

Abstract
We examined the distributions of short tandemly repeated DNAs (microsatellites) in nine complete microbial genomes (Saccharomyces cerevisiae, Archaeoglobus fulgidus, Escherichia coli, Haemophilus influenzae, Helicobacter pylori, Methanococcus jannaschii, Mycoplasma pneumoniae, M. genitalium, and Synechocystis PCC6803.) These repeats contribute differently to the global features of these genomes, and we explore the evolutionary implications of these differences by empirical examination of length polymorphisms at 20 long triplet-repeats repeats in S. cerevisiae, and by comparison of observed and expected repeat distributions. All of a sample of 20 microsatellites found in S. cerevisiae are highly polymorphic in length, suggesting that mutation pressure overcomes overall selection for small genome size that will tend to shorten or eliminate unnecessary DNA. By comparison, prokaryotes have fewer long repeats than expected, except for a few statistically improbable repeats that appear to function in gene regulation. Finally, we find that in all these genomes there is an excess of repeats shorter than those traditionally considered to be microsatellites. This finding suggests that even in prokaryotes these repeats are being generated by mutational pressures. These results have important potential implications for understanding genome stability and evolution in these microbial species.
Microsatellite loci, short tandemly repeated motifs of 1–6 bases, form one of the most biologically interesting patterns in eukaryotic DNA (1). Such tracts are a major component of higher organism DNAs and are hypervariable in length (2, 3) as a result of replication slippage processes (4, 5). Thus, microsatellites have become extremely popular molecular markers (6, 7). Whereas most of them are presumed to evolve neutrally, the most widely studied exceptions are the growing number of triplet-repeat loci that cause genetic diseases in humans (8). In prokaryotes, strong positive selective pressures are associated with highly mutable microsatellite tracts that control pathogenicity (9).
Little is currently known about microsatellites in simple organisms (10). The recent sequencing of nine complete microbial genome sequences affords a novel opportunity to investigate microsatellite evolution in small genomes in which absolute abundances of repeated patterns can be calculated (11). We find here that short tandem repeats contribute very differently to the global features of these genomes, and we explore the implications of these differences through the empirical examination of length polymorphisms at 20 repeats in Saccharomyces cerevisiae, and by generating expected distributions of repeats based on genome size and base content.
MATERIALS AND METHODS
Yeast Strains.
Seven strains of S. cerevisiae, and a single strain of each of five closely related species, were obtained from the American Type Culture Collection: S. cerevisiae (ATCC nos. 9763, 38976, 7752, 4098, and 32701), S. c. ellipsoideus (834 and 4108), S. pastorianus subsp. arbignensis (2366), S. diasticus (36902), S. bayanus (36022), S. ilicis (2341), and Zygosaccharomyces prioranus (2601).
PCR.
PCR primers (Research Genetics, Huntsville, AL) were manually designed (Table 1) to amplify products of 100–400 nucleotides. Loci were typed by PCR and scored as previously described (12) or were scored by using an Applied Biosystems 373 automated sequencer and Genescan software.
Table 1.
Twenty polymorphic microsatellite loci in the yeast genome
| Nuclear loci | Primers (forward and reverse) | Locus | Gene and function | Chr. | Repeat | A. A | No. alleles | No. het. |
|---|---|---|---|---|---|---|---|---|
| RMP2 | cccttttaaggaagagcaagcc ccccaataagctgagagtgg | SCPTSY7 | 66 bp 3′ of ribo-nuclease P (RMP2) | XIII | (aat)33 | — | 11 (7/5) | 4 (2/2) |
| 3′ORF1 | ggacagtgggaggagggaaatgg gctttacgcactagattgtgcgg | SC8358 | 37 bases 3′ of ORF similar to a.a. permeases | IV | (tat)24 | — | 5 (4/3) | 0 |
| ORF1 | gcagcgaagctaaacctgttgg caagcattccgaaattgtggg | YBR150c | Potential permease | II | (cat)21 | (D) | 5 (4/2) | 1 (0/1) |
| ORF2 | ggtgactctaacggcagagtgg cccgtatactgcaagtagatcc | YOR267C | Potential permease | XV | (caa)20 | (Q) | 8 (6/3) | 4 (2/2) |
| SSN6 | cagcatcctgctcaacaaacgcc gcagctgttgtcttggtaggggc | YSCSSN6 | SSN6 protein kinase; repressor of transcription | II | (caa)20 | (Q) | 7 (5/5) | 1 (0/1) |
| 3′ORF2 | gctacagcacttgctgaacataagc ccaatcctggatctagttttccc | SCCHRIX | 93 bp 3′ of YIL130W: putative regulatory protein | IX | (taa)19 | — | 5 (2/3) | 1 (0/1) |
| 3′ORF3 | ggcagcgagattgctcttgt cctcccgactgtggcattggcg | SCORFTAN | 51 bp 3′ of J1545; unknown function | X | (taa)16 | — | 6 (4/3) | 3 (0/1) |
| ORF3 | ctgctcaacttgtgatgggttttgg cctcgttactatcgtcttcatcttgc | SC8132X | ORF of unknown function | XVI | (gaa)16 | (E) | 11 (10/4) | 6 (5/1) |
| FAB1 | ctacaattccaaaggtccttcgc cgtgccattgtcgtttgaggg | U01017 | FAB1 kinase; essential for vacuole function | VI | (aat)15 | (N) | 6 (6/3) | 2 (2/2) |
