, controlled and monitored by a DPM-1B transducer (Bio-Tek, Winooski, VT). Typically, −30 or −40 mmHg was sufficient to elicit mechanosensitive responses. To test for reversibility and obtain difference currents, protocols were structured as before/during/after stretch (B/D/A) sets. Data were discarded when rundown of Nav1.5 current led to poor signal/noise ratios.
Resolution of currents
The best way to reversibly stretch a voltage-clamped area of membrane is to apply suction to a gigasealed patch. A caveat: stimulus intensity is known only qualitatively (i.e., stronger pipette suction means higher membrane tension) unless the patch is visualized to enable application of Laplace's law (which relates patch curvature, applied pressure, and membrane tension). For multiple reasons, visualization would be incompatible with resolving Nav1.5 channel activation kinetics. Low-noise, mechanically robust seals require thick-walled, strongly tapered, fire-polished, sylgard-coated borosilicate pipettes (7), as used here. Patch imaging requires thin-walled pipettes with little taper and no sylgard. For Nav1.5 channels, however, despite the fact that we always used sylgard-coated thick-walled pipettes, our working bandwidth RC-filtered the rising phase of macroscopic Nav1.5 currents and attenuated fast-rising peaks at voltages >∼15 mV above the foot of the g(V). Inactivation kinetics were not a problem. We interpret our data within these acknowledged limitations.
RESULTS
Nav 1.5 current and before/during/after-stretch protocols
For the irreversible responses of Nav1.4 to membrane stretch (11,12) and the reversible ones of Cav channels (40), the classic piece of VGC data—the peak current I/V plot—provides key information, but for Nav1.5 channels it proved more useful to focus on the I(t) data. (I(t) data are also most useful for Kv1 and HCN channels; rate-limiting depolarization-dependent transitions are stretch sensitive (41,42,49) in Kv1 and HCN, but not in Cav channels). For steeply voltage dependent channels, slight acceleration of the rate-limiting activation step substantially increases current amplitude near the foot of the g(V) but will be inconsequential near the top of the g(V). The slope factor for Nav1.5 activation is about e-fold per ∼6 mV (50) and Nav1.5 inactivation is also steeply voltage-dependent (2,51,52), further complicating peak INa/V plots. We therefore used INa/V data only for locating the foot region of the g(V) and we monitored INa(t) using B/D/A protocols. This enabled us to look for reversible kinetic changes over a range of voltages, paying attention to any filtering limitations.
Stretch acceleration of activation and inactivation
Fig. 1 A plots a Nav1.5 channel current family (15 mV increments) without (black) then with (red) stretch (n = 4 runs/voltage). Stretch caused current to develop (activate) faster and to decay (inactivate) faster. For the peak-current I/V plot (Fig. 1 B), four families without stretch (n = 16 runs/voltage) interspersed with three families with stretch (n = 12 runs/voltage) were averaged. The plot perforce includes B/D/A information, but seen from the perspective of this plot, the effect of stretch on Nav1.5 channels is unimpressive and could be summarized as “a slight increase in the negative-resistance voltage region, a slight decrease at more depolarized voltages—overall, not much.”
FIGURE 1.

Membrane stretch and Nav1.5 currents: families, I/V plots and use of B/D/A protocols. (A) Currents (running averages, n = 4) for steps from Vhold = −110 mV in 15-mV increments to successively more depolarized voltages. black, no-stretch controls; red, during stretch (using −30 mmHg). Here and throughout, imperfectly subtracted capacitive transients mark the start and end of voltage steps. For reference, the −50 mV current pair is labeled. Early current is time-expanded at right. (B) For the same patch, the average peak current (16 control, 12 stretch runs) obtained from alternating no-stretch/stretch families (n = 16 and 12 runs for 0 and −30 mmHg, respectively). The −50 mV points are labeled and +10 mV currents are shown as an inset (arrow). (C) For the same patch as in A and B, n = 40 B/D/A protocols were done next (using −30 mmHg applied continuously for the during-stretch traces) in the order −50 mV, −60 mV, −70 mV, −30 mV (the −30 mV expansion shows that stretch accelerated current onset). B/D/As are plotted black, red, and gray, respectively, here and elsewhere. (D) Stretch difference currents for these B/D/A sets; difference current above the x axis represents “stretch inhibited” (SI) and that below “stretch augmented” (SA) INa. (E) Finally, the beginning of a dose-response at −70 mV (n = 40) obtained in the order −30 mmHg, 0 mmHg, −40 mmHg, 0 mmHg (pink, black, red, gray, respectively). The patch ruptured during an attempt at −45 mmHg.
Fig. 1 C shows B/D/A data from this patch in the form of I(t), with larger sample sizes to diminish the stochastic noise. Since −80 mV was below the activation threshold (Fig. 1 B), we began at −70 mV. For this preparation (meaning, for the particular combination of patch size, rate of current rundown, inherent patch noise, efficacy of gadolinium inhibition of endogenous mechanosensitive cation channels, Nav1.5 expression level, 60 Hz interference, pipette RC properties), n = 40 (or n = 120 for the B/D/A) handled the noise at the smallest test step (−70 mV) and cleanly resolved the stretch-accelerated activation at the largest step (−30 mV; here, the no-stretch activation was clearly not filtered by the pipette RC). At −70 mV, −60 mV, and −50 mV, the combination of stretch-accelerated activation and stretch-accelerated inactivation resulted in larger currents that peaked sooner. The larger currents do not mean that stretch increased the number of channels in the patch; rather, they reflect changed kinetics in a fixed population of channels. Changed kinetics are starkly evident where stretch traces cross no-stretch traces during inactivation (−60 mV, −50 mV). Further, at large depolarizations, stretch did not increase gmax (i.e., peak current at −30 mV did not increase; the good signal/noise ratio here, in the %gmax(V) ≈ 1 region, results from intrinsically minimal channel noise plus the still-large driving force on Na+). Thus, even as g(V) saturated, stretch-accelerated activation and inactivation were both evident. This suggests that stretch accelerated the Nav1.5 voltage sensor motions that trigger the two processes.
