

Abstract
Colicins reach their targets in susceptible Escherichia coli strains through two envelope protein systems: the Tol system is used by group A colicins and the TonB system by group B colicins. Colicin E2 (ColE2) is a cytotoxic protein that recognizes the outer membrane receptor BtuB. After gaining access to the cytoplasmic membrane of sensitive Escherichia coli cells, ColE2 enters the cytoplasm to cleave DNA. After binding to BtuB, ColE2 interacts with the Tol system to reach its target. However, it is not known if the entire colicin or only the nuclease domain of ColE2 enters the cell. Here I show that preincubation of ColE2 with Escherichia coli cells prevents binding and translocation of pore-forming colicins of group A but not of group B. This inhibition persisted even when cells were incubated with ColE2 for 30 min before the addition of pore-forming colicins, indicating that ColE2 releases neither its receptor nor its translocation machinery when its nuclease domain enters the cells. These competition experiments enabled me to estimate the time required for ColE2 binding to its receptor and translocation.
Colicins are plasmid-encoded antibacterial proteins that are secreted as part of the stress response system of Escherichia coli to kill other closely related bacteria (7). They have various types of lethal activity. The largest group of colicins (A, E1, N, B, Ia, Ib, 5, and 10 [ColA, ColE1, etc.]) form ion channels in the cytoplasmic membrane of sensitive cells (23). Others degrade nucleic acids of target cells (E2 to E10 and D) (20). Colicin M cleaves peptidoglycan precursors between the undecaprenyl and 1-pyrophospho-MurNAc moieties (16).
In common with many toxins, colicins are organized into structural domains that perform various functions: an N-terminal part required for translocation across the outer membrane of the target cell, a central domain necessary for binding to a cell surface receptor, and a C-terminal domain responsible for the lethal function (1). Colicins enter the cell by interacting with various proteins in the E. coli outer envelope, including an outer membrane receptor and one or more periplasmic proteins. For example, the E colicins (colicins E1 to E9) and ColA bind to a minor component of the outer membrane (OM)—the BtuB receptor protein—which is an essential element of the high-affinity uptake system for vitamin B12 in E. coli (8). After binding to specific receptors, colicins translocate across the OM in a process mediated by a group of membrane and periplasmic proteins of the Tol or the Ton system. Colicins are classified into two groups (A and B) depending on the translocation system they use to enter cells. Group A colicins (ColA and ColE1 to -E9) use the Tol system (composed of TolA, -B, -Q, and -R), whereas group B colicins (ColB and ColIa, -Ib, -D, -M, -5, and -10) use the Ton system (TonB, ExbB, and ExbD) (9, 10). Both the Ton and Tol systems are coupled to the proton motive force across the inner membrane, acting as energy transducers for active transport (Ton) or maintenance of outer envelope integrity (Tol) (24). Most colicins also require porins for their transport across the OM. Colicins N and A use OmpF as a receptor and for translocation, whereas colicins E2 to E9 require OmpF, OmpC, or PhoE for translocation (8, 19, 22).
The lethal function of nuclease colicins indicates that these toxins must cross the inner membrane to reach the cytoplasm. However, it is not clear whether the entire colicin or only the C-terminal domain penetrates the cytoplasm. Various authors have suggested recently that ColD and ColE7 are cleaved, liberating their nuclease domains in the cytoplasm (11, 30). It was also reported that the DNase domains of colicins E9 and E2 exhibit channel-forming activity in planar lipid bilayers in vitro and thus appear to be involved in translocation of the cytotoxic domains of these colicins across the inner membrane of E. coli (25). However, it is unknown whether the rest of the molecule remains fixed to the import machinery when the nuclease domain enters the cytoplasm. Pore-forming colicins do not need to completely translocate across the outer membrane to kill sensitive cells. These colicins remain in contact with their receptors and translocation machinery when the pore has formed in the inner membrane (4, 14).
In this study, competition and protease accessibility experiments were used to revisit the question of nuclease colicin translocation across the cell envelope. I show that ColE2 simultaneously occupied the reception and translocation sites of group A colicins and thus spanned the whole envelope. ColE2 spanning the envelope prevented binding and translocation of group A but not group B colicins. Our findings also indicate that ColE2 remained in contact with its receptor and import machinery even when its nuclease domain reached the cytoplasm. This clearly suggests that only the C-terminal domain of ColE2 penetrated the cytoplasm, attached or not to the rest of the toxin.