| SIS2 | gtaaatatgctgcgtgaatttgcc caaaatcgttatgaaattgggtggg | SCYKR072C | SIS2; aspartic acid-rich protein | XI | (gac)13 | (D) | 6 (6/5) | 5 (2/3) |
| ORF4 | gctcgcagggagaaatctgcttcc cttcatcggtatccgttccactagg | SC8337 | Unknown function | XIII | (gat)11 | (D) | 4 (4/2) | 0 |
| SRP40 | gaaaattaaagttgacgaagtgcc gatccactggagctagagtcgg | YSCSRP40X | SRP40; RNA polymerase I & III supressor | XI | (agc)11 | (S) | 4 (4/3) | 3 (3/0) |
| NAB3 | cgatggaatcgaatttgacgcccc cctcatcctcaccgtcttcagcggc | SCU05314 | NAB3; polyadenylated RNA-binding gene | XVI | (gaa)10 | (E) | 5 (5/3) | 2 (1/1) |
| CCP | ctgggcagaaccgcccataagagg gacctccctttttcgacagaggcg | YSCCCP | CCP; cytochrome c peroxidase precursor | XI | (gct)9 | (A) | 6 (5/2) | 3 (3/0) |
| TFA1 | gaatgattactacgctgctttggc cggaccatatcaaacgtcctc | SCU12825 | TFAI; TFIIE large subunit | XI | (tta)10 | (N) | 6 (5/3) | 2 (1/1) |
| FUN12 | cgcaagaatccaccgcaagcc gtcttaccggtatcgacatgaccc | YSCFUN12A | FUN12; essential gene | I | (gaa)9 | (E) | 5 (4/2) | 2 (1/1) |
| NGR1 | ccaataagattatcatggggacgcc gcaccgtcttgttcgatatacggg | SCNGR1 | NGR1; negative growth regulatory gene | II | (cag)9 | (Q) | 3 (3/2) | 0 |
| SNF5 | gcaacgacaccaacagttactgagg cgctggagctaagggcacttgacc | YSCSNF5 | SNF5; transcriptional activator | II | (caa)9 | (Q) | 3 (3/2) | 3 (1/2) |
| GRR | gctgcacccacctgatatacatcc cgttgcatccctaacctcacttgc | YSCGRR1 | GRR; required for glucose repression | X | (cag)9 | (Q) | 3 (3/1) | 1 (1/0) |
| 3′ORF4 | gcaacccatgcttggttcaactcc gctttaaccattaagctaagagagc | YSCMTCG03 | intergenic region of trna-lys and trna-arg | MT | (taa)10 | — | 4 | Haploid |
A total of 12 yeast strains were genotyped, seven S. cerevisiae and five strains of closely related species. All loci examined, without exception, were found to be length-polymorphic, with 3-11 alleles. Both intra- and interspecific variation was found. If the numbers of alleles and heterozygotes found in the seven strains of S. cerevisiae and the five additional Saccharomyces species differ, they are given in parentheses. Primer sites were conserved across all strains and species except for the loci NAB3, NGR1, and GRR, which failed to amplify in Zygosaccharomyces, the most distantly related yeast strain. MT, mitochondrial.
Genome Analysis.
Six completed genome sequences (Table 2) were obtained by anonymous FTP from GenBank (13–17). Helicobacter pylori (18) and Archaeoglobus fulgidus were obtained from The Institute for Genomic Research (personal communication). A search algorithm was written in True Basic for the Macintosh and used to search for mono- to hexanucleotide repeats. Although these expected numbers are based on a very simple model of genome size and base composition, excesses generally can be accepted as indicative of a replication slippage process (1).
Table 2.
The nine evolutionarily diverse microbial species surveyed in this study
| Species | Genome size (Mb) | A+T | Evolutionary domain | Type of organism |
|---|---|---|---|---|
| M. genitalium | 0.6 | 61% | Bacteria | Obligate pathogen |
| M. pneumoniae | 0.8 | 60% | Bacteria | Obligate pathogen |
| M. jannaschii | 1.6 | 68% | Archea | Methanogenic autotroph |
| H. pylori | 1.7 | 60% | Bacteria | Obligate pathogen |
| H. influenzae | 1.8 | 61% | Bacteria | Obligate pathogen |
| A. fulgidus | 2.2 | 52% | Archea | Chemoautotroph |
| Synechocystis PCC6803 | 3.6 | 52% | Bacteria | Autotroph |
| E. coli | 4.6 | 50% | Bacteria | Heterotroph |
| S. cerevisiae | 13.1 | 61% | Eukarya | Saprophyte |
Expected repeat frequencies were determined for mononucleotide repeats by an analytical solution based on the probabilities of the occurrence of A, C, G, and T in each genome. For a genome of Ng nucleotides, the probability of obtaining a repeat of a given length is given by: Prepeat = f(B)y, where f(B) is the frequency in the genome of base or sequence B, and y is the number of repeats. We began by determining the total of all subunits involved in repeats up to length j, where j is sufficiently long that the probability of obtaining a repeat of this length or longer is very small. For the present calculations, we chose a j of 20, because little error is introduced by neglecting higher values of j. T, the total of all subunits, is obtained from:
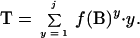 |
The fraction of this total consisting of repeats of length y is Ty = (f(B)y⋅y)/T, and the total number of repeats of length y is (Ty⋅Ng)/y. Division by y adjusts for the fact that long repeats will constitute a larger fraction of T per repeat than shorter repeats. These calculations were reiterated for each repeat type—that is, separate calculations were carried out for each of the four bases. The summed totals are presented here. More detailed results for each of the bases will be given elsewhere (unpublished work). Expected frequencies for di- through hexanucleotide repeats were obtained by artificially generating genomes of the same size and with the same frequencies of A, C, G, and T as those seen in the actual genomes.