Stretch difference currents (Fig. 1 D) were obtained by subtracting stretch traces from the averaged before/after traces. Note that these difference currents, considered over a range of voltages that would be experienced during an action potential, show that in a freely firing excitable cell, stretch-modulated Nav1.5 channel current would produce first stretch-augmented (SA) then stretch-inhibited (SI) sodium current. It is important to note that SA current would dominate near the resting potential, and then SI current would rapidly come to dominate as the cell depolarized. In a repetitively firing cell, the alternating actions should facilitate higher frequencies, unless stretch also decreases the rate of recovery from inactivation, a point on which we have no information as yet.
Before the patch broke we managed to compare −70 mV current at 0 mmHg (black), −30 mmHg (pink), and −40 mmHg (red) (Fig. 1 E). Nav1.5 currents ran down gradually, so the control (0 mmHg) amplitudes were less than earlier (Fig. 1 C, −70 mV). Reversible stretch sensitivity was, nevertheless intact, and a dose effect was observed, with activation and inactivation both more strongly accelerated at −40 mmHg than at −30 mmHg.
Currents for the Fig. 2 A patch (sequence: −60 mV, −65 mV, −70 mV) illustrate that reversible acceleration of activation and inactivation was unequivocal even when rundown was apparent within a B/D/A (as at −60 mV). Stretch-accelerated inactivation was always clear-cut, but activation, the faster process, was problematic because of filtering, as seen in B/D/As for another patch (Fig. 2 B). Even without stretch, activation at −50 mV was filtered by the pipette RC (likewise the Fig. 1 B inset for currents at +10 mV, which has filtered activation but unequivocal stretch-accelerated inactivation).
FIGURE 2.

Nav1.5 current activation as seen in different patches. (A) A patch in which activation (no stretch) was well-resolved at the voltages tested. Three B/D/A sets (n = 40 runs) obtained in the order -60 mV, -65 mV, -70 mV (the -70 mV “after” trace is missing because the patch ruptured at run 11 destroying the running average, but visual monitoring till that point showed currents at the control amplitude). (B) A patch in which two B/D/A sets (n = 40) were obtained in the order -50 mV, -65 mV. The RC properties of the recording pipette filtered current activation, even at -65 mV, likely because the sylgard-coating, which unavoidably varies from pipette to pipette, was insufficient.
Briefer stretch stimuli
In the B/D/As above, stretch was applied continuously for >40 s (and where n = 80 was used, stretch was sustained twice as long), but briefer stretch stimuli also reversibly accelerated Nav1.5 kinetics, as seen in Fig. 3 (a B/D/A for which n = 1). Fig. 3, A and B, shows the no-stretch and experimental sets, respectively, for the same patch; reversible stretch acceleration is evident despite stochastic noise and other shortcomings. At −60 mV, it is clear that stretch accelerated current onset, that it increased peak INa (inactivation was overly noisy), and that during the −40 mV train, inactivation was faster with stretch. In general, however, Nav channel currents from patches required more extensive sampling. This contrasts with HCN and Kv1 channel currents, where we found stochastic noise to be less problematic (41,42) or where use of pulse trains is more feasible (41,42,49).
Elevated tension and patch curvature
When applied pipette pressure acts on channels by stretching the membrane, elevated membrane tension achieved by suction (negative pipette pressure) or by “blowing” (positive pipette pressure) should be equally effective. The B/D/A data of Fig. 4 (for which n = 80) show that negative and positive pressure acted in the same fashion. Thus, it is safe to assume that elevated membrane tension, and not some perturbation that differs with the sign of membrane curvature (convex versus concave), was the effective stimulus. B/D/As at various other stretch intensities (Supplementary Material) showed that both −15 mmHg and +16 mmHg were too small to generate effective tension.
FIGURE 4.

Reversible stretch effects with negative or positive pipette pressure. Three B/D/A sets (n = 80) from a patch during steps to −40 mV. Endogenous stretch-activated cation channel activity in this oocyte batch was little affected by the usual level of gadolinium, so a higher level (nominally 120 μM) was used. This right-shifted the g(V), putting −40 mV very near threshold. Other dose response data from this patch are provided as Supplementary Material. When current rundown became excessive, pipette pressure was slowly increased until, at −79 mmHg, the patch ruptured. Pressures shown here were, thus, moderate.
Temporal scaling of normalized currents yields a single stretch acceleration factor
Nav1.5 is kinetically too complex for us to fit our data sets to a kinetic scheme, but we can ask by what numerical factor stretch accelerated the overall processes of activation and inactivation. Fig. 5 shows that for high-quality B/D/A data sets (i.e., sets showing well-resolved, nonattenuated peak current both with stretch and without stretch), amplitude normalization (to the peak) followed by time scaling caused stretch and no-stretch activation phases to coincide. Strikingly, this simultaneously resulted in complete overlap of the current's inactivation phase. In every instance where activation was well resolved, this double overlap was found. This result signifies two important points: 1), stretch elicited no new rate-determining transitions; and 2), stretch accelerated the preexisting rate-limiting steps in activation and inactivation to precisely the same extent. Table 1 lists the temporal scaling factors that produced this kind of overlap (that is, the stretch acceleration factor). The overlapped traces are shown in Fig. 5 or in Supplementary Material.
FIGURE 5.

Stretch acceleration factors from B/D/A sets. The peak amplitude of stretch traces was normalized to the peak amplitudes of before and after traces. Then the time base of the stretch traces was expanded until the activation traces overlapped. This caused the inactivation trajectories to overlap as well. The cases illustrated (top to bottom) are from Fig 1 C, −60 mV; Fig. 2 A, −65 mV, Fig. 2 A, −60 mV; and Fig. 2 B, −65 mV (entries 2, 8, 9,and 10 of Table 1) and had stretch acceleration factors (rescaling of time axis) of 1.40, 1.40, 1.45, and 1.35, respectively. The overall data quality (noise, stationarity for before and after, minimal RC filtering of activation time course during stretch) was best in the top set and least good in the bottom set where, during stretch, activation is assumed to have been somewhat filtered.
TABLE 1.
