

Abstract
We have previously shown that diabetes significantly enhances apoptosis of osteoblastic cells in vivo and that the enhanced apoptosis contributes to diabetes impaired new bone formation. A potential mechanism is enhanced apoptosis stimulated by advanced glycation endproducts (AGEs). To investigate this further, an advanced glycation product, carboxymethyl lysine modified collagen (CML-collagen) was injected in vivo and stimulated a 5 fold increase in calvarial periosteal cell apoptosis compared to unmodified collagen. It also induced apoptosis in primary cultures of human or neonatal rat osteoblastic cells or MC-3T3-E1 cells in vitro. Moreover, the apoptotic effect was largely mediated through RAGE receptor. CML-collagen increased p38 and JNK activity 3.2 and 4.4 fold, respectively. Inhibition of p38 and JNK reduced CML-collagen stimulated apoptosis by 45% and 59% and by 90% when used together (P<0.05). The predominant apoptotic pathway induced by CML-collagen involved caspase-8 activation of caspase-3 and was independent of NF-κB activation. When osteoblastic cells were exposed to a long-term low dose incubation with CML-collagen there was a higher degree of apoptosis compared to short term incubation. In more differentiated osteoblastic cultures apoptosis was enhanced even further. These results indicate that advanced glycation endproducts, which accumulate in diabetic and aged individuals may promote apoptosis of osteoblastic cells and contribute to deficient bone formation.
Keywords: AGE, apoptosis, bone, cell death, diabetes, hyperglycemia
INTRODUCTION
Advanced glycation endproducts form when glucose reacts with the proteins to form unstable Schiff bases, which then undergo further modification to form Amadori products (44, 50). Additional rearrangements or modifications may occur giving rise to advanced glycation endproducts seen in prolonged hyperglycemia or aged individuals. A common modification is generated by oxidative cleavage of Amadori intermediates to form (N-epsilon-(carboxymethyl) lysine (CML) structures. Proteins with long half lives such as collagen are particularly susceptible to formation of advanced glycation end-products (AGEs) (36).
AGEs accumulate as a normal process of aging and as a result of hyperglycemia, particularly in long-lived molecules such as collagen. AGEs have been shown to be etiologic factors in diabetes-induced nephropathy, retinopathy, neuropathy, and diabetes-accelerated atherosclerosis (40, 44, 50). AGEs are also thought to contribute to many of the complications of aging including disorders such as osteoarthritis, cataract formation and changes observed in myocardial dysfunction (6, 30, 39).
Advanced glycation end products accumulate in bone and may play a functional role in the development of osteoporosis associated with diabetes and aging (53). In support of this concept, serum levels of AGEs are elevated in individuals with osteoporosis (18). In addition advanced glycation end products may lead to damage of articular surfaces and enhance inflammation thereby contributing to osteoarthritis (39). AGEs have also been implicated in the most common form of pathologic bone loss, periodontal disease (26).
AGEs may exert a negative effect on bone by interfering with osteoblast differentiation and the production of matrix proteins such as collagen and osteocalcin (13, 33, 38, 54). It has recently been reported that AGEs stimulate apoptosis of human mesenchymal stem cells which could limit the formation of adipose tissue, cartilage and bone (25). Other parameters may also be affected including AGE-inhibited proliferation and differentiation (33). Although not formally linked, it is possible that problems with fracture repair associated with type 1 diabetes (10, 14, 41) are directly or indirectly related to the effects of AGEs on osteoblasts. Since enhanced osteoblast apoptosis may represent an important mechanism through which bone formation is limited (19) we investigated the capacity of glycated collagen to stimulate apoptosis of osteoblastic cells and investigated the apoptotic pathways that were stimulated.
MATERIALS AND METHODS
CML-collagen
CML-collagen was prepared by chemical modification of acid soluble bovine skin collagen (Sigma), as previously described (23, 38). Briefly, 50 μg of collagen was dissolved 1 mM HCl followed by incubation in sodium cyanoborohydride and glyoxylic acid in PBS. Control collagen was prepared at the same time, except that no glyoxylic acid was added. All samples were then incubated at 37° C for 24 hours and then dialyzed against distilled water or PBS. CML-collagen was highly reactive on Western blots with anti-CML monoclonal antibody 6D12 (Wako, Richmond, VA), whereas control collagen was not reactive. In total, 3% to 8% of lysine residues in CML-collagen were converted to CML, as determined by the TNBS (trinitrobenzenesulfonic acid) assay (15). The per cent modification of collagen is less than the amount used in a recent report to assess CML binding and activation of NF-κB (23) and only a small amount higher than that found in the skin of aged or diabetic individuals (12). The amount of endotoxin contamination was measured by measured by Pyrochrome Limulus Amebocyte Lysate (LAL) assay (Associates of Cape Cod, Inc, Woods Hole, MA) and found to be very low, approximately 0.09 ng/μg (endotoxin/protein) for both control collagen and CML-collagen.
Cell Culture
Unless otherwise stated, experiments were performed with primary cultures of human adult osteoblastic cells purchased from Cambrex Company (Walkersville, MD) and maintained in Dulbecco’s Modified Eagle’s Medium (Cambrex) supplemented with 10% fetal bovine serum, Gentamicin (100μg/ml) and Amphotericin B (100ng/ml) at 37°C in a humidified atmosphere of 5% CO2 in air. The murine osteoblastic cell line, MC3TC-E1, subclone 14 (CRL-2594) was obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured in α-MEM supplemented as above. Neonatal osteoblastic cells were derived from the cells lining the calvarium of 2 day old rats as described in (8). In some experiments, neonatal calvarial osteoblasts or MC3T3-E1 cells were cultured in the presence or absence of 50 μg/ml ascorbic acid (Sigma) to promote osteoblast differentiation. Experiments with soluble CML-collagen or unmodified collagen were performed in culture medium supplemented with 0.5% fetal bovine serum. Assays were performed when the cultures reached 75%– 85% confluence.
Apoptosis Assay
For apoptosis assays cells were plated at a density of 20,000 osteoblasts/cm2 and incubated until they were approximately 75–85% confluent, incubated for 2 days in culture medium supplemented with 0.5% FBS and then changed to assay medium. For most experiments cells were incubated with CML-collagen (200μg/ml) or unmodified collagen (200μg/ml) for 24 hours. Apoptosis of osteoblasts was determined by ELISA measuring cytoplasmic histone-associated-DNA fragments (Roche Applied Science, Indianapolis, IN) as described F (2, 3). In some experiments cells were preincubated for 2 hrs with p38 (20 μM) or JNK-1 (20 μM) inhibitors which were also incubated in assay medium. p38 inhibitor, SB203580 [4-(4-fluorophenyl)-2- (4-methylsulfinylphenyl)-5-(4-pyridyl) 1 H-imidazole], and JNK-1 inhibitory peptide H-GRKKRRQRRRPPRPKRPTTLNLFPQVPRSQDT-NH2 [ L-HIV-TAT48–57 PP-JBD20] were purchased from Calbiochem (LaJolla, CA). The concentration of inhibitors, 20μM was selected based on published studies (7, 20). In other experiments cells were treated with 50μM inhibitors to caspase-3 (Z-DEVD-FMK), caspase-8 (Z-IETD-FMK) or caspase-9 (Z-LEHD-FMK) at the time of CML-collagen stimulation. Inhibitors were purchased from R&D Systems (Minneapolis, MN). To test the effect of NF-κB on apoptosis, osteoblastic cells were incubated the specific NF-κB inhibitor, SN50 (100 μg/ml) (Biomol, Plymouth Meeting, PA) in assay media with CML-collagen or control collagen. To study the role of the receptor for AGE (RAGE) primary cultures of human adult osteoblastic cells were incubated with CML-collagen (200 μg/ml) in presence of polyclonal anti-RAGE antibody specific for the extracellular domain of RAGE (10 μg/ul) or non-immune serum (10 μg/ul) (Biocompare, San Francisco, CA). For each experiment cell number was assessed in separate wells at the end of the experiment to normalize ELISA values. Each experiment was performed at least 3 times with similar results. Statistical significance was established by ANOVA with Tukey’s post-hoc test.