Significance Levels for Observed and Expected Distributions of Mononucleotide Repeats in Coding and Noncoding Regions Were Generated By Simulation.
Each genome was divided into coding and noncoding regions based on GenBank documentation. Within each region, bases were shuffled to produce sets of sequences with base compositions identical to the original sequences but in randomized order (19). Runs of mononucleotides were determined in each of these shuffled sequences, and these runs were summed over the entire genome. The observed number of runs of each length was compared with the distribution of simulated runs of that same length by a t test. In Table 3, t-values significant at the 0.01 and 0.001 levels are represented by + and ∗, respectively. If none of the simulations produced runs of a given length, the t test could not be performed for that length. All programs are available from dfield@ucsd.edu.
Table 3.
Mononucleotide repeat distributions compared with random expectations
| bp | Yeast noncoding
|
Yeast coding
|
E. coli
|
Synechocystis
|
A. fulgidus
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| OBS. | EXP. | OBS. | EXP. | OBS. | EXP. | OBS. | EXP. | OBS. | EXP. | |
| 1 | 1,740,821* | 1.850e6 | 4,379,591* | 4.748e6 | 2.51 e6* | 2.60 e6 | 1.56 e6* | 2.01 e6 | 1,121,996* | 1,224,660 |
| 2 | 444,997* | 477,245 | 1,278,285* | 1.205e6 | 678,930* | 651,915 | 548,381* | 502,282 | 323,227* | 306,189 |
| 3 | 136,534* | 133,286 | 349,532* | 320,168 | 163,340* | 163,033 | 170,787* | 125,918 | 83,639* | 76,616 |
| 4 | 50,567* | 39,464 | 107,038* | 88,627 | 42,901* | 40,782 | 58,969* | 31,632 | 25,168* | 19,188 |
| 5 | 19,400* | 12,141 | 32,748* | 25,399 | 13,837* | 10,204 | 216.7* | 7,963 | 8,441* | 4,809 |
| 6 | 7,134* | 3,823 | 9,798* | 7,484 | 4,123* | 2,554 | 6,779* | 2,008 | 2,247* | 1,206 |
| 7 | 3,867* | 1,220 | 3,135* | 2,253 | 1,000* | 639 | 1,596* | 508 | 338 | 303 |
| 8 | 1,680* | 392 | 978* | 690 | 217* | 160 | 325* | 129 | 25* | 76 |
| 9 | 962* | 127 | 301 | 214 | 22* | 40 | 56* | 33 | 3+ | 19 |
| 10 | 663* | 41 | 91+ | 67 | 1* | 10 | 15 | 8 | 5 | |
| 11 | 440* | 13.3 | 41* | 21 | 0+ | 3 | 1 | 2 | 1.2 | |
| 12 | 318* | 4.3 | 26* | 6.7 | 0.6 | 0 | 0.5 | |||
| 13 | 235* | 1.4 | 9+ | 2.1 | ||||||
| 14 | 122* | 0.45 | 8 | 0.7 | ||||||
| 15 | 91* | 0.15 | 2 | 0.2 | 1 | |||||
| 16 | 64* | 0.05 | 2 | 0.07 | ||||||
| 17 | 48 | 0.01 | 3 | 0.02 | ||||||
| 18 | 28 | 0.005 | 1 | 0.007 | ||||||
| 19 | 31 | 0.001 | ||||||||
| 20 | 23 | 5.2e-4 | ||||||||
| 21 | 16 | 1.7e-4 | ||||||||
| 22 | 15 | 5.6e-5 | ||||||||
| 23 | 14 | 1.8e-5 | ||||||||
| 24 | 18 | 5.9e-6 | ||||||||
| 25 | 7 | 1.9e-6 | 1 | 2.7e-6 | ||||||
| 26 | 9 | 6.2e-7 | 1 | 8.9e-7 | ||||||
| 27 | 4 | 2.0e-7 | ||||||||
| 28 | 3 | 6.5e-8 | 1 | 9.4e-8 | ||||||
| 29 | 3 | 2.1e-8 | ||||||||
| 30 | 1 | 6.9e-9 | ||||||||
| 31 | 4 | 2.2e-9 | ||||||||
| 32 | 0 | 7.2e-10 | 1 | 1.1e-9 | ||||||
| 33 | 1 | 2.3e-10 | ||||||||
| 34 | 1 | 7.6e-11 | ||||||||
| 35 | 2 | 2.5e-10 | ||||||||
| 36 | 1 | 8.0e-12 | ||||||||
| 37 | 1 | 2.6e-12 | ||||||||
| 42 | 1 | 9.3e-15 | ||||||||
| bp |
H. influenzae
|
H. pylori
|
M. jannaschii
|
M. pneumoniae
|
M. genitalium