MATERIALS AND METHODS
Construction of colicin E2S37A.
The Quik Change system (Stratagene) was used to introduce the single mutation S37A of ColE2 into pBRE2 (3).
Protein purification.
Colicins A, B, E1, and E2 and ColE2(R544A H575A) were purified from E. coli W3110 as previously described (13, 15, 26, 29). Colicin multiplicity was defined as the number of colicin molecules per cell.
Measurement of colicin-induced K+ efflux.
Variations in the cytoplasmic K+ content of cells induced by the colicins were determined by measuring the changes in the K+ concentration in the external medium with a K+/valinomycin-selective electrode as previously described (5). The E. coli [araΔ(lac pro) thi (F′ lac pro)] strain (33) was grown at 37°C to an A650 of 0.5 (5 × 108 cells per ml) in 2.3 BT medium (5 g Bacto tryptone per liter, 8 g nutrient broth per liter, 5 g NaCl per liter, 10 mM Tris-HCl [pH 7.2], 1 mM CaCl2). Cells were washed and resuspended in 100 mM sodium phosphate buffer (pH 7.2) and kept on ice at a density of 5 × 1010 cells per ml. They were used within 2 h of preparation. Cells (2 × 109/ml) were incubated for 5 min at 37°C in 100 mM sodium phosphate buffer (pH 7.2) containing glucose (0.2% [wt/vol]) and 0.5 mM KCl; under these conditions, cells accumulated K+ by the Trk system to a steady-state level of 400 to 450 mmol/mg (dry weight) of cells (5). Colicins were added at the multiplicities (numbers of molecules per cell) indicated below, and the initial rate of K+ efflux was determined in the linear part of the K+ efflux curve as previously described (6).
Competition experiments.
Cells were prepared as described above. ColE2, ColE2(R544A H575A), or ColE2(S37A) was added to E. coli at various multiplicities, and the cells were incubated for 5 min or 15 min to allow complete reception or translocation of colicins. These colicins were added at appropriate multiplicities, and the initial rates of K+ efflux were determined as described above. These rates 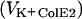 were compared with those determined without preincubation of the cells with ColE2
were compared with those determined without preincubation of the cells with ColE2 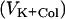 . Relative inhibition of K+ efflux is defined as
. Relative inhibition of K+ efflux is defined as 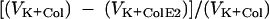 .
.
Trypsin accessibility experiments.
Cells were incubated for the indicated times at 37°C in 100 mM sodium phosphate buffer (pH 7.2) with ColE2 at a multiplicity of 800. Trypsin (250 μg/ml) was then added, and proteolysis was stopped after 1 min by addition of trypsin inhibitor (120 μg/ml). ColA was added 2 min after the trypsin inhibitor, and the initial rate of K+ efflux was measured. Control experiments were preformed without trypsin.
RESULTS
Colicin E2 inhibits the reception of colicins A and E1.
Colicins can be divided into two groups according to their killing activity: pore-forming and enzymatic colicins. In vivo activity of pore-forming colicins was determined from the measurements of the initial rate of cytoplasmic K+ efflux induced by these toxins, as previously described (6). The initial rates of K+ efflux induced by colicins A, E1, and B increased with the number of colicins added and were saturated at a multiplicity (number of colicin molecules per cell) of approximately 400 (Fig. 1A). However, the initial rate of K+ efflux induced by colicin B at saturation was three times lower than that of colicins A and E1. This difference in the initial rate of K+ efflux is likely due to a different translocation pathway used by colicin B, as previously described (14). Although ColE2 has been shown to be able to form channels in planar lipid bilayers (25), this nuclease colicin did not induce a K+ efflux, whatever the multiplicity tested (Fig. 1A). The cells were incubated with various multiplicities of ColE2 for 5 min, which is more than enough time to bind its receptor (27), to determine whether ColE2 can inhibit importation of pore-forming colicins into the cell envelope. Then, colicins A, E1, and B were added at a multiplicity of 200, at which the K+ efflux began to saturate. Inhibition of the K+ efflux as a function of ColE2 multiplicity was observed (Fig. 1B). We calculated the percentage of inhibition as described in Materials and Methods. As suspected, inhibition for group A colicins only was observed. Maximal inhibition of the K+ efflux induced by colicins A and E1 was achieved with addition of 400 to 800 ColE2 molecules. The maximal inhibition is probably due to a limitation in the number of receptors. Similar to ColA and ColE1, ColE2 uses BtuB (there are 200 to 400 molecules of BtuB per cell) as its receptor. The 400 to 800 molecules of ColE2 may occupy these 400 receptors on the cell, inhibiting binding of colicins A and E1 to the receptors.