Caspase Activity
Human primary osteoblastic cells were incubated with CML-collagen as described above and changed to apoptosis assay medium. Caspase-3, -8 and -9 activities were measured from 200μg of total protein per data point using a fluorimetric kit purchased from R&D Systems. Caspase-3/7 activity was detected by using the specific caspase-3 fluorogenic substrate, DEVD peptide conjugated to 7-amino-4-trifluoromethyl coumarin (AFC). Caspase-8 activity was detected by using the specific caspase-8 fluorogenic substrate, IETD-AFC. Caspase-9 activity was detected by using the specific caspase-9 fluorogenic substrate, LEHD-AFC. Measurements were made on a fluorescent microplate reader using filters for excitation (400 nm) and detection of emitted light (505 nm). In some assays cells were incubated with caspase-8 and-9 inhibitors prior to assay for caspase-3 activity.
In vivo experiments
Eleven week old CD1 mice were purchased from Charles River Laboratories (Waltham, MA). CML-collagen or unmodified collagen was inoculated adjacent to the periosteum at a point on the midline of the skull located between the ears. Injection at this anatomic site can be reproducibly achieved. For each data point there were 6 mice (n=6). Animals were euthanized 24 hours after injection by decapitation and the heads were fixed for 72 hours in cold 4% paraformaldehyde and decalcified by incubation of cold Immunocal (Decal Corporation, Congers, NY) for approximately 12 days, with solution changed daily. Paraffin embedded sagittal sections were prepared at a thickness of 5 microns. Apoptotic cells were detected by an in situ TUNEL assay by with a TACS 2 TdT-Blue Label kit purchased from Trevigen (Gaithersburg, MD) and cells were counterstained with fast red following the manufacturer’s instructions. TUNEL positive cell counts were made by one examiner and confirmed by a second independent examiner. Students t-test was used to determine significant differences between the experimental and control groups at the p<0.05 level. All procedures involving mice were approved by the Boston University Medical Center Institutional Animal Care and Use Committee.
EMSA
Adult human primary osteoblast cultures were incubated for 1 hour with 200 μg/ml of CML-collagen or unmodified collagen in presence or absence of a specific NF-κB inhibitor, SN50 (100 ug/ml) (Biomol, Plymouth Meeting, PA). Nuclear proteins were extracted using protein extraction kit (Pierce, Rockford, Illinois) following the manufacturer’s instructions and the concentration measured using a BCA protein assay kit (Pierce, Rockford, Illinois). Interaction between NF-κB in the protein extract and DNA probe was investigated using electrophoretic-mobility shift assay (EMSA) kit from Panomics (Redwood City, CA) following the manufacturer’s instruction.
RESULTS
To test whether AGEs stimulated apoptosis of bone-lining cells in vivo, CML-collagen and control collagen were injected adjacent to the scalp periosteum and apoptosis of periosteal cells was measured by the TUNEL assay as we have previously described (16). CML-collagen stimulated a 5 fold increase in apoptosis of these bone lining cells compared to unmodified collagen (Fig. 1A). The capacity to induce apoptosis in different osteoblastic cell cultures, primary human and rat neonatal calvarial cells and MC3T3-E1 osteoblastic cells was tested in vitro (Fig 1B). A similar level of apoptosis was induced in all three in vitro cultures, with CML-collagen stimulating apoptosis 3.5 to 4 fold compared to unmodified collagen (P<0.05).
Figure 1. CML-collagen induces apoptosis in bone-lining cells in vivo and osteoblastic cells in vitro.
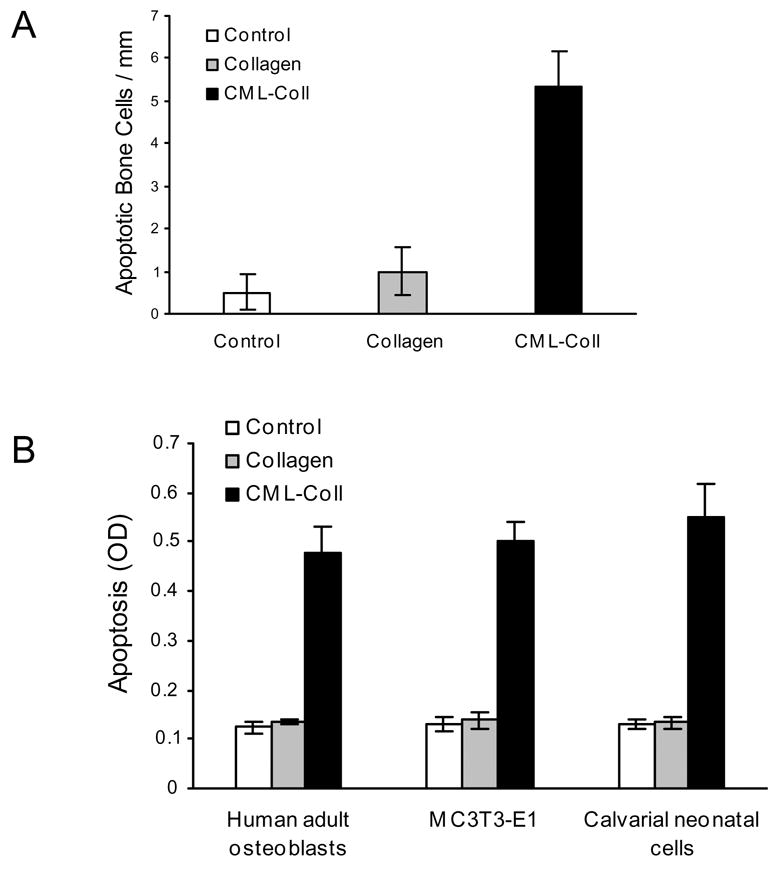
A. CML-collagen (100 μg) or control unmodified collagen (100 μg) was injected into the scalp as described in Materials and Methods and mice were euthanized 24 hours later. Apoptotic cells were identified in paraffin sections by the TUNEL assay. The number of TUNEL-positive bone-lining periosteal cells was counted at 1000x magnification between the occipital and coronal sutures. Each value represents the mean of 6 specimens ± SEM. B. CML-collagen (200 μg/ml) or an equal amount of unmodified control collagen were incubated with human, MC3T3-E1 or neonatal rat calvarial osteoblastic cells for 24 hours and compared to controls which were incubated in assay medium alone. Each value represents the mean of 4 replicates ± SEM and each experiment was performed three times with similar results.
Dose response and time course experiments were carried out with the primary cultures of human osteoblastic cells (Fig. 2A). At all doses unmodified collagen was similar to the zero time point (P>0.05). CML-collagen stimulated a dose-dependent increase in apoptosis that plateaued at 0.4 μg/ml CML-collagen, which induced a four fold increase in programmed cell death. In time-course experiments a small but significant increase in apoptosis was noted at 6 hours, but was much less than the four fold increase that was noted at 24 hours (Fig 2B). Similar results were obtained with MC3T3-E1 and rat neonatal calvarial cells (data not shown).
Figure 2. CML-collagen stimulates a dose and time dependent increase in apoptosis of osteoblastic cells.