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| OBS. | EXP. | OBS. | EXP. | OBS. | EXP. | OBS. | EXP. | OBS. | EXP. | |
| 1 | 873,540* | 997,261 | 756,016* | 912,277 | 746,961* | 864,585 | 369,878* | 448,959 | 255,010* | 301,952 |
| 2 | 262,733* | 254,222 | 198,402* | 231,986 | 235,355* | 227,410 | 125,332* | 113,776 | 82,649* | 79,295 |
| 3 | 78,444* | 68,284 | 80,370* | 61,803 | 77,898* | 67,123 | 37,389* | 29,954.2 | 25,491* | 23,310 |
| 4 | 27,927* | 19,145 | 35,390* | 17,121 | 28,410* | 21,310 | 12,113* | 8,152.4 | 10,534* | 7,365 |
| 5 | 10,398* | 5,545 | 15,315* | 4,890 | 11,655* | 7,038.5 | 4,730* | 2,279.7 | 4,594* | 2,422 |
| 6 | 3,892* | 1,643 | 6,459* | 1,428 | 5,072* | 2,371.2 | 1,455* | 651 | 1,855* | 813 |
| 7 | 1,045* | 494 | 1,872* | 424 | 1,469* | 806.4 | 360 | 188.8 | 798* | 275 |
| 8 | 145 | 150 | 361* | 127 | 96* | 275.5 | 30 | 55.4 | 170* | 94 |
| 9 | 16* | 46 | 56+ | 38 | 8* | 94.3 | 6 | 16.4 | 10* | 32 |
| 10 | 2+ | 14 | 6+ | 12 | 1* | 32.3 | 4.9 | 0+ | 10.9 | |
| 11 | 4.3 | 2 | 3.5 | 0* | 11.1 | 1.5 | 3.7 | |||
| 12 | 1.3 | 5* | 1.1 | 0+ | 3.8 | 0.4 | 1.3 | |||
| 13 | 0.4 | 6* | 0.3 | 1.3 | 0.4 | |||||
| 14 | 10* | 0.1 | 0.5 | |||||||
| 15 | 7* | 3.1 e-2 | 1 | |||||||
| 16 | 3* | 9.4 e-3 | 2 | |||||||
| 17 | ||||||||||
| 18 | ||||||||||
| 19 | 1 | |||||||||
| 20 | ||||||||||
| 21 | ||||||||||
| 22 | ||||||||||
| 23 | ||||||||||
| 24 | 1 | |||||||||
OBS are the observed and EXP the expected numbers of repeats of a given length, based on genome size and overall base composition (see Materials and Methods). Expected numbers greater than observed numbers are given in boldface. Significant deviations from expected values at the 0.01 and 0.001 levels are indicated by + and ∗, respectively. The genomic locations of three of the long monoculeotide repeats were identified by blast searches: (G)19: 77 bp 5′ to M. genitalium polC DNA polymerase III (U39681); (A)16: 211bp 5′ to M. pneumoniae: putative lipoprotein (MPAE000039); (T)16: 143 bp 3′ to M. penumoniae putative lipoprotein (MPAE000002).
RESULTS
Excess Repeats Found in the Yeast Genome Are Found in Both Coding and Noncoding Regions and Are Highly Polymorphic in Length, Suggesting Strong Mutational Pressures That Are Not Completely Overcome By Selection on Small Genome Size.
Although specific loci containing microsatellite DNA have been known to exist in the yeast genome for many years, a systematic analysis of the locations and nature of these repeats has not yet been conducted, nor has the degree of length polymorphism at these loci between strains been examined (10, 11, 20–23). All mono- through trinucleotide tracts that can be classified as traditional microsatellites —that is, loci with ≥8 repeats (3) that have a high probability of being polymorphic, are presented on a map of the yeast genome (Fig. 1). These loci are distributed throughout the genome and do not show a marked tendency to be concentrated in particular regions of chromosomes. The average distance between these repeats is about 25 kb, compared with about 6 kb in humans (5). Twelve of these triplet-repeats are among the longest repeat loci that have been found in a survey of GenBank (10).
Figure 1.