FIG. 1.

ColE2 preincubated with sensitive cells inhibits reception of group A colicins. (A) Initial rate of K+ efflux induced by various colicins at various multiplicities (number of colicin molecules per cell). Cells (2 × 109 per ml) were incubated at 37°C in sodium phosphate buffer (100 mM [pH 7.2]) containing 0.2% glucose and 0.5 mM KCl. Colicins A (diamonds), B (triangles), E1 (squares), and E2 (circles) were added at time zero. The initial rates were measured in the linear part of the efflux curves. (B) Effect of colicin E2 on the activities of group A and B colicins. Colicin E2 was added to the cells at various multiplicities at time zero. Colicins A (diamonds), E1 (squares), and B (triangles) were added at a multiplicity of 200 5 min after introduction of colicin E2. The initial rates were measured and correlated to the numbers of colicin molecules reaching their targets. The percentage of inhibition was calculated as described in Materials and Methods. (C) Colicin E2 was added to the cells at the multiplicity of 800 at time zero. Colicin A was added at a multiplicity of 200 at various times after introduction of colicin E2. The initial rates were measured and correlated to the number of colicin molecules reaching their targets. The percentage of inhibition was calculated as described in Materials and Methods. Each experiment was performed in triplicate.
ColE2 remains in contact with its receptor after translocation.
ColA was added at various times after addition of ColE2 to gain insight into the kinetics of ColE2 reception and to test whether ColE2 remains in contact with its receptor. ColE2 inhibited the K+ efflux induced by colicin A, even if the two proteins were added at the same time, suggesting that the binding kinetics of the two colicins are similar (Fig. 1C). This inhibition persisted even if colicin A was added 30 min after ColE2, which is more than enough time for its full translocation (34). This finding suggests that translocated ColE2 did not release its receptor. The same experiment was conducted with an inactive form of ColE2, which carries a double mutation (R544A H575A) in its C-terminal domain (31), in order to exclude the possibility that the DNase activity of ColE2 was responsible for inhibition of ColA. ColE2(R544A H575A) inhibited the K+ efflux induced by ColA with kinetics similar to those of wild-type ColE2 (data not shown). Moreover, wild-type ColE2 did not inhibit the initial rate of K+ efflux induced by ColB, which used another receptor and import machinery (data not shown).
ColE2 blocks the translocation of colicins A and E1.
We tested whether ColE2 can block translocation of ColA by conducting the same experiments (as those described above) with an E. coli strain carrying pAG1, which carries the btuB gene and allows overproduction of the BtuB receptor (18). BtuB is a major component of the outer membrane in pAG1-bearing cells, comprising 20 to 40% of the outer membrane proteins. Thus, receptor binding is not the limiting factor for colicin activity in pAG1-containing cells. The limiting step in these cells is at the level of the translocation machinery. The K+ efflux induced by colicins A and E1 saturated at a multiplicity of about 1,000 in cells overproducing BtuB protein. This number approximately corresponded to the number of functional Tol complexes, as previously described (14). The percentage of K+ efflux inhibition induced by colicins A, B, and E1 relative to the multiplicity of ColE2 was determined (Fig. 2A). ColE2 did not inhibit ColB activity but inhibited activities of colicins A and E1. Maximal inhibition was obtained by addition of 1,000 to 1,600 ColE2 molecules, whereas 400 to 800 ColE2 molecules was sufficient for complete inhibition of group A colicins in an E. coli strain that did not contain pAG1. We obtained the same results with ColE2(R544A H575A) (data not shown). These findings suggest that ColE2 inhibits translocation of group A colicins. The mutation S37A was introduced into the TolB box of ColE2 to confirm these results. The tol-dependent colicins have been reported to contain a pentapeptide sequence designated as the TolB box (DGS37GW; single-letter amino acid code) close to the N terminus (17). Mutation of Ser37 to Ala has been shown previously to abolish translocation of the homologous ColE9 without affecting its receptor binding (17). ColE2S37A was inactive in vivo, as expected (data not shown). ColE2S37A added to wild-type cells at a multiplicity of 400 to 800 inhibited ColA activity, whereas ColE2S37A added to pAG1-bearing cells at the multiplicity of 1,600 had no effect (Fig. 2B). Comparison of these findings with those obtained with wild-type ColE2 indicates that ColE2S37A inhibited colicin A reception but not colicin A translocation. This clearly confirms that wild-type ColE2 inhibited colicin A translocation in pAG1-bearing cells.