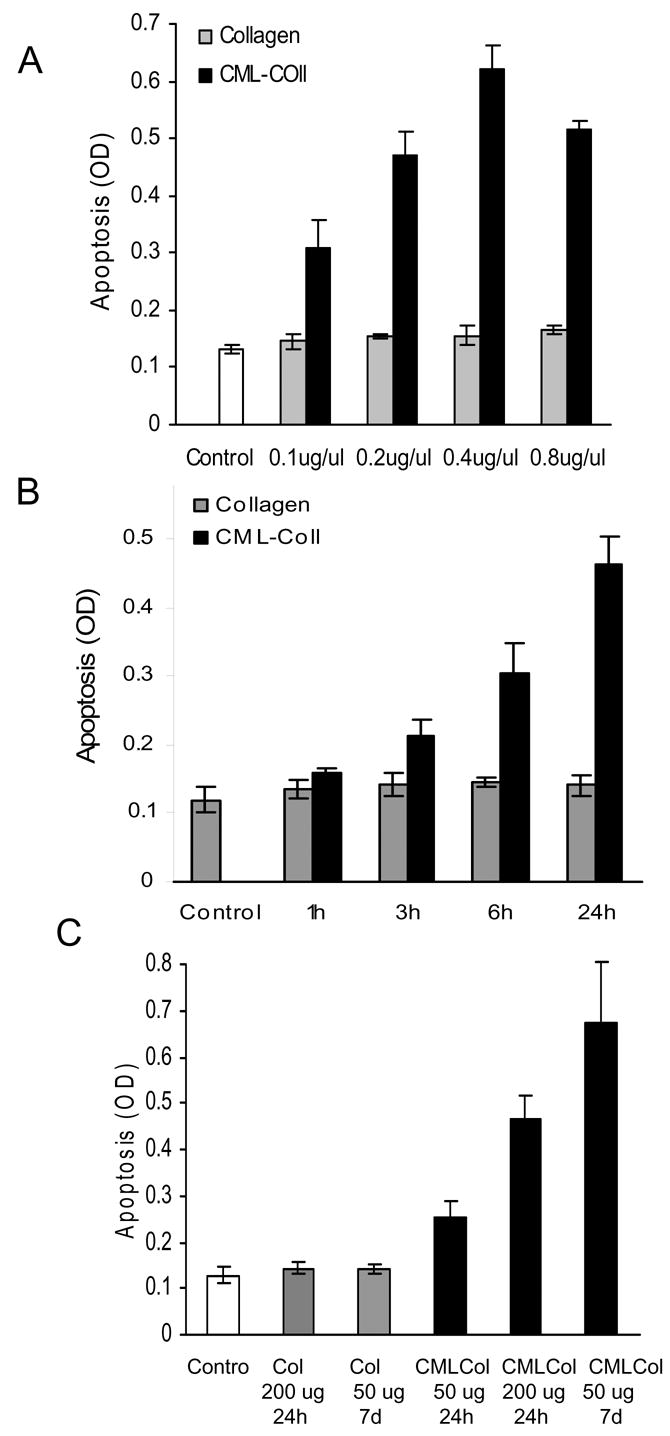
A: Human osteoblastic cells were incubated with different concentrations of CML- collagen or unmodified collagen for 24 hours. The extent of apoptosis was determined by ELISA. B. Human osteoblasts were incubated with AGE-collagen or unmodified collagen (200μg/ml) as indicated. Apoptosis was determined by ELISA. Each value represents the mean of 4 replicates ± SEM and each experiment was performed three times with similar results. C. Primary human osteoblasts were incubated with CML-collagen (50 or 200 μg/ml) or unmodified collagen for 24 hours or 1 week. As an additional control cells were incubated with medium alone. At the end of each incubation period apoptosis was determined by ELISA. The ELISA values were normalized by cell number. Each value represents the mean of 4 replicates ± SEM. The experiment was performed three times with similar results.
The above studies provide evidence that AGEs are relatively potent inducers of osteoblast apoptosis. In vivo cells are more likely to experience long-term exposure to AGEs rather than short-term stimulation. To determine whether a long-term exposure to AGEs even at low doses could induce osteoblast apoptosis experiments were carried out in which cells were incubated with 50 μg/ml of CML-collagen or unmodified-collagen for 1 week and compared to stimulation with 50 or 200 μg/ml for 24 hours (Fig. 2C). Incubation with unmodified collagen for both time periods did not affect osteoblast apoptosis (P>0.05). Long-term low dose exposure to CML-collagen (50 μg/ml) stimulated a 4.7 fold increase and a short-term stimulation with 50 or 200 μg/ml CML-collagen induced a 1.8 and 3.6 fold increase, respectively. Similar results were obtained with MC3T3-E1 and rat neonatal calvarial cells (data not shown).
Studies were undetaken to identify whether signalling occurred via the well characterized receptor for AGE, RAGE (40). When primary human osteoblasts were incubated with CML-collagen in the presence of antibody specific for the extracellular domain of RAGE there was a significant decrease in apoptosis (P<0.05) (Fig 3). That there was no difference between baseline levels and incubation of CML-collagen with anti-RAGE antibody (P>0.05) indicates that RAGE signalling is required for apoptosis. Matched pre-immune serum had no effect on CML-collagen induced apoptosis demonstrating specificity and cells incubated with RAGE antibody or pre-immune serum in the absence of CML-collagen had no change in baseline levels of apoptosis. One of the primary signalling pathways through which apoptosis is induced is the MAP kinase pathway. It has been shown that advanced glycation endproducts induce apoptosis in Schwann and mesangial cells partly through p38 map kinase signalling (28, 43). Another member of the MAP kinase family, JNK, has been shown to mediate apoptosis of other mediators including TNF-α (29, 48). To determine whether CML-collagen induced two of the principal pro-apoptotic signalling components in this pathway, activation of p38 and JNK were measured (Fig 4). CML-collagen stimulated a 3.2 fold increase in p38 activity while it induced a 4.4 fold increase in JNK1/2 activity. The response to p38 peaked earlier than that of JNK1/2. The functional significance of MAP kinase signalling in apoptosis was then assessed (Fig 4C). Inhibition of p38 with the specific inhibitor, SB203580, reduced CML-stimulated apoptosis by 45% percent (P<0.05). Incubation with the JNK inhibitor [L-HIV-TAT48–57 PP-JBD20] caused a 59% percent reduction in the level of apoptosis, which was significant. However, when both inhibitors were used simultaneously CML-collagen stimulated apoptosis was reduced by 90%, which was greater than either inhibitor alone (P<0.05). Thus, both p38 and JNK arms of this signalling pathway are involved in CML-collagen induced apoptosis. Similar results were obtained with MC3T3-E1 and rat neonatal calvarial cells (data not shown).
Figure 3. CML-collagen inducedCollagen stimulates apoptosis is mediated through RAGE.
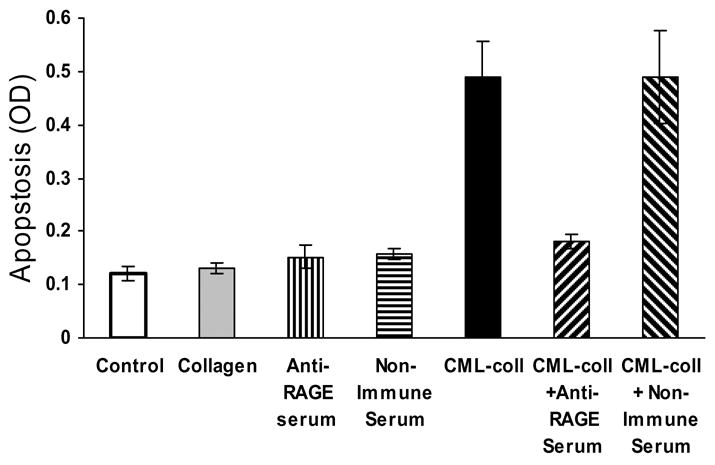
Primary cultures of human adult osteoblastic cells were incubated with CML-collagen (200μg/ml) in presence or absence of antibody specific for the extra-cellular domain of RAGE (10 μg/ul) or non-immune serum (10 μg/ul) for 24 hours. In some cases cells were incubated with antiserum or non-immune serum alone. The extent of apoptosis was determined by ELISA. Each value represents the mean of 4 replicates ± SEM. The experiment was performed three times with similar results.