The distribution of the >455-long mono-, di-, and trinucleotide repeats on the chromosomes of S. cerevisiae. Mononucleotides greater than 20 subunits, and di- and trinucleotides greater than 8 subunits in length are shown. Each horizontal bar represents one repeat locus. One hundred thirty mononucleotides were found, all of which are poly(A/T), 233 dinucleotides (201 = AT/TA, 24 = CA/TG, 8 = CT/GA), 92 trinucleotides predominantly composed of five triplets: ATT, 16; GAA, 14; CAA, 14; CAG, 12; and CAT, 12. These code for N, E, Q, Q, D when found in coding regions.
Long mono- and dinucleotide repeats are found almost exclusively in nontranslated regions, whereas long trinucleotide repeats are found in both the translated and nontranslated regions (Tables 3 and 4). These mono- and dinucleotide repeats are extremely A+T biased, a tendency that is also present but less pronounced in trinucleotide repeats. The yeast genome is 61% A+T, but this is not sufficient to explain the overrepresentation of poly (A/T) and (AT/TA) tracts (24).
Table 4.
Cumulative numbers of di- to hexa-repeats with eight or more units (and maximum lengths found for each repeat class)
| Species | Di | Tri | Tetra | Penta | Hexa | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| S. c. COD | 12 | (16) | 72 | (24) | 0 | (7) | 0 | (3) | 0 | (6) |
| Non-COD | 253 | (32) | 20 | (33) | 1 | (13) | 0 | (7) | 1 | (8) |
| E. coli | 0 | (6) | 0 | (5) | 0 | (4) | 0 | (2) | 0 | (3) |
| Synecho | 0 | (5) | 0 | (5) | 0 | (4) | 0 | (3) | 0 | (3) |
| A. fulgidus | 0 | (5) | 0 | (4) | 0 | (4) | 0 | (4) | 0 | (3) |
| H. influ. | 0 | (5) | 1 | (5) | 11 | (6) | 1 | (4) | 0 | (4) |
| H. pylori | 7 | (11) | 0 | (5) | 0 | (5) | 0 | (4) | 0 | (4) |
| M. jannaschii | 0 | (5) | 0 | (5) | 0 | (4) | 0 | (3) | 0 | (3) |
| M. pneumo. | 1 | (5) | 0 | (6) | 0 | (3) | 0 | (3) | 0 | (3) |
| M. genital. | 0 | (4) | 8 | (7) | 0 | (4) | 0 | (3) | 0 | (4) |
Shown is the cumulative count of repeats traditionally considered to be microsatellites (e.g., more than 8 repeat units) in each genome, and given is the maximum length of each type of repeat found. COD and Non-COD refer to repeats found in coding and noncoding regions in yeast. The locations of the long triple-repeats found in M. genitalium were identified. (ACA)11 codes a polythreonine run in a putative lipoprotein (HIU32768). The remaining triplet-repeats are found within coding [(AGT)15, (AGT)11, (AGT)10, (AGT)9] and noncoding [(CIT)16, (ACA)11, (CTA)9] regions of the multiple copy MgPA virulence operon (MYCMGP).
It is a striking feature of the yeast genome that so many triplet-repeats are found within coding regions. This is especially interesting because coding region triplet-repeats play a role in a growing number of genetic diseases identified in humans (8) and may influence gene regulation (25). We selected 16 translated and four untranslated triplet-repeat loci, including one in the yeast mitochondrion, for amplification by PCR. These repeats include nine of the longest triplet-repeats in the yeast genome and range over a wide variety of repeat motifs (11) and lengths (8–36 units). All of these loci showed length polymorphisms, both within the seven strains of S. cerevisiae and among the five additional closely related yeast species (Table 1). This amount of length variability suggests that these repeats experience strong mutation pressures.
An Excess of Short Repeats Is Present in Both the Prokaryote and Eukaryote Genomes.
In both yeast and prokaryotes there is an excess of short repeats, but in prokaryotes, in contrast to yeast, this excess is confined to mono- and trinucleotide repeats (data not shown). Table 3 shows that there is a significant excess of the short mononucleotide repeats lying between length 2 and approximately lengths 7–8 in all the prokaryote genomes, except for H. pylori in which the excess begins with runs of length 3. A similar excess is seen in the yeast genome, beginning with length 2 in the coding regions and with length 3 in the noncoding regions. Although not shown here, this excess is seen primarily in A’s and T’s, even in E. coli and Synechocystis in which the A+T content at 50% is the lowest of any of the microbial genomes.
Despite This Excess of Very Short Repeats, Long Repeats Are Actively Selected Against in Prokaryotes, Except in Cases of Positive Selection Associated with Gene Regulation of Virulence Factors.
In prokaryotes, longer repeats are not found in any abundance, except for a very few extremely long repeats that fall far outside the range of lengths predicted to occur at random. This deficiency is most clearly seen in the mononucleotide repeats, in which there is often a highly significant “cutoff” effect. When observed vs. expected frequencies are compared, there is a highly significant switch from excesses of observed numbers among repeats shorter than length 7 or 8, to deficiencies of observed numbers in repeats longer than this threshold (Table 3). These “cutoffs” can be detected in trinucleotide repeats as well, and are present to approximately the same extent in both coding and noncoding regions (data not shown). Synechocystis and H. pylori are exceptions to this otherwise conserved pattern among prokaryotic genomes. Among the longest mononucleotides (length 10 to 11), observed frequencies match expectations in Synechocystis. H. pylori is the most unusual prokaryote examined here with regard to distributions of mono- and dinucleotide repeats, because it appears to have long tails of both of these types of repeats, making it more like yeast than like the other prokaryotes.