FIG. 2.

ColE2 preincubated with sensitive cells inhibits translocation of group A colicins. (A) Effect of colicin E2 on the translocation of group A and B colicins. Cells carrying pAG1 were prepared as described in the legend to Fig. 1. Colicin E2 was added to the cells at various multiplicities at time zero. Colicins A (diamonds), B (triangles), and E1 (squares) were added at a multiplicity of 800 15 min after introduction of colicin E2. The initial rates of K+ efflux were measured and correlated to the numbers of colicins reaching their targets. The percentage of inhibition was calculated as described in Materials and Methods. (B) Effect of colicin E2S37A on reception and translocation of colicin A. Colicin E2 S37A was added to wild-type cells (dark gray diamonds) or pAG1-bearing cells (light gray diamonds) at various multiplicities at time zero. Colicin A was added to the cells at a multiplicity of 200 (dark gray diamonds) or 800 (light gray diamonds) 5 min or 15 min, respectively, after introduction of colicin E2S37A. (C) Colicin E2 was added to the cells at a multiplicity of 1,600 at time zero. Colicin A was added at a multiplicity of 800 at various times after introduction of colicin E2. The initial rates were measured and correlated to the number of colicins reaching their targets. The percentage of inhibition was calculated as described in Materials and Methods. Each experiment was performed in triplicate.
ColA was added at various times after addition of ColE2 to pAG1-bearing cells to test whether ColE2 remains in contact with its translocation machinery. It took 20 s for ColE2 to inhibit half of the ColA translocation (half-life = 20 s), slightly more time than that needed to inhibit ColA reception (half-life = 2 s) (Fig. 2C). This inhibition persisted even when ColA was added 30 min after ColE2, indicating that the toxin remained in contact with its translocation machinery after its DNase domain entered the cytoplasm.
ColE2 is not released from its receptor once digested by trypsin.
Greater survival of colicin-treated cells was observed when trypsin was added to the cell suspension shortly after colicin had bound to the cell surface. For example, trypsin added 1 min after colicin A stopped the efflux of K+ induced by the toxin, suggesting that trypsin inactivated the toxin (4). Digested ColA has been reported to release its receptor and translocation machinery (14). In this study, we used trypsin to investigate how long ColE2 can inhibit ColA and whether digested ColE2 releases its receptor and import machinery. K+ efflux induced by ColA on trypsin-treated cells (Fig. 3A, right panel) was similar to that obtained with untreated cells (Fig. 3A, left panel), indicating that trypsin did not damage any bacterial component necessary for binding or translocation. However, trypsin completely inactivated ColA (Fig. 3A, central panel). We determined the effects of trypsin and trypsin inhibitor on ColE2 inhibition of ColA (Fig. 3B). Trypsin and ColE2 were added simultaneously (Fig. 3B, central panel) or trypsin was added 2 min after ColE2 (Fig. 3B, right panel). Proteolysis was stopped 1 min later by adding trypsin inhibitor and added ColA after another 2 min. The K+ efflux induced by ColA was not inhibited by the presence of ColE2 when trypsin and ColE2 were added simultaneously (Fig. 3B, central panel). This indicates that ColE2 was completely inactivated by trypsin and was unable to inhibit ColA activity. Previous in vitro protease accessibility experiments of the endonuclease colicin E9 indicate that its receptor-binding domain is not protected from protease digestion (35). Thus, the ColE2 inactivation described above is probably caused by trypsin digestion of its receptor-binding domain. ColA activity was maximally inhibited (Fig. 3B, right panel) when trypsin was added 2 min after ColE2. This indicates that the receptor-binding domain of ColE2 was protected from trypsin 2 min after ColE2 addition. Trypsin was added at various times to investigate the kinetics of ColE2 digestion. The percentage of inhibition of K+ efflux induced by colicin A relative to the time that trypsin was introduced after ColE2 addition was determined. Maximal inhibition occurred when trypsin was introduced 60 s after ColE2 addition. If trypsin was introduced before 60 s, digested ColE2 did not bind BtuB and it released its receptor. If trypsin was introduced after 60 s, intact or cleaved ColE2 remained associated with import machinery (Fig. 4).