Figure 4. CML-Collagen stimulates apoptosis through p38 and JNK MAP kinase activity.
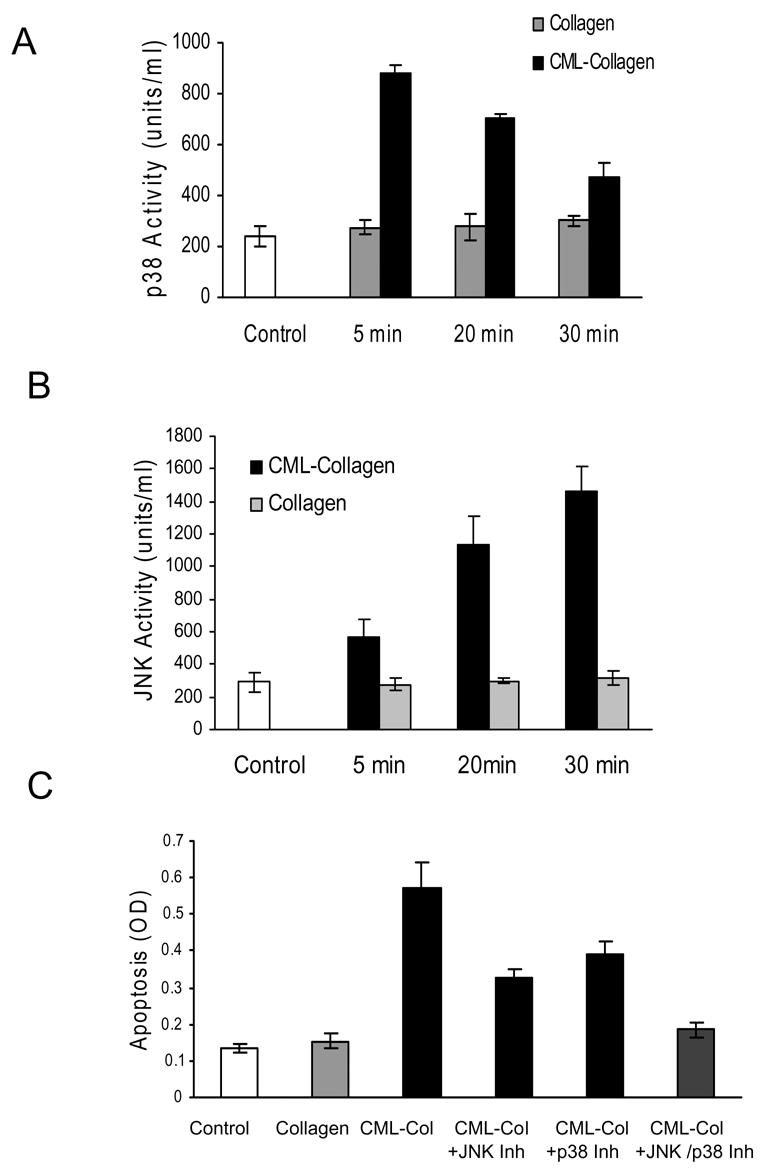
A Primary human osteoblasts were incubated for 0–30 mins with 200 μg/ml of CML-collagen or unmodified collagen. Cytoplasmic extracts were tested for activated (A) p38 and (B) JNK activity using a phosphosite-specific ELISA. (C) Cells were pre-incubated for 2 hours with a specific inhibitors for p38, SB203580 or JNK inhibitory peptide, H-GRKKRRQRRRPPRPKRPTTLNLFPQVPRSQDT-NH2. The inhibitors were also present in assay media. At the end of the incubation period the number of cells was counted and apoptosis was determined by ELISA. The ELISA values were normalized by cell number. For Panels A and B each value represents the mean of 3 replicates ± SEM and experiments were performed three times with similar results. For Panel C each value represents the mean of 4 replicates ± SEM and the experiment was performed four times with similar results.
The capacity of CML-collagen to activate initiator and executioner caspases was measured in human adult osteoblastic cells following stimulation by CML-collagen or unmodified collagen. CML-collagen stimulated a 4.3 fold increase in caspase-3 activity, a 3.4 fold increase in caspase-8 and a 2.2 fold increase in caspase-9 activity, all of which were significant (P<0.05) (Fig 5A). Since caspase-3 is the principal effector caspase through which the mitochondrial and cytosolic pathways induce apoptosis, the dependence of caspase-3 activity on caspase-8 and -9 was measured (Fig 5B). A caspase-8 inhibitor blocked CML-collagen activation of caspase-3 by 79% while a caspase-9 inhibitor reduced it by 37%. The difference between caspase-8 and caspase-9 inhibitors was significant (P<0.05). The functional significance in mitochondrial and cytosolic pathways in mediating CML-collagen induced apoptosis was measured in Fig 5C. Caspase-3 inhibitor reduced CML-collagen stimulated apoptosis by 86%, while caspase-8 and -9 inhibitor reduced it 63% and 33%, respectively, all of which were significant (P<0.05). These results are consistent with the degree to which CML-collagen induced caspase activity associated with their respective apoptotic signalling pathway.
Figure 5. CML-collagen stimulates caspase activity in osteoblasts.
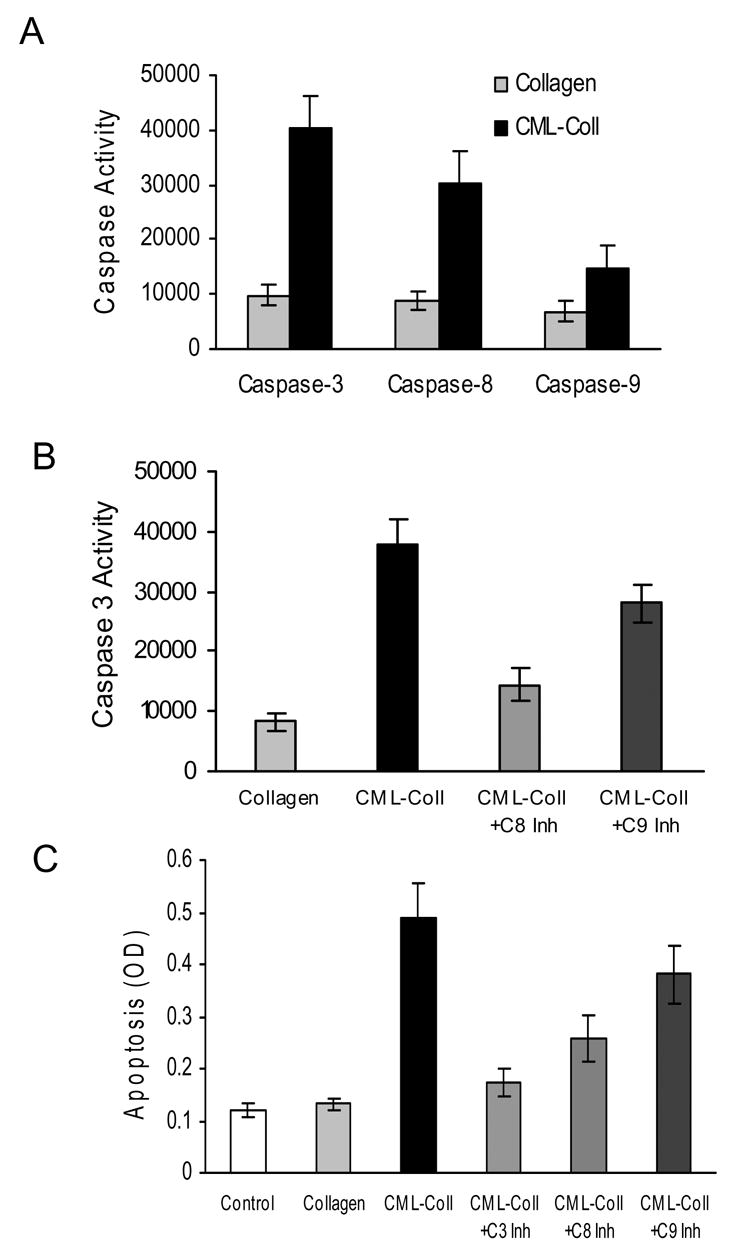
A. Human osteoblasts were incubated with CML- collagen with or without different caspase inhibitors for 24 hours. A. Caspase-3, -8 and -9 activity was measured by fluorimetric assay in lysates from cell cultures obtained after 24 hours stimulation with CML-collagen (200 μg/ml). B. Caspase-3 activity was measured using a fluorimetric assay in lysates from cell cultures obtained after 24 hours stimulation with CML-collagen (200 μg/ml) with or without caspase-8 or -9 inhibitors. (C) Cells were incubated with control collagen (200μg/ml) or CML- collagen (200 μg/ml) with or without inhibitors to caspase-3, -8 or -9. Apoptosis was measured 24 hours later by ELISA. For each panel the value represents the mean of 4 replicates ± SEM and experiments were performed three times with similar results.