There is now an extensive literature on the involvement of hypermutable microsatellite loci as translational and transcriptional “switches” in a variety of pathogenic prokaryotes (reviewed in ref. 16). In addition to the 12 repeats already identified in known or suspected virulence factors in H. influenzae (26–30), we found 17 unusually long repeats in the prokaryotes investigated here (excluding the long mono- and dinucleotide repeats of H. pylori) (Tables 3 and 4). blast searches (31) revealed that 10 of these additional repeats can be shown to be associated with genes involved in virulence. These genes include homologues to known lipoprotein genes, which in other pathogenic prokaryotes are known to be antigenic determinants controlled by repeats (32).
DISCUSSION
Future Empirical and Genomic Studies Aimed at a Better Understanding of These Nonrandom Patterns Are Required.
Although our expected distributions are based on a very simple model that takes into account only genome size and base content, comparisons between observed and expected numbers were highly informative in identifying switches between significant excesses and deficiencies of various length classes in the distributions of single repeat types. These switches include a deficiency of repeats of length 1 and 2, an excess of repeats lying between lengths 3 and 8, and a“cutoff” effect in most prokaryotes above lengths 8–9. Yeast shows the most highly significant deficiency of mononucleotides of lengths 1 and 2, and in contrast to the prokaryotes shows a strong excess of mononucleotide repeats at all observed lengths greater than 2. Similar patterns also are seen in this organism for di- and trinucleotides (data not shown). These patterns provide evidence for very different equilibria between mutation and selection forces in these prokaryote and eukaryote genomes.
Examination of these patterns reveals significant information about genome organization and stability. There is evidence that at the biochemical level mutational pressures associated with replication slippage are roughly equivalent among prokaryotes and lower and higher eukaryotes (33). Slippage rates in extrachromosomal tracts in E. coli (34), S. cerevisiae (35), and mammalian cell lines (36) are comparable, and the genes involved in repair of these mutations (4, 37) are highly conserved (33). But there may be significant differences in the presence or absence of genes involved in the replication and repair of DNA among prokaryotes. It is unclear, for example, whether H. pylori has unusually long mono- dinucleotide repeats because they are functional (18) or because this genome lacks mismatch repair (38). The present study found no statistical evidence, in sharp contrast to analysis of genomes like H. influenzae, that these repeats are under selection. Rather, they seem merely to be one end of an underlying distribution of shorter repeats that are statistically in excess.
It is clear that examination of the evolutionary implications of all types of repetitive DNA, with regard to genome instability and function, will be greatly aided by rapidly growing number of newly sequenced genomes (39). It will be useful to compare these patterns to those found in higher eukaryotic genomes and prokaryotic genomes larger than E. coli. Microbes also offer the advantages of large population sizes and rapid growth, which facilitate experimental manipulation.
Strong Mutation Pressures Have Generated Long Repeats in S. cerevisiae Despite Strong Selection for Small Genome Size, Making This Organism a Useful Model System in which to Study Genomic Microsatellite Evolution.
Microsatellites are abundant enough in the yeast genome to provide targets for direct experimentation. Such studies will greatly complement past and present studies of artificial extrachromosomal tracts in S. cerevisiae (4, 40) and may shed new insight into the mutational dynamics and biological significance, if any, of these loci. Further, the identification of these highly mutable molecular markers that are inherited in a Mendelian fashion substantially expands the potential for genetic and evolutionary studies of yeasts. This will be especially relevant in studies of yeast mating structure, about which very little currently is known. Low heterozygosities are seen at these microsatellite loci despite high allelic diversity, presumably because yeast is primarily a selfing organism. Therefore, the heterozygous loci that are seen in this yeast may be the result of sufficiently high rates of mutation during mitotic reproduction to allow mutations to accumulate in the intervals between sexual cycles. Alternatively, they may be the result of occasional outcrossing events.
Microsatellite loci currently are not being used extensively in the study of microbial evolution. Yet it is clear from this and past studies (10, 12) that microsatellites could be used to complement studies that use traditional methods, such as RAPDs and DNA fingerprinting using various repetitive DNA repeat probes, by which evolutionarily related strains can be distinguished and grouped.
Strong Mutational Pressures Act to Shape Even the Shortest Iterated Tracts, and This Finding Has Significant Implications for Understanding Genome Stability.
The excesses of short iterations suggest that mutational pressures are acting on these sequences. Little is known about the nature of slippage at extremely short repeats, because these repeats are not usually studied in higher eukaryotes. However, slippage mutations are known to occur frequently at runs of 3–4 bases in bacteria (41, 42). In addition, extremely short coding-region repeats (2–5 units long) are polymorphic in length and sequence composition in the yeast Candida albicans (12). We also have identified repeats that are 4–5 integral units in length in the 0.2 MB complete genome sequence of cytomegalovirus. These repeats also show length polymorphisms (unpublished results). These studies suggest that repeats that are shorter than those traditionally defined as microsatellites (>8 integral units) undergo appreciable rates of mutation in genomes that have been selected for small size. This suggests the need for further empirical studies on these and other microbial genomes.