The stretch acceleration factor
| Entry | Patch | Vm (mV) | Pipette pressure (mmHg) | Stretch acceleration factor* | Original traces in… | Scaled traces in… | Notes |
|---|---|---|---|---|---|---|---|
| 1 | A | −70 | −30 | 1.40 | Fig. 1 C | Fig. S2 | |
| 2 | A | −60 | −30 | 1.40 | Fig. 1 C | Fig. 5 | |
| 3 | A | −50 | −30 | 1.35 | Fig. 1 C | Fig. S2 | |
| 4 | A | −30 | −30 | 1.30 | Fig. 1 C | Fig. S2 | |
| 5 | A | −70 | −30 | 1.55 | Fig. 1 E | Fig. S2 | 2nd time at −70 mV …and a higher pressure “after” not obtained |
| 6 | A | −70 | −40 | 1.85 | Fig. 1 E | Fig. S2 | |
| 7 | B | −70 | −30 | 1.40 | Fig. 2 A | Fig. S2 | |
| 8 | B | −65 | −30 | 1.40 | Fig. 2 A | Fig. 5 | |
| 9 | B | −60 | −30 | 1.45 | Fig. 2 A | Fig. 5 | |
| 10 | C | −65 | −30 | 1.35 | Fig. 2 B | Fig. 5 | activation likely filtered |
| 11 | D | −40 | −37 | 1.50 | Fig. 4 | Fig. S2 | |
| 12 | D | −40 | +31 | 1.50 | Fig. 4 | Fig. S2 | |
| 13 | D | −35 | −27 | 1.45 | Fig. S1 B† | Fig. S2 | |
| 14 | D | −15 | −27 | 1.30 | Fig. S1 B | Fig. S2 | patch lysed at −79 mmHg |
Rounded to the nearest 0.05.
Supplementary data figure.
A given pressure yields the same tension in different patches only if, under pressure, they have the same radius of curvature. With that proviso, what emerges from Table 1 is that for our typical stimulus (−30 mmHg), stretch accelerated both activation and inactivation by ∼1.4-fold. The “outlier” (scale factor 1.85) is for a larger stretch stimulus. If we think of the Nav channel as a simple two-state (closed, open) channel, then the observed temporal scaling can be summarized as
 |
and this equality directly relates the scaling factor (the stretch acceleration factor, which in effect is the ratio of activation time constants with/without stretch) to two important parameters for voltage gated channels, namely the g(V) midpoint and the g(V) slope factor in the following way. If g(V) relations are taken to be Boltzmann relations, a Nav channel g(V) midpoint reflects the work needed to change the closed/open ratio in the absence of voltage (53). A left-shifted g(V) signifies a destabilized closed state that opens sooner on depolarization. In Kv1 (Shaker), stretch accelerates the rate-limiting activation step and left-shifts the g(V) with no change in the slope factor (45). Assuming a comparable scenario for Nav1.5, stretch acceleration will relate to the ratio left-shift/slope factor according to
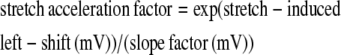 |
(e.g., see equations 1.25 and 1.28 in Jackson (54), and see, in Conti et al. (57), in the context of hyperbaric pressure, how a comparable use is made of the ratio of sodium channel activation time constants). Since the e-fold slope factor for the Nav1.5 channel g(V) is ∼6 mV (50), observed stretch-acceleration factors of, say, 1.4 and 1.9 would correspond to g(V) left-shifts of 2 mV and 4 mV, respectively. This is small, but for Nav channels operating near the foot of the g(V), this much shift could matter. Comparing a stretch trace at −70 mV with no-stretch traces at -65 mV (Fig. 2 A) illustrates this point: stretch at −70 mV (stretch acceleration factor, 1.4) had almost half the effect of 5 mV depolarization. In rhythmically active excitable cells, a 2- to 3-mV depolarization—or its stretch equivalent—would not be inconsequential for Nav channel activity.
DISCUSSION
The oocyte patch preparation
Human heart Nav1.5 channels were expressed in oocytes, yielding small (<150 pA) macroscopic currents in cell-attached patches. Within-patch comparisons were made of currents before, during, and after patch stretch. Importantly, patch “history” effects (i.e., consistent and irreversible stretch-induced changes in gating behavior) were not a factor. This contrasts sharply with both the oocyte's endogenous stretch-activated cation channels (55) (now classified as TRPC1-containing channels (46)) and with Nav1.4 α-subunits expressed in oocytes (11,12). The microstructural changes behind those patch history effects would have occurred in our patches, but for Nav1.5 channel responses they were not relevant.
Acceleration of Nav1.5 channel kinetics by stretch
After a voltage step, the time course of voltage-dependent ionic current is, by definition, shaped by the rate-limiting voltage-dependent transition. If that transition is also mechanosensitive, this will be seen in the time course of the ionic current. Stretch elicited no current at the holding potential (−110 mV), but during depolarizing steps (from the foot to the head of the g(V) relation and beyond), both activation and inactivation were accelerated reversibly by stretch. Effects of stretch were dose-dependent: in a given patch at a given potential, the extent of acceleration increased with increased stretch intensity.
Thus, for Nav1.5 channels (as we have argued for Kv1 and HCN channels) the simplest explanation for our data sets is that voltage sensor motions accelerate with stretch. In Kv1, stretch accelerates the independent depolarization-driven activation motions without changing the amount of charge moved (44). In HCN2 channels, too, the rate-limiting depolarization-driven process (in that case, deactivation (56)) accelerates with stretch (42). As with Kv1 (i.e., Shaker (44)), Nav1.5 activation and inactivation accelerated the same-fold with stretch; the full overlap of normalized-then-scaled Nav1.5 traces is a strong indicator that (as in Kv1) stretch introduced no novel transitions.