Osteoblastic cells in culture, when incubated in the presence of ascorbic acid, undergo a sequence of events that involve proliferation, differentiation and formation of a calcifying extracellular matrix over a two to three week period (8). Thus, the cells exhibit a more differentiated phenotype with time. To investigate the impact of differentiated phenotype on the apoptotic response to CML-collagen, MC3T3-E1 cells were cultured in presence of ascorbic acid and stimulated with CML-Collagen (200μg/ml) for 24 hours (Fig 6A). Cells incubated with ascorbic acid for one week and then stimulated with CML-collagen exhibited a five fold increase in apoptosis compared to four fold increase in the absence of ascorbic acid, with the difference between them being significant (p<0.05). However, the level of apoptosis was increased to 6.5 fold when cells had been incubated for two weeks which was significantly higher than cells incubated with ascorbic acid for less time or not at all (P<0.05). When rat neonatal calvarial osteoblasts were tested, CML-collagen stimulated a 4.2 fold increase in apoptosis in cells not incubated with ascorbic acid (Fig 6B). After 1 and 2 week exposure to ascorbic acid a 5.3 and 6.6 fold increases in apoptosis in response to CML-Collagen was observed, respectively. Thus, apoptosis increased by approximately 50% after 2 weeks exposure to ascorbic acid in both differentiated osteoblastic cell cultures compared with less differentiated cells not incubated with ascorbic acid (p<0.05).
Figure 6. Apoptotic effect of CML-Collagen increased in differentiated osteoblasts.
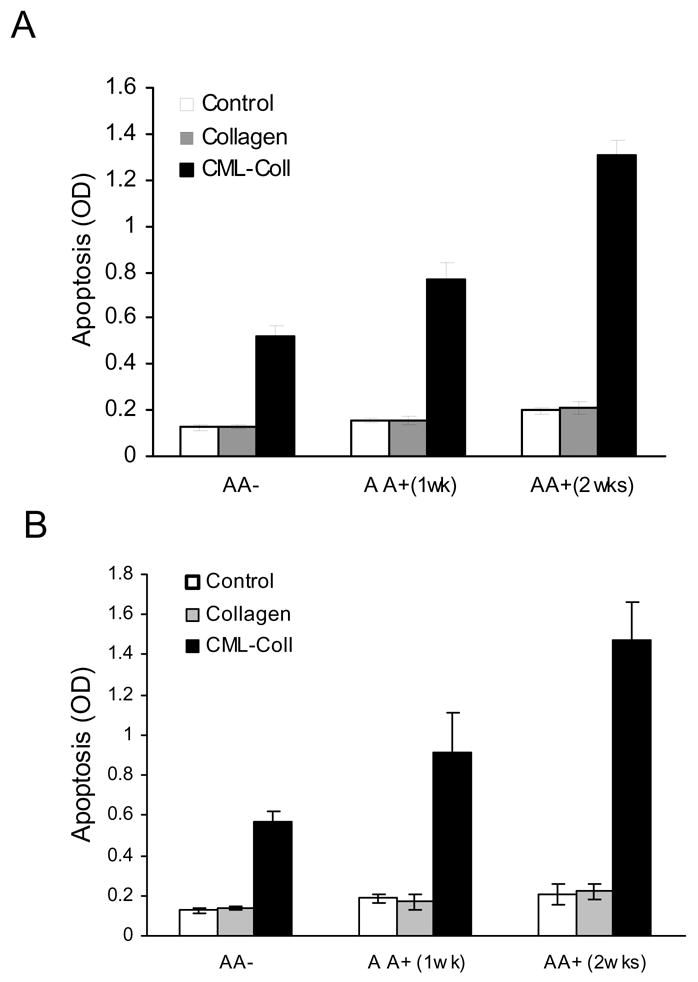
MC3T3-E1 cells (A) or primary neonatal calvarial osteoblastic cells (B) were cultured in the presence or absence of ascorbic acid (50 μg/ml) for one or two weeks. Cells were then incubated with CML-collagen or unmodified collagen (200 μg/ml) for 24h. Control cells were incubated in assay medium alone. Apoptosis was determined by ELISA and the values were normalized by cell number. Each value represents the mean of 4 replicates ± SEM. The experiment was performed three times with similar results.
Since AGEs have previously been reported to stimulate NF-κB in osteoblastic cells (46), we examined the impact of NF-κB on AGE-induced apoptosis by using the specific NF-κB inhibitor, SNF-50. Adult human osteoblasts were incubated with CML-collagen or unmodified collagen for 1 hour. CML-collagen but not unmodified collagen induced NF-κB activation as determined by electrophoretic mobility shift assay (Fig 7A). When apoptosis was measured (Fig 7B), inhibition of NF-κB with the specific inhibitor, SN50, enhanced CML-collagen induced apoptosis by 51%, which was statistically significant (P<0.05).
Figure 7. AGEs stimulate osteoblast apoptosis through an NF-κB-independent mechanism.