The implications of these excess numbers of short iterated repeats could be extremely important not only for genomic stability, but also with regard to the evolution of additional genomic features such as codon usage. For example, the potential to form iterative mononucleotides may vary dramatically depending on codon usage. Arginine-glycine (RG) might be coded by the highly iterative codons cggggg or the less iterative agaggt, and glutamine-lysine (QK) can be coded for by caaaaa or cagaag.
We have begun to investigate the relationship between short iterated mononucleotide repeats and selection on coding regions and noncoding regions of these genomes, by computer simulation involving randomization. If genes are randomized by shuffling all the bases, the observed numbers of repeats found in the randomized sequences closely match expected values at all repeat lengths. In contrast, preliminary results show that if genes are randomized by shuffling codons rather than bases, the excess of short repeats remains, albeit somewhat reduced. In addition, the “cutoff” effect seen in many of the prokaryote genomes is lessened. Because nonrandom codon usage is preserved in this randomization procedure, it appears that such usage or overall amino acid usage or both play a large role in determining the short-repeat excess and the selection against long repeats. These preliminary studies indicate that the coding regions of these prokaryotic genomes have accommodated this apparent mutation pressure through nonrandom codon use. Essentially, codons appear to be arranged to minimize the generation of long random iterations.
The Action of Negative and Positive Selection and the Evolutionary Potential of Microsatellite Loci in Microbial Genomes.
Strong selection for rapid replication should act to remove repetitive, unnecessary DNA. Such negative selection acts with differential strength on microsatellites located in genomic regions with different functions (43). For example, triplet-repeats are better tolerated than dinucleotides in coding regions (43).
In the yeast genome a disproportionate number of mono- through trinucleotide repeats have accumulated in noncoding regions, and abundant trinucleotide repeats also have accumulated in coding regions. This finding is particularly striking in view of the virtual absence of such repeats in the prokaryotic genomes and provides evidence that the intensity of negative selection has been relaxed in yeast. Repeat abundances therefore are not only a positive function of increased genome size and increased quantities of noncoding DNA (43), but also involve differential selective pressures acting on underlying rates of slippage mutation.
We have found no statistical evidence in this analysis for positive selection on yeast microsatellites. It is unclear whether repeats acting as molecular switches would be selected for in a diploid eukaryote, because switches will be most effective in haploids, but it is possible that in eukaryotes microsatellite loci influence adaptation in more subtle ways (44, 45).
All functional microsatellites in bacteria have been found to be involved in gene regulation of virulence factors. In this survey, two repeats were found in nonpathogenic species, a (G)24 in M. jannaschii, and an (A)15 in A. fulgidus. Although it is possible that both loci are maintained by simple mutational pressures—particularly, because of its shorter length, the (A)15 in A. fulgidus—it is tempting to speculate that there are additional selective pressures that are strong enough to maintain such loci.
Acknowledgments
We thank The Institute for Genomic Research for making available sequence data before publication. This paper benefited from valuable discussions with R. Moxon, N. Saunders, D. Hood, J. Peden, P. Morin, M. Tanaka, and S. Ptak. Thanks for technical expertise to D. Ingrande and the Scripps Automated Sequencing Core Facility. This work was supported by grants from the National Science Foundation (to D.F.) and the U.S. Department of Energy (to C.W.). D.F. is supported by a Lucille P. Markey Fellowship.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Tautz D, Trick M, Dover G. Nature (London) 1986;322:652–656. doi: 10.1038/322652a0. [DOI] [PubMed] [Google Scholar]
- 2.Tautz D. Nucleic Acids Res. 1989;17:6463–6471. doi: 10.1093/nar/17.16.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber J L. Genomics. 1990;524:524–530. doi: 10.1016/0888-7543(90)90195-z. [DOI] [PubMed] [Google Scholar]
- 4.Strand M, Prolla T, Liskay R, Petes T. Nature (London) 1994;365:274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- 5.Tautz D, Schlotterer C. Curr Opin Genet Dev. 1994;4:832–837. doi: 10.1016/0959-437x(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 6.Ashley M, Dow B. Exs. 1994;69:185–201. doi: 10.1007/978-3-0348-7527-1_10. [DOI] [PubMed] [Google Scholar]