Gating current in the context of stretch has not been studied, but hyperbaric pressure has been used. This is important because hyperbaric pressure laterally compresses (thickens) the bilayer by increasing the orderliness and packing density of the hydrocarbon tails (39), whereas stretch has the opposing effect. For squid axon Na channels, hyperbaric pressure decelerates both ionic and gating current (57,67)). Moreover, amplitude-time normalization (comparable to Fig. 5) of the squid data at any given voltage yields superimposed high versus low pressure (i.e., atmospheric) traces. We postulate that stretch-accelerated Nav kinetics and hyperbaric-pressure-decelerated Nav kinetics represent responses along a bilayer mechanical continuum from abnormally extended to abnormally compressed. The bacterial stretch-activated channel, MscS, provides a precedent for this interpretation: under elevated pressure (magnitudes like those used on squid axon) versus atmospheric pressure, MscS kinetics indicate that high pressure favors the closed state in a way that points to lateral compression of the bilayer as the operative factor (58).
Are activation and inactivation independently stretch-sensitive processes?
In Nav1.5, activation and inactivation transitions depend directly on different voltage sensor motions (2,51,52). We can unequivocally assert that at least one activation transition (i.e., the rate-limiting one) accelerates with stretch. The observation that inactivation continued to show stretch-acceleration at voltages supramaximal for activation is consistent with the possibility that, as with Kv1 (44), Nav1.5 inactivation is an independently stretch-sensitive transition. If not, then kinetic coupling could explain the same-fold stretch acceleration of the two Nav1.5 processes (likewise for squid axon sodium channels and hyperbaric pressure).
A nonspecific effect?
If activation in all VGCs accelerated with stretch, it might be argued that Nav 1.5 acceleration was nonspecific, akin to raising the temperature. But stretch-acceleration of depolarization-dependent activation is not universal among VGCs. The rate-limiting transition for activation does not accelerate with stretch in N-type or L-type Cav channels (40). Nor does it do so in Kv3 channels (Shaw2 F335A) (45). In Kv3 and Cav channels, stretch increases peak current magnitude without affecting the rate of activation and, thus, increases peak current at all voltages; consequently, for those channels, the effect of stretch is clearly evident when plotting peak I/V relations with/without stretch (contrary, as we saw, to the case for Nav1.5). In Shaker ILT (a Kv1 mutant in which a concerted voltage-dependent step before pore opening limits the rate of current activation), stretch slows the rate of activation.
The mechanostimulus was elevated bilayer tension
Small stretch-induced increases in current magnitude (e.g. <2-fold) can raise the question of whether stretch somehow augmented the area of channel-bearing membrane (see (7)). The kinetic signature of Nav1.5 (accelerated currents) plus unequivocal reversibility obviated this concern here. Moreover, prolonged stretch (> 1 min) had the same effect as brief stretch (a few seconds). If stretch acted via an increased area of channel-bearing membrane, peak current would increase at all voltages (not just near the foot of the g(V) relation) and amplitude scaling alone (i.e., with no time scaling) would yield trace overlap; this was never the case. Increased area clearly could not explain, say, Fig. 3 B where, during a single stretch stimulus early current at -60 mV increased and late current at -40 mV decreased; kinetic changes are needed to explain this. Also, with voltage saturation, peak current increase “saturated” (Fig. 1 C, at −30 mV) even as stretch accelerated both activation and inactivation. Finally, the sign of long-range membrane curvature was irrelevant: stressing the patch concavely versus convexly had the same effect (accelerated activation and inactivation). Thus, the relevant “membrane deformation” was elevated tension in the plane of the membrane.
Possible consequences of stretch modulation of Nav channels
Nav1.5 is the principal mediator of cardiac INa. Various cardiac mechanoelectric feedback phenomena are inhibited by gadolinium. Nav channels (like all VGCs and many other channels) are inhibited by gadolinium (59). To assess whether inhibition of Nav1.5 might contribute to gadolinium's antiarrhythmic effect on stretch-induced ectopic cardiac activity, Li and Baumgarten (60) examined ventricular myocyte INa. They concluded that gadolinium acting on INa could suppress stretch-induced arrhythmias (and hence, that these arrhythmias do not necessarily implicate stretch-activated cation channels). Now that we have evidence for stretch-modulation of Nav1.5 channels, it will be important to test whether cardiomyocyte INa contributes to mechanoelectric feedback phenomena. We have preliminary unpublished data (C. E. Morris and W. R. Giles) for rat ventricular myocyte Nav channels showing stretch responses like those of the recombinant channels studied here.
Nociceptive nerve endings that lack functional Nav1.7 channels make humans completely unable to sense pain (61). It seems urgent, therefore, to determine whether INa in mechanosensory and nociceptive nerve endings responds to membrane stretch, especially since electrophysiological (3) and immunocytochemical evidence (4) suggest that Pacinian corpuscle Nav channels participate in tactile mechanotransduction. During repetitive activity, the responses we report would “tune up” action potentials, allowing for both earlier firing and repolarization. In nociceptive endings, the inherent mechanosensitivity of Nav channels might contribute to allodynia (hypermechanosensitivity) in neuropathic pain; Nav1.8 channels of damaged sensory axons are implicated in ectopic mechanosensitivity (62). Allodynia in nociceptive nerve endings during inflammatory cytokine signaling has proved to be unrelated to TRPV1 (a putative mechanosensory cation channel) (63); instead the hypermechanosensitivity correlates with elevated INa density. If nociceptive nerve ending Nav channels are stretch-modulated, increased INa density (relative to IK) could lower the mechanical threshold for action potential trains.
Based on whole-cell recordings, Nav1.5 channels in interstitial cells of Cajal detect shear stress (26). To revisit that preparation using membrane patches would be particularly worthwhile given the new evidence that these cells are mechanosensitive pacemakers for the gut (28).
The lateral pressure profile and Nav channel side effects
Nav channels, like Kv1 channels, presumably have four peripherally disposed voltage sensor domains (64). The resulting cruciform cross section, unlike earlier (“conventional”) VGC models (65), makes for extensive lipid-protein interactions that would include mobile voltage sensor residues. This may be why, like other VGCs (43), Nav channel gating is modulated by many lipid stress agents. Stretch, we have now shown, is one such “agent” for Nav channels. Other physical lipid stress agents are elevated pressure and reduced temperature; both yield more ordered (and thicker and stiffer) bilayers and both slow Nav channel activation and inactivation (66–68). For Nav channels, as for other VGCs, bilayer mechanics appear to modulate both activation and inactivation motions. For this reason, it is likely that many side effects of lipophilic drugs on Nav channels are, literally, side (lateral-pressure) effects (31–33).