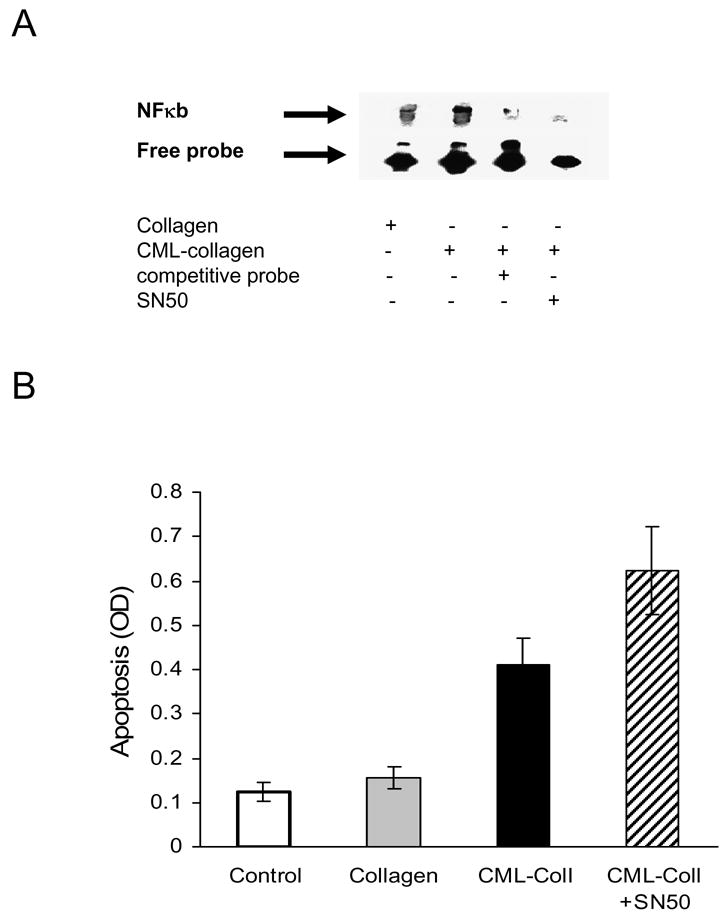
A. Primary human osteoblasts were incubated for 24 hours with 200 μg/ml of CML-collagen or unmodified collagen in presence or absence of NF-κB inhibitor SN50 (100μg/ml). Nuclear extracts were isolated and incubated with biotinylated NF-κB probe or probe plus excess unlabelled probe. EMSA was carried out as described in Materials and Methods. The arrow points to the NFκB band in lane 2. The experiment was performed three times with similar results. B. Primary human osteoblasts were incubated with CML-collagen (200 μg/ml) with or without NF-κB inhibitor SN50 (100μg/ml) or unmodified collagen (200 μg/ml). Apoptosis was measured 24 hrs later by ELISA. Each value represents the mean of 5 replicates ± SEM. The experiment was performed three times with similar results.
It has been reported that AGEs may be synergistic with other factors to enhance apoptosis (9). Therefore we compared the effect of CML-collagen with another well defined apoptotic factor, TNF-α and tested for synergistic interaction (Figure 8). When tested under the same conditions TNF-α (5–20 ng/ml) increased osteoblast apoptosis to a greater extent the CML-collagen (100–400μg/ml) (Fig 8A). To investigate potential synergy between CML-collagen and TNF-α cells were incubated with CML-collagen (100μg/ml) and increasing amounts of TNF-α (Fig 8B). A lower concentration of CML-collagen, 100μg/ml was selected in order to detect potential synergistic effects. The addition of TNF-α to CML-collagen only slightly increased apoptosis. Similar results were obtained when varying concentrations of CML-collagen were incubated with a fixed sub-maximal dose of TNF-α (data not shown). Thus, there was no evidence of synergy.
Figure 8. CML-Collagen compared to TNF-induced apoptosis in primary human osteoblasts.
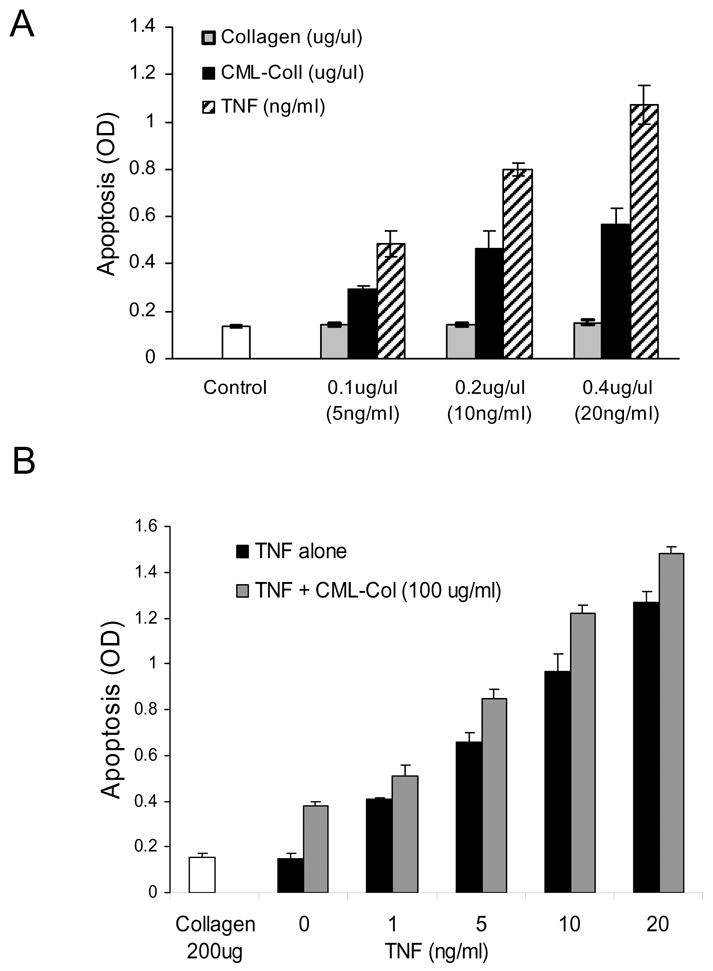
A. Cells were incubated with different concentration of TNF or CML-collagen for 24h. The extent of apoptosis was determined by ELISA. B. Cells were incubated with a suboptimal dose of CML-collagen (100 μg/ml) and increasing concentrations of TNF-α (10–20ng/ml). The extent of apoptosis was determined by ELISA. The experiments were performed two (B) or three times (A) with similar results. Each value represents the mean of 3 or 4 replicates ± SEM.
DISCUSSION
Diabetes and aging are both characterized by diminished bone formation (1, 42). There are multiple avenues through which this could occur, one of which is reduced numbers of osteoblastic cells or their precursors. For example, premature aging caused by ablation of the klotho gene is associated with higher levels of osteoblast and osteocyte apoptosis (45). We have recently shown that diabetes causes enhanced apoptosis of osteoblastic cells following a bacterial stimulus (16). When apoptosis is inhibited there is a significant improvement in the capacity to form new bone demonstrating that diabetes-enhanced apoptosis is functionally important (5).
One of the mechanisms through which diabetes or aging could induce apoptosis is through stimulation by advanced glycation endproducts. Data presented here demonstrate that CML-collagen, one of the predominant advanced glycation endproducts found in bone and serum of individuals with osteoporosis (17, 18) stimulates apoptosis of bone-lining cells in vivo and in various osteoblastic cell cultures and that signalling is mediated through RAGE. This agrees well with previous reports that advanced glycation endproducts stimulate oxidative stress in osteoblastic cells (33). The degree of glycosylation of collagen used in the experiments is only slightly higher than that found in various tissues in vivo (12, 37) and is likely to be biologically relevant, particularly since long-term low dose exposure was especially effective in stimulating apoptosis. However, CML-collagen did not induce apoptosis to the same extent as TNF-α and there was no synergy between them.
To investigate mechanisms by which CML-collagen induced apoptosis we examined the MAP kinase signalling pathway. CML-collagen stimulated both p38 and JNK activity in osteoblasts and inactivation of both with inhibitors significantly reduced apoptosis. That the highest degree of inhibition was achieved when p38 and JNK inhibitors were used together indicates that both arms participate in CML-collagen stimulated cell death. Both p38 and JNK are frequently involved in upstream signalling of apoptosis (21, 24). Although there have been no reports on advanced glycation products stimulating apoptosis in osteoblasts through this pathway, hypoxia, nitric oxide and integrin-mediated osteoblast apoptosis appear to involve the MAP kinase pathway (22, 47). However, there are MAP kinase-independent pathways through which osteoblast apoptosis is induced by other mediators (27).