- 7.Dib C, Fauve S, Eizames C, Samson D, Drouot N, et al. Nature (London) 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 8.Sutherland G, Richards R. Proc Natl Acad Sci USA. 1995;92:3636–3641. doi: 10.1073/pnas.92.9.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moxon E, Rainey P, Nowak M, Lenski R. Curr Biol. 1994;4:24–33. doi: 10.1016/s0960-9822(00)00005-1. [DOI] [PubMed] [Google Scholar]
- 10.Field D, Wills C. Proc R Soc London. 1996;263:209–215. doi: 10.1098/rspb.1996.0033. [DOI] [PubMed] [Google Scholar]
- 11.Karlin S, Mrazek J, Campbell A M. J Bacteriol. 1997;179:3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Field D, Metzgar D, Eggert L, Rose R, Wills C. FEMS Lett. 1996;15:73–79. doi: 10.1111/j.1574-695X.1996.tb00056.x. [DOI] [PubMed] [Google Scholar]
- 13.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 14.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 15.Fraser C M, Gocayne J D, White O, Adams M D, Clayton R A, et al. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 16.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li B C, Herrmann R. Nucleic Acids Research. 1996;24:4420–2249. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeuchi M. Tanpakushitsu Kakusan Koso. 1996;41:2579–2583. [PubMed] [Google Scholar]
- 18.Tomb J, White O, Kerlavage A R, Clayton R A, Sutton G G, et al. Nature (London) 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 19.Wills C, Condit R, Foster R, Hubbell S P. Proc Natl Acad Sci USA. 1997;94:1252–1257. doi: 10.1073/pnas.94.4.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamada H, Petrino M, Kakunaga T. Proc Natl Acad Sci USA. 1982;79:6465–6469. doi: 10.1073/pnas.79.21.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Soriano J, Wong W, Ryk D V, Nazar R. J Mol Biol. 1991;217:629–635. doi: 10.1016/0022-2836(91)90521-7. [DOI] [PubMed] [Google Scholar]
- 22.Karlin S, Blaisdell B, Sapolsky R, Cardon L, Burge C. Nucleic Acids Res. 1993;21:703–711. doi: 10.1093/nar/21.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valle G. Yeast. 1993;9:753–759. doi: 10.1002/yea.320090709. [DOI] [PubMed] [Google Scholar]
- 24.Hancock J M. J Mol Evol. 1995;41:1038–1047. doi: 10.1007/BF00173185. [DOI] [PubMed] [Google Scholar]
- 25.Gerber H, Seipel K, Georgiev O, Hoffener M, Hug M, Rusconi S, Schaffner W. Science. 1994;263:808–811. doi: 10.1126/science.8303297. [DOI] [PubMed] [Google Scholar]
- 26.Weiser J, Love J, Moxon E. Cell. 1989;59:657–656. doi: 10.1016/0092-8674(89)90011-1. [DOI] [PubMed] [Google Scholar]
- 27.vanHam S, vanAlphen L, Mooi F, vanPutten J. Cell. 1993;73:1187–1196. [Google Scholar]
- 28.High N, Deadman M, Moxon E. Mol Microbiol. 1993;9:1275–1282. doi: 10.1111/j.1365-2958.1993.tb01257.x. [DOI] [PubMed] [Google Scholar]
- 29.Jarosik G P, Hansen E J. Infect Immun. 1994;62:4861–4867. doi: 10.1128/iai.62.11.4861-4867.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hood D W, Deadman M E, Jennings M P, Bisercic M, Fleischmann R D, Venter J C, Moxon E R. Proc Natl Acad Sci USA. 1996;93:11121–11125. doi: 10.1073/pnas.93.20.11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul S T, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Yogev D, Rosengarten R, Watson-McKown R, Wise K. EMBO J. 1991;10:4069–4079. doi: 10.1002/j.1460-2075.1991.tb04983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Modrich P, Lahue R. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 34.Levinson G, Gutman G. Nucleic Acids Res. 1987;15:5323–5338. doi: 10.1093/nar/15.13.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henderson S, Petes T. Mol Cell Biol. 1992;12:2749–2757. doi: 10.1128/mcb.12.6.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farber R A, Petes T D, Dominska M, Hudgens S S, Liskay R M. Hum Mol Genet. 1994;3:253–256. doi: 10.1093/hmg/3.2.253. [DOI] [PubMed] [Google Scholar]
- 37.Heale S, Petes T. Cell. 1995;83:539–45. doi: 10.1016/0092-8674(95)90093-4. [DOI] [PubMed] [Google Scholar]
- 38.Eisen J A, Kaiser D, Myers R M. Nat Med. 1997;3:1076–1078. doi: 10.1038/nm1097-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strauss S J, Falkow S. Science. 1997;276:707–711. doi: 10.1126/science.276.5313.707. [DOI] [PubMed] [Google Scholar]
- 40.Strand M, Earley M C, Crouse G F, Petes T D. Proc Natl Acad Sci USA. 1995;92:10418–10421. doi: 10.1073/pnas.92.22.10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foster P L, Trimarchi J M. Science. 1994;265:407–409. doi: 10.1126/science.8023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenberg S M, Longerich S, Gee P, Harris R S. Science. 1994;265:405–407. doi: 10.1126/science.8023163. [DOI] [PubMed] [Google Scholar]
- 43.Hancock H H. BioEssays. 1996;18:421–425. doi: 10.1002/bies.950180512. [DOI] [PubMed] [Google Scholar]
- 44.King D. Science. 1994;263:595–596. [PubMed] [Google Scholar]
- 45.King D G, Soller M, Kashi Y. Endeavour. 1997;21:36–40. [Google Scholar]