Table 2 lists lipidic agents and drugs that modulate Nav channels. Other VGCs respond to many of these same agents (e.g., see (42,43)). Do Nav1.5 channels (and other Nav, HCN, Kv, and Cav channels, etc.) have “low-affinity binding sites” for resveratrol, ethanol, halothane, canabinoids, propofol, etc.? We suggest that they do not. Since all VGCs respond to membrane stretch (with different rate-limiting steps determining the precise nature of any given channel's response), a more appealing view (43) of the channel-agent interactions listed in Table 2 is one that recognizes that Nav channels, like most membrane proteins (69), are modulated by bilayer mechanics. We suggest that low-affinity binding-site models (e.g. (70,71)) and bilayer mechanical models converge when the nanostructure of the channel-bilayer interface is part of the energetic equation. Both physical and chemical lipid stress agents alter that lateral-pressure profile of Nav channels, and in both cases, this translates to modulated gating.
TABLE 2.
Side effects: Nav channel modulation by bilayer mechanical agents
| Halothane and isoflurane |
| Human heart Nav1.5: halothane and isoflurane accelerate inactivation, stabilize inactivated states (72). |
| Nav1.2, Nav1.4, Nav1.6 (not Nav1.8): isoflurane inhibits at clinical levels (73). |
| Rat nerve terminal INa: isoflurane (0.8 mM) left-shifts inactivation, slows recovery from inactivation; halothane slows recovery from fast inactivation (74). |
| Propofol |
| Rat nerve terminal INa: propofol (5 μM) left-shifts inactivation, delays recovery from inactivation (74). |
| Neuronal INa: propofol analogue 2,6 di-tert-butylphenol inhibits (more potent than propofol) (75). |
| Short-chain alkanols and complex alcohols |
| Human heart Nav1.5: ethanol, 44 mM, reduces single-channel open probability (76). |
| Nav1.2: perfluorinated heptanol [CF(3)(CF(2))(5)CH(2)OH] inhibits, as do other alcohols (ethanol, heptanol, CF(3)CH(2)OH) (73). |
| Cyclobutanes |
| Nav1.2: fluorocyclobutane (1-chloro-1,2,2-trifluorocyclobutane) inhibits (73). |
| DRG neuron INa: 1-chloro-1,2,2-trifluorocyclobutane inhibits peak INa, left-shifts inactivation (77). |
| Fatty acids |
| Human heart Nav1.5: n-3 polyunsaturated acids inhibit (78). |
| Human breast cancer cell INa: n-3 polyunsaturated acids inhibit (79). |
| Human bronchial smooth muscle INa: eicosapentaenoic acid (ED50 2 μM) left-shifts inactivation. Docosahexaenoic, arachidonic, stearic, oleic acids – similar, less potent (80). |
| Rat DRG neuron INa: lysophosphatidic acid left-shifts activation and inactivation. (81). |
| Cholesterol, ceramide and amphiphiles |
| Nav1.4: nonphysiological amphiphiles that reduce bilayer stiffness (β-octyl-glucoside, Genapol X-100, Triton X-100) left-shift inactivation. Depletion of cholesterol (decreases bilayer stiffness), left-shifts inactivation (32). |
| Rat sensory neuron INa: ceramide left-shifts activation, increases peak INa (82). |
| Resveratrol, menthol, thymol, capsaicin, capsazepine, anandamide, nicotine |
| Human heart Nav1.5: resveratrol (cardioprotective polyphenol in red grapes), 26 μM, inhibits late current in long-QT mutant, R1623Q; in wild-type Nav1.5, it inhibits late current and diminishes peak INa with IC50 = 77 μM (83). |
| Nav1.2, Nav1.4: thymol (<l50 μM) and menthol (<600 μM): voltage-dependent block (84). |
| Nav 1.4: capsaicin, capsazepine left-shift inactivation. Amphiphiles promoting the opposite lipid monolayer curvature also left-shift inactivation (33). |
| Rat DRG neuron INa: anandamide (endogenous cannabinoid) suppresses INa (85). |
| Rat trigeminal ganglion nociceptor INa: nicotine inhibits (nAChRs not involved) (86). |
Acknowledgments
We thank Cicely Gu and Wei Lin for technical help and Dorothy Hanck and Chris Miller for helpful discussions.
This work was supported by a grant to C.E.M. from the Canadian Institutes of Health Research.
Editor: Francisco Bezanilla.