We previously established that CML-collagen stimulated fibroblast apoptosis largely through caspase-3, which was activated through both cytoplasmic (caspase-8 dependent) and mitochondrial (caspase-9) pathways (4). Similar pathways are stimulated in osteoblastic cells as fluorimetric assays indicated that caspase-3, -8 and -9 were induced by CML collagen. Caspase-3 and -8 were induced at higher levels than caspase-9 suggesting that the cytosolic pathway was relatively more important. That there was an 86% decrease in apoptosis when caspase-3 was inhibited demonstrate that it plays a significant role in AGE-induced apoptosis. As was shown for fibroblasts, the inhibition of caspase-8 had a greater effect on CML-collagen stimulated apoptosis than inhibition of caspase-9, agreeing with activity measurements that the cytosolic pathway was the predominant apoptotic mechanism in osteoblasts. However, there was still a small but statistically significant contribution by the mitochondrial pathway since inhibition of caspase-9 reduced both caspase-3 activation and apoptosis. This results demonstrate that while caspase-8 plays as dominant pathway, role of caspase-9 can not be ignored.
AGEs have previously been reported to stimulate activation of NF-κB (46). When NF-κB was inhibited, apoptosis in response to CML-collagen was further enhanced. Thus, CML-collagen simultaneously induces a pro-apoptotic pathway involving MAP kinase signalling and an anti-apoptotic pathway involving NF-κB. The latter agrees well with the reported anti-apoptotic activity of NF-κB (51). The net effect though is increased apoptosis indicating that the pro-apoptotic pathway predominates. Thus, the results presented here provide information on the signalling pathways through which AGEs, associated with aging or diabetes, may lead to osteoblast cell death and diminish the capacity to form bone. This is consistent with a report that in diabetic mice there are fewer bone lining cells (49).
Studies in soft tissue wounds indicate that diabetic mice have increased levels of apoptosis, which may interfere with the capacity for wound healing (11, 32, 46). Thus, diabetes may have a general effect of increasing apoptosis of matrix producing cells, which limits the repair of injured hard or soft connective tissue. This tendency of enhanced apoptosis may be due in part to the formation of advanced glycation end products. To our knowledge there are no studies that have investigated whether long term AGEs contribute to osteopenia in diabetic or aged individuals through inducing osteoblast apoptosis. We have recently reported that when caspase inhibitors are applied in vivo there is significantly improved reparative bone formation following bacteria-induced injury in diabetic animals (5). Interestingly, we found that CML-collagen induced apoptosis to a greater extent in more differentiated osteoblastic cells. Given the potential to shorten the lifespan of mature osteoblasts as well mesenchymal stem cells (25) AGE-stimulated apoptosis may represent an avenue through which osteogenesis is impaired in aged and diabetic individuals. However, AGEs do not represent the only mechanism for diabetes-enhanced osteoblast apoptosis as inhibition of TNF in vivo also reduces apoptosis of periosteal cells and promotes new bone formation (31). These and other reports underscore the potential importance of osteoblast apoptosis as a mechanism for impaired bone formation in diabetic and aged individuals (5, 34, 35).
Acknowledgments
We would like to thank Alicia Ruff for help in preparing this manuscript. This work was supported by NIH grants P01AR49920 and R01DE14066.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Akin O, Gol K, Akturk M, Erkaya S. Evaluation of bone turnover in postmenopausal patients with type 2 diabetes mellitus using biochemical markers and bone mineral density measurements. Gynecol Endocrinol. 2003;17:19–29. [PubMed] [Google Scholar]
- 2.Alikhani M, Alikhani Z, Graves D. FOXO1 functions as a master switch that regulates gene expression necessary for TNF-induced fibroblast apoptosis. J Biol Chem. 2005;280:12096–102. doi: 10.1074/jbc.M412171200. [DOI] [PubMed] [Google Scholar]
- 3.Alikhani M, Alikhani Z, Raptis M, Graves D. TNF-alpha in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol. 2005;201:341–8. doi: 10.1002/jcp.20067. [DOI] [PubMed] [Google Scholar]
- 4.Alikhani Z, Alikhani M, Boyd C, Nagao K, Trackman P, Graves D. Advanced glycation endproducts enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J Biol Chem. 2005;280:12087–95. doi: 10.1074/jbc.M406313200. [DOI] [PubMed] [Google Scholar]
- 5.Al-Mashat HA, Kandru S, Liu R, Behl Y, Desta T, Graves DT. Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing. Diabetes. 2006;55:487–95. doi: 10.2337/diabetes.55.02.06.db05-1201. [DOI] [PubMed] [Google Scholar]
- 6.Argirov O, Lin B, Orotwerth B. 2-ammonio-6-(3-oxidopyridinium-1-yl)hexanoate(OP-lysine) is a newly identified advanced glycation end product in cataractous and aged human lenses. J Biol Chem. 2004;279:6487–95. doi: 10.1074/jbc.M309090200. [DOI] [PubMed] [Google Scholar]
- 7.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–97. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 8.Bellows C, Aubin J, Heersche J, Antosz M. Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif Tissue Int. 1986;38:143–154. doi: 10.1007/BF02556874. [DOI] [PubMed] [Google Scholar]
- 9.Boulanger E, Wautier MP, Gane P, Mariette C, Devuyst O, Wautier JL. The triggering of human peritoneal mesothelial cell apoptosis and oncosis by glucose and glycoxydation products. Nephrol Dial Transplant. 2004;19:2208–16. doi: 10.1093/ndt/gfh277. [DOI] [PubMed] [Google Scholar]
- 10.Carneavale V, Romagnoli E, D’Erasmo E. Skeletal involvement in patients with diabetes mellitus. Diabetes Metab Res Rev. 2004;20:196–20. doi: 10.1002/dmrr.449. [DOI] [PubMed] [Google Scholar]
- 11.Darby I, Bisucci T, Hewitson T, MacLellan D. Apoptosis is increased in a model of diabetes-impaired wound healing in genetically diabetic mice. Int J Biochem Cell Biol. 1997;29:191–200. doi: 10.1016/s1357-2725(96)00131-8. [DOI] [PubMed] [Google Scholar]
- 12.Dyer D, Dunn J, Thorpe S, Bailie K, Lyons T, McCance D, Baynes J. Accumulation of Maillard reaction products in skin collagen in diabetes and aging. J Clin Invest. 1993;91:2463–9. doi: 10.1172/JCI116481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong Y, Edelstein D, Wang E, Brownlee M. Inhibition of matrix-induced bone differentiation by advanced glycation end-products in rats. Diabetologia. 1993;36:802–7. doi: 10.1007/BF00400353. [DOI] [PubMed] [Google Scholar]
- 14.Gooch HL, Hale JE, Fujioka H, Balian G, Hurwitz SR. Alterations of cartilage and collagen expression during fracture healing in experimental diabetes. Connect Tissue Res. 2000;41:81–91. doi: 10.3109/03008200009067660. [DOI] [PubMed] [Google Scholar]
- 15.Habeeb A. Determination of free amino groups in proteins by trinitrobenezenesilfonic acid. Ana Biochem. 1966;14:328–336. doi: 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- 16.He H, Liu R, Desta T, Leone C, Gerstenfeld L, Graves D. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology. 2004;145:447–52. doi: 10.1210/en.2003-1239. [DOI] [PubMed] [Google Scholar]