References
- 1.Darbon, P., C. Yvon, J. C. Legrand, and J. Streit. 2004. INaP underlies intrinsic spiking and rhythm generation in networks of cultured rat spinal cord neurons. Eur. J. Neurosci. 20:976–988. [DOI] [PubMed] [Google Scholar]
- 2.Maltsev, V. A., and A. I. Undrovinas. 2006. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 69:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolanowski, S. J., Jr. 1984. Intensity and frequency characteristics of pacinian corpuscles. III. Effects of tetrodotoxin on transduction process. J. Neurophysiol. 51:831–839. [DOI] [PubMed] [Google Scholar]
- 4.Pawson, L., and S. J. Bolanowski. 2002. Voltage-gated sodium channels are present on both the neural and capsular structures of Pacinian corpuscles. Somatosens. Mot. Res. 19:231–237. [DOI] [PubMed] [Google Scholar]
- 5.Fedida, D., P. M. Orth, J. C. Hesketh, and A. M. Ezrin. 2006. The role of late I and antiarrhythmic drugs in EAD formation and termination in Purkinje fibers. J. Cardiovasc. Electrophysiol. 17(Suppl 1):S71–S78. [DOI] [PubMed] [Google Scholar]
- 6.Kahlig, K. M., S. N. Misra, and A. L. George, Jr. 2006. Impaired inactivation gate stabilization predicts increased persistent current for an epilepsy-associated SCN1A mutation. J. Neurosci. 26:10958–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris, C. E., P. F. Juranka, W. Lin, T. J. Morris, and U. Laitko. 2006. Studying the mechanosensitivity of voltage-gated channels using oocyte patches. Methods Mol. Biol. 322:315–329. [DOI] [PubMed] [Google Scholar]
- 8.Hilgemann, D. W. 2004. Biochemistry. Oily barbarians breach ion channel gates. Science. 304:223–224. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt, D., Q. X. Jiang, and R. MacKinnon. 2006. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 444:775–779. [DOI] [PubMed] [Google Scholar]
- 10.Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- 11.Tabarean, I. V., P. Juranka, and C. E. Morris. 1999. Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys. J. 77:758–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shcherbatko, A., F. Ono, G. Mandel, and P. Brehm. 1999. Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys. J. 77:1945–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abriel, H., and R. S. Kass. 2005. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 15:35–40. [DOI] [PubMed] [Google Scholar]
- 14.Lei, M., S. A. Jones, J. Liu, M. K. Lancaster, S. S. Fung, H. Dobrzynski, P. Camelliti, S. K. Maier, D. Noble, and M. R. Boyett. 2004. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J. Physiol. 559:835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baruscotti, M., D. DiFrancesco, and R. B. Robinson. 2000. Na+ current contribution to the diastolic depolarization in newborn rabbit SA node cells. Am. J. Physiol. Heart Circ. Physiol. 279:H2303–H2309. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez, J. L., F. Aimond, P. Lorente, and G. Vassort. 2000. Late post-myocardial infarction induces a tetrodotoxin-resistant Na+current in rat cardiomyocytes. J. Mol. Cell. Cardiol. 32:1169–1179. [DOI] [PubMed] [Google Scholar]
- 17.Yarbrough, T. L., T. Lu, H. C. Lee, and E. F. Shibata. 2002. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ. Res. 90:443–449. [DOI] [PubMed] [Google Scholar]
- 18.Cronk, L. B., B. Ye, T. Kaku, D. J. Tester, M. Vatta, J. C. Makielski, and M. J. Ackerman. 2007. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 4:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, C. J., S. D. Dib-Hajj, M. Renganathan, T. R. Cummins, and S. G. Waxman. 2003. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 278:1029–1036. [DOI] [PubMed] [Google Scholar]
- 20.Hartmann, H. A., L. V. Colom, M. L. Sutherland, and J. L. Noebels. 1999. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat. Neurosci. 2:593–595. [DOI] [PubMed] [Google Scholar]
- 21.Wu, L., K. Nishiyama, J. G. Hollyfield, and Q. Wang. 2002. Localization of Nav1.5 sodium channel protein in the mouse brain. Neuroreport. 13:2547–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scornik, F. S., M. Desai, R. Brugada, A. Guerchicoff, G. D. Pollevick, C. Antzelevitch, and G. J. Perez. 2006. Functional expression of “cardiac-type” Nav1.5 sodium channel in canine intracardiac ganglia. Heart Rhythm. 3:842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teener, J. W., and M. M. Rich. 2006. Dysregulation of sodium channel gating in critical illness myopathy. J. Muscle Res. Cell Motil. 27:291–296. [DOI] [PubMed] [Google Scholar]
- 24.Ou, Y., S. J. Gibbons, S. M. Miller, P. R. Strege, A. Rich, M. A. Distad, M. J. Ackerman, J. L. Rae, J. H. Szurszewski, and G. Farrugia. 2002. SCN5A is expressed in human jejunal circular smooth muscle cells. Neurogastroenterol. Motil. 14:477–486. [DOI] [PubMed] [Google Scholar]
- 25.Ou, Y., P. Strege, S. M. Miller, J. Makielski, M. Ackerman, S. J. Gibbons, and G. Farrugia. 2003. Syntrophin gamma 2 regulates SCN5A gating by a PDZ domain-mediated interaction. J. Biol. Chem. 278:1915–1923. [DOI] [PubMed] [Google Scholar]
- 26.Strege, P. R., Y. Ou, L. Sha, A. Rich, S. J. Gibbons, J. H. Szurszewski, M. G. Sarr, and G. Farrugia. 2003. Sodium current in human intestinal interstitial cells of Cajal. Am. J. Physiol. Gastrointest. Liver Physiol. 285:G1111–G1121. [DOI] [PubMed] [Google Scholar]
- 27.Faussone-Pellegrini, M. S. 2005. Interstitial cells of Cajal: once negligible players, now blazing protagonists. Ital. J. Anat. Embryol. 110:11–31. [PubMed] [Google Scholar]
- 28.Won, K. J., K. M. Sanders, and S. M. Ward. 2005. Interstitial cells of Cajal mediate mechanosensitive responses in the stomach. Proc. Natl. Acad. Sci. USA. 102:14913–14918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke 3rd, G. R., M. J. Ackerman, A. R. Zinsmeister, P. Thapa, and G. Farrugia. 2006. Gastrointestinal symptoms in families of patients with an SCN5A-encoded cardiac channelopathy: evidence of an intestinal channelopathy. Am. J. Gastroenterol. 101:1299–1304. [DOI] [PubMed] [Google Scholar]