- 17.Hein G, Weiss C, Lehmann G, Niwa T, Stein G, Franke S. Advanced glycation end product modification of bone proteins and bone remodelling: hypothesis and preliminary immunohistochemical findings. Ann Rheum Dis. 2006;65:101–4. doi: 10.1136/ard.2004.034348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hein G, Wiegand R, Lehmann G, Stein G, Franke S. Advanced glycation end-products pentosidine and N epsilon-carboxymethyllysine are elevated in serum of patients with osteoporosis. Rheumatology (Oxford) 2003;42:1242–6. doi: 10.1093/rheumatology/keg324. [DOI] [PubMed] [Google Scholar]
- 19.Boyce BF, Xing L, Jilka RL, Bellido T, Weinstein RS, Parfitt AM, Manolagas SC. Apoptosis in bone cells. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles Bone Biol. Principles of bone biology. 2 Vol. 1. Vol. 1. Academic Press; 2002. pp. 151–168. [Google Scholar]
- 20.Iwasaki S, Iguchi M, Watanabe K, Hoshino R, Tsujimoto M, Kohno M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogenetic protein-2. J Biol Chem. 1999;274:26503–10. doi: 10.1074/jbc.274.37.26503. [DOI] [PubMed] [Google Scholar]
- 21.Johnson G, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y, Chae S. JNK/SAPK is required in nitric oxide-induced apoptosis in osteoblasts. Arch Pharm Res. 2003;26:937–42. doi: 10.1007/BF02980203. [DOI] [PubMed] [Google Scholar]
- 23.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–9. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–26. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 25.Kume S, Kato S, Yamagishi S, Inagaki Y, Ueda S, Arima N, Okawa T, Kojiro M, Nagata K. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J Bone Miner Res. 2005;20:1647–58. doi: 10.1359/JBMR.050514. [DOI] [PubMed] [Google Scholar]
- 26.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105:1117–24. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemonnier J, Hay E, Delannoy P, Fromigue O, Lomri A, Modrowski D, Marie PJ. Increased osteoblast apoptosis in apert craniosynostosis: role of protein kinase C and interleukin-1. Am J Pathol. 2001;158:1833–42. doi: 10.1016/S0002-9440(10)64139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B, Miyata S, Hirota Y, Higo S, Miyazaki H, Funkunaga M, Hamada Y, Ueyama S, Muramoto O, Uriuhara A, Kasuga M. Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int. 2003;63:947–57. doi: 10.1046/j.1523-1755.2003.00829.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Masurekar M, Vatner D, Jyothirmayi G, Regan T, Vatner S, Meggs L, Malhotra A. Glycation end-product cross-link breaker reduces collagen and improves cardiac function in aging diabetic heart. Am J Physiol Heart Circ Physiol. 2003;285:H2587–91. doi: 10.1152/ajpheart.00516.2003. [DOI] [PubMed] [Google Scholar]
- 31.Liu R, Bal HS, Desta T, Behl Y, Graves DT. Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am J Pathol. 2006;168:757–64. doi: 10.2353/ajpath.2006.050907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu R, Desta T, He H, Graves D. Diabetes alters the response to bacteria by enhancing fibroblast apoptosis. Endocrinology. 2004;145:2997–3003. doi: 10.1210/en.2003-1601. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy A, Etcheverry S, Bruzzone L, Lettieri G, Barrio D, Cortizo A. Non-enzymatic glycosylation of a type I collagen matrix: effects of osteoblastic development and oxidative stress. BMC Cell Biol. 2001;2:16. doi: 10.1186/1471-2121-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J, Su H, Tong X, Amizuka N, Gupta A, Genant HK, Kronenberg HM, Goltzman D, Karaplis AC. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J Clin Invest. 2005;115:2402–11. doi: 10.1172/JCI24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–41. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 36.Paul R, Bailey A. Glycation of collagen: the basis of its central role in the late complications of ageing and diabetes. Int J Biochem Cell Biol. 1996;28:1297–1310. doi: 10.1016/s1357-2725(96)00079-9. [DOI] [PubMed] [Google Scholar]
- 37.Reddy S, Bichler J, Wells-Knecht K, Thorpe S, Baynes J. N-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry. 1995;34:10872–10878. doi: 10.1021/bi00034a021. [DOI] [PubMed] [Google Scholar]
- 38.Santana RB, Xu L, Chase HB, Amar S, Graves DT, Trackman PC. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes. 2003;52:1502–10. doi: 10.2337/diabetes.52.6.1502. [DOI] [PubMed] [Google Scholar]
- 39.Saudek D, Kay J. Advanced glycation endproducts and osteoarthritis. Curr Rheumatol Rep. 2003;5:33–40. doi: 10.1007/s11926-003-0081-x. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt A, Yan S, Yan S, Stern D. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498:99–111. doi: 10.1016/s0167-4889(00)00087-2. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz A. Diabetes Mellitus: Does it Affect Bone? Calcif Tissue Int. 2003;73:515. doi: 10.1007/s00223-003-0023-7. [DOI] [PubMed] [Google Scholar]
- 42.Seeman E. Pathogenesis of bone fragility in women and men. Lancet. 2002;359:1841–50. doi: 10.1016/S0140-6736(02)08706-8. [DOI] [PubMed] [Google Scholar]
- 43.Sekido H, Suzuki T, Jomori T, Takeuchi M, Yabe-Nishimura C, Yagihashi S. Reduced cell replication and induction of apoptosis by advanced glycation end products in rat Schwann cells. Biochem Biophys Res Commun. 2004;320:241–8. doi: 10.1016/j.bbrc.2004.05.159. [DOI] [PubMed] [Google Scholar]
- 44.Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia. 2001;44:129–46. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki H, Amizuka N, Oda K, Li M, Yoshie H, Ohshima H, Noda M, Maeda T. Histological evidence of the altered distribution of osteocytes and bone matrix synthesis in klotho-deficient mice. Arch Histol Cytol. 2005;68:371–81. doi: 10.1679/aohc.68.371. [DOI] [PubMed] [Google Scholar]
- 46.Takagi M, Ksayama S, Yamamoto T, Motomura T, Hashimoto K, Yamamoto H, Sato B, Okada S, Kishimoto T. Advanced glycation endproducts stimulate interleukin-6 production by human bone-derived cells. J Bone Miner Res. 1997;12:439–46. doi: 10.1359/jbmr.1997.12.3.439. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka Y, Nakayamada S, Fujimoto H, Okada Y, Umehara H, Kataoka T, Minami Y. H-Ras/mitogen-activated protein kinase pathway inhibits integrin-mediated adhesion and induces apoptosis in osteoblasts. J Biol Chem. 2002;277:21446–52. doi: 10.1074/jbc.M202238200. [DOI] [PubMed] [Google Scholar]
- 48.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–7. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 49.Verhaeghe J, Suiker A, Nyomba B, Visser W, Einhorn T, Dequeker J, Bouillon R. Bone mineral hoeostasis in spontaneously diabetic BB rats. II. Impaired bone turnover and decreased osteocalcin synthesis. Endocrinology. 1989;124:573–82. doi: 10.1210/endo-124-2-573. [DOI] [PubMed] [Google Scholar]
- 50.Vlassara H, Palace M. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 51.Wang C, Mayo M, Korneluk R, Goeddel D, Baldwin AJ. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 52.Weinstein R, Jilka R, Parfitt A, Manolagas S. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocoriticoids. Potential mechanisms of their deleterious effects of bone. J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamagishi S, Nakamura K, Inoue H. Possible participation of advanced glycation end products in the pathogenesis of osteoporosis in diabetic patients. Med Hypotheses. 2005;65:1013–5. doi: 10.1016/j.mehy.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto T, Ozono K, Miyauchi A, Kasayama S, Kojima Y, Shima M, Okada S. Role of advanced glycation end products in dynamic bone disease in patients with diabetic nephropathy. Am J Kidney. 2001;Dis 38:S161–4. doi: 10.1053/ajkd.2001.27428. [DOI] [PubMed] [Google Scholar]