- 30.Fraser, S. P., J. K. Diss, L. J. Lloyd, F. Pani, A. M. Chioni, A. J. George, and M. B. Djamgoz. 2004. T-lymphocyte invasiveness: control by voltage-gated Na+ channel activity. FEBS Lett. 569:191–194. [DOI] [PubMed] [Google Scholar]
- 31.Leifert, W. R., E. J. McMurchie, and D. A. Saint. 1999. Inhibition of cardiac sodium currents in adult rat myocytes by n-3 polyunsaturated fatty acids. J. Physiol. 520:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundbaek, J. A., P. Birn, A. J. Hansen, R. Sogaard, C. Nielsen, J. Girshman, M. J. Bruno, S. E. Tape, J. Egebjerg, D. V. Greathouse, G. L. Mattice, R. E. Koeppe 2nd, and O. S. Andersen. 2004. Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of micelle-forming amphiphiles and cholesterol. J. Gen. Physiol. 123:599–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lundbaek, J. A., P. Birn, S. E. Tape, G. E. Toombes, R. Sogaard, R. E. Koeppe 2nd, S. M. Gruner, A. J. Hansen, and O. S. Andersen. 2005. Capsaicin regulates voltage-dependent sodium channels by altering lipid bilayer elasticity. Mol. Pharmacol. 68:680–689. [DOI] [PubMed] [Google Scholar]
- 34.Cantor, R. S. 2002. Size distribution of barrel-stave aggregates of membrane peptides: influence of the bilayer lateral pressure profile. Biophys. J. 82:2520–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gullingsrud, J., and K. Schulten. 2004. Lipid bilayer pressure profiles and mechanosensitive channel gating. Biophys. J. 86:3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiggins, P., and R. Phillips. 2005. Membrane-protein interactions in mechanosensitive channels. Biophys. J. 88:880–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yifrach, O., and R. MacKinnon. 2002. Energetics of pore opening in a voltage-gated K+ channel. Cell. 111:231–239. [DOI] [PubMed] [Google Scholar]
- 38.Simon, S. A., S. Advani, and T. J. McIntosh. 1995. Temperature dependence of the repulsive pressure between phosphatidylcholine bilayers. Biophys. J. 69:1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scarlata, S., H. McBath, and H. C. Haspel. 1995. Effect of lipid packing on the conformational states of purified GLUT-1 hexose transporter. Biochemistry. 34:7703–7711. [DOI] [PubMed] [Google Scholar]
- 40.Calabrese, B., I. V. Tabarean, P. Juranka, and C. E. Morris. 2002. Mechanosensitivity of N-type calcium channel currents. Biophys. J. 83:2560–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu, C. X., P. F. Juranka, and C. E. Morris. 2001. Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys. J. 80:2678–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin, W., U. Laitko, P. Juranka, and C. Morris. 2007. Dual stretch responses of mHCN2 pacemaker channels: accelerated activation, accelerated deactivation. Biophys. J. 92:1559–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris, C. E., and P. F. Juranka. 2006. Lipid stress at play: mechanosensitivity of voltage-gated channels. Hamill, O., S. Simon, and D. Benos, editors. Mechanosensitive Ion Channels, Part B. Current Topics in Membranes, Volume 59. 297–337. [DOI] [PubMed]
- 44.Laitko, U., and C. E. Morris. 2004. Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J. Gen. Physiol. 123:135–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laitko, U., P. F. Juranka, and C. E. Morris. 2006. Membrane stretch slows the concerted step prior to opening in a Kv channel. J. Gen. Physiol. 127:687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maroto, R., A. Raso, T. G. Wood, A. Kurosky, B. Martinac, and O. P. Hamill. 2005. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 7:179–185. [DOI] [PubMed] [Google Scholar]
- 47.Makita, N., P. B. Bennett, Jr., and A. L. George, Jr. 1996. Multiple domains contribute to the distinct inactivation properties of human heart and skeletal muscle Na+ channels. Circ. Res. 78:244–252. [DOI] [PubMed] [Google Scholar]
- 48.Gil, Z., S. D. Silberberg, and K. L. Magleby. 1999. Voltage-induced membrane displacement in patch pipettes activates mechanosensitive channels. Proc. Natl. Acad. Sci. USA. 96:14594–14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabarean, I. V., and C. E. Morris. 2002. Membrane stretch accelerates activation and slow inactivation in Shaker channels with S3–S4 linker deletions. Biophys. J. 82:2982–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McNulty, M. M., and D. A. Hanck. 2004. State-dependent mibefradil block of Na+ channels. Mol. Pharmacol. 66:1652–1661. [DOI] [PubMed] [Google Scholar]
- 51.Clancy, C. E., and Y. Rudy. 1999. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 400:566–569. [DOI] [PubMed] [Google Scholar]
- 52.Irvine, L. A., M. S. Jafri, and R. L. Winslow. 1999. Cardiac sodium channel Markov model with temperature dependence and recovery from inactivation. Biophys. J. 76:1868–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hille, B. 2001. Ionic Channels of Excitable Cells. Sinauer Associates, Sunderland, MA.
- 54.Jackson, M. 2006. Molecular and Cellular Biophysics. Cambridge University Press, New York.
- 55.Hamill, O. P., and D. W. McBride, Jr. 1997. Induced membrane hypo/hyper-mechanosensitivity: a limitation of patch-clamp recording. Annu. Rev. Physiol. 59:621–631. [DOI] [PubMed] [Google Scholar]
- 56.Mannikko, R., F. Elinder, and H. P. Larsson. 2002. Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. Nature. 419:837–841. [DOI] [PubMed] [Google Scholar]
- 57.Conti, F., R. Fioravanti, J. R. Segal, and W. Stuhmer. 1982. Pressure dependence of the sodium currents of squid giant axon. J. Membr. Biol. 69:23–34. [DOI] [PubMed] [Google Scholar]
- 58.Macdonald, A. G., and B. Martinac. 2005. Effect of high hydrostatic pressure on the bacterial mechanosensitive channel MscS. Eur. Biophys. J. 34:434–441. [DOI] [PubMed] [Google Scholar]
- 59.Elinder, F., and P. Arhem. 1994. Effects of gadolinium on ion channels in the myelinated axon of Xenopus laevis: four sites of action. Biophys. J. 67:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li, G. R., and C. M. Baumgarten. 2001. Modulation of cardiac Na+ current by gadolinium, a blocker of stretch-induced arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 280:H272–H279. [DOI] [PubMed] [Google Scholar]
- 61.Cox, J. J., F. Reimann, A. K. Nicholas, G. Thornton, E. Roberts, K. Springell, G. Karbani, H. Jafri, J. Mannan, Y. Raashid, L. Al-Gazali, H. Hamamy, E. M. Valente, S. Gorman, R. Williams, D. P. McHale, J. N. Wood, F. M. Gribble, and C. G. Woods. 2006. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 444:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roza, C., J. M. Laird, V. Souslova, J. N. Wood, and F. Cervero. 2003. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol. 550:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin, X., and R. W. Gereau 4th. 2006. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J. Neurosci. 26:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Long, S. B., E. B. Campbell, and R. Mackinnon. 2005. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908. [DOI] [PubMed] [Google Scholar]
- 65.Tombola, F., M. M. Pathak, and E. Y. Isacoff. 2005. How far will you go to sense voltage? Neuron. 48:719–725. [DOI] [PubMed] [Google Scholar]
- 66.Collins, C. A., and E. Rojas. 1982. Temperature dependence of the sodium channel gating kinetics in the node of Ranvier. Q. J. Exp. Physiol. 67:41–55. [DOI] [PubMed] [Google Scholar]
- 67.Conti, F., I. Inoue, F. Kukita, and W. Stuhmer. 1984. Pressure dependence of sodium gating currents in the squid giant axon. Eur. Biophys. J. 11:137–147. [DOI] [PubMed] [Google Scholar]
- 68.Jonas, P. 1989. Temperature dependence of gating current in myelinated nerve fibers. J. Membr. Biol. 112:277–289. [DOI] [PubMed] [Google Scholar]
- 69.McIntosh, T. J., and S. A. Simon. 2006. Roles of bilayer material properties in function and distribution of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 35:177–198. [DOI] [PubMed] [Google Scholar]
- 70.Kang, J. X., and A. Leaf. 1996. Evidence that free polyunsaturated fatty acids modify Na+ channels by directly binding to the channel proteins. Proc. Natl. Acad. Sci. USA. 93:3542–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shahidullah, M., T. Harris, M. W. Germann, and M. Covarrubias. 2003. Molecular features of an alcohol binding site in a neuronal potassium channel. Biochemistry. 42:11243–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stadnicka, A., W. M. Kwok, H. A. Hartmann, and Z. J. Bosnjak. 1999. Effects of halothane and isoflurane on fast and slow inactivation of human heart hH1a sodium channels. Anesthesiology. 90:1671–1683. [DOI] [PubMed] [Google Scholar]
- 73.Shiraishi, M., and R. A. Harris. 2004. Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J. Pharmacol. Exp. Ther. 309:987–994. [DOI] [PubMed] [Google Scholar]
- 74.Ouyang, W., G. Wang, and H. C. Hemmings, Jr. 2003. Isoflurane and propofol inhibit voltage-gated sodium channels in isolated rat neurohypophysial nerve terminals. Mol. Pharmacol. 64:373–381. [DOI] [PubMed] [Google Scholar]
- 75.Haeseler, G., and M. Leuwer. 2003. High-affinity block of voltage-operated rat IIA neuronal sodium channels by 2,6 di-tert-butylphenol, a propofol analogue. Eur. J. Anaesthesiol. 20:220–224. [DOI] [PubMed] [Google Scholar]
- 76.Klein, G., A. Gardiwal, A. Schaefer, B. Panning, and D. Breitmeier. 2007. Effect of ethanol on cardiac single sodium channel gating. Forensic Sci. Int. In press. [DOI] [PubMed]
- 77.Ratnakumari, L., T. N. Vysotskaya, D. S. Duch, and H. C. Hemmings, Jr. 2000. Differential effects of anesthetic and nonanesthetic cyclobutanes on neuronal voltage-gated sodium channels. Anesthesiology. 92:529–541. [DOI] [PubMed] [Google Scholar]
- 78.Xiao, J., T. V. Nguyen, K. Ngui, P. J. Strijbos, I. S. Selmer, C. B. Neylon, and J. B. Furness. 2004. Molecular and functional analysis of hyperpolarisation-activated nucleotide-gated (HCN) channels in the enteric nervous system. Neuroscience. 129:603–614. [DOI] [PubMed] [Google Scholar]
- 79.Isbilen, B., S. P. Fraser, and M. B. Djamgoz. 2006. Docosahexaenoic acid (omega-3) blocks voltage-gated sodium channel activity and migration of MDA-MB-231 human breast cancer cells. Int. J. Biochem. Cell Biol. 38:2173–2182. [DOI] [PubMed] [Google Scholar]
- 80.Jo, T., H. Iida, S. Kishida, H. Imuta, H. Oonuma, T. Nagata, H. Hara, K. Iwasawa, M. Soma, Y. Sato, T. Nagase, R. Nagai, and T. Nakajima. 2005. Acute and chronic effects of eicosapentaenoic acid on voltage-gated sodium channel expressed in cultured human bronchial smooth muscle cells. Biochem. Biophys. Res. Commun. 331:1452–1459. [DOI] [PubMed] [Google Scholar]
- 81.Seung Lee, W., M. P. Hong, T. Hoon Kim, Y. Kyoo Shin, C. Soo Lee, M. Park, and J. H. Song. 2005. Effects of lysophosphatidic acid on sodium currents in rat dorsal root ganglion neurons. Brain Res. 1035:100–104. [DOI] [PubMed] [Google Scholar]
- 82.Zhang, Y. H., M. R. Vasko, and G. D. Nicol. 2002. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J. Physiol. 544:385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wallace, C. H., I. Baczko, L. Jones, M. Fercho, and P. E. Light. 2006. Inhibition of cardiac voltage-gated sodium channels by grape polyphenols. Br. J. Pharmacol. 149:657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haeseler, G., D. Maue, J. Grosskreutz, J. Bufler, B. Nentwig, S. Piepenbrock, R. Dengler, and M. Leuwer. 2002. Voltage-dependent block of neuronal and skeletal muscle sodium channels by thymol and menthol. Eur. J. Anaesthesiol. 19:571–579. [DOI] [PubMed] [Google Scholar]
- 85.Kim, H. I., T. H. Kim, Y. K. Shin, C. S. Lee, M. Park, and J. H. Song. 2005. Anandamide suppression of Na+ currents in rat dorsal root ganglion neurons. Brain Res. 1062:39–47. [DOI] [PubMed] [Google Scholar]
- 86.Liu, L., W. Zhu, Z. S. Zhang, T. Yang, A. Grant, G. Oxford, and S. A. Simon. 2004. Nicotine inhibits voltage-dependent sodium channels and sensitizes vanilloid receptors. J. Neurophysiol. 91:1482–1491. [DOI] [PubMed] [Google Scholar]