FIG. 3.

Protease accessibility of ColE2. (A) Trypsin-inactivated colicin A. Cells were prepared as described in the legend to Fig. 1. Colicin A added at a multiplicity of 200 induced a decrease in the cytoplasmic K+ content of the cells (left panel). The initial rate of K+ efflux was measured as Vi = 280 nmol K+/min/mg. Trypsin (Ti [250 μg/ml]) was preincubated with sensitive cells for 1 min. Trypsin inhibitor (Ti inh [120 μg/ml]) was added 1 min after introduction of trypsin, and colicin A (multiplicity of 200) was added 3 min after introduction of trypsin (right panel). The initial rate of K+ efflux was measured as Vi = 280 nmol K+/min/mg. Trypsin (250 μg/ml) was added to cells 15 s before colicin A addition (multiplicity of 200 [central panel]). The initial rate of K+ efflux was measured as Vi = 0 nmol K+/min/mg. (B) Colicin E2 did not inhibit colicin A. Colicin E2 was added at a multiplicity of 800 to sensitive cells at time zero. Colicin A (multiplicity of 200) was added 2 min later (left panel). The initial rate of K+ efflux was measured as Vi = 20 nmol K+/min/mg. Colicin E2 was added at a multiplicity of 800 to sensitive cells at time zero. Trypsin (250 μg/ml) was added 5 s (central panel) or 2 min (right panel) later. Trypsin inhibitor (120 μg/ml) was added 2 min after introduction of trypsin, and colicin A (multiplicity of 200) was added 4 min after introduction of trypsin. The initial rate of K+ efflux was measured as Vi = 20 nmol K+/min/mg in the right panel and Vi = 280 nmol K+/min/mg in the central panel.
FIG. 4.

Kinetics of ColE2 digestion by trypsin. Cells were prepared as described in the legend to Fig. 1. Trypsin (250 μg/ml) was added at various times after introduction of colicin E2. Trypsin inhibitor (120 μg/ml) and colicin A (multiplicity of 200) were added 2 and 4 min after introduction of trypsin, respectively. The initial rates of K+ efflux were measured and correlated to the number of colicins reaching their targets. The percentage of inhibition was calculated as described in Materials and Methods.
DISCUSSION
Since the discovery of ColE2 activity, it has been clear that part of this toxin must reach the cytoplasm to find its target, DNA (28). However, there have been no reports showing whether the entire ColE2 or only its C-terminal domain enters the cytoplasm. A pore-forming colicin, colicin A, was used to examine this question. Measurement of K+ efflux induced by ColA is a rapid and sensitive method to study translocation and reception of pore-forming colicins (12). I used this sensitive assay for competition experiments to study reception and translocation of the nuclease colicin E2. Although ColE2 forms channels in planar lipid bilayers, it does not induce K+ efflux and thus can be used as a competitor against pore-forming colicin activity. Moreover, ColA and ColE2 use BtuB and OmpF as receptors and the Tol machinery for translocation.
Our findings from competition experiments indicate that addition of ColE2 to sensitive cells prevented K+ efflux induced by group A pore-forming colicins. These competition experiments showed that 400 to 800 molecules of ColE2 was required to fully inactivate group A pore-forming colicin activity in wild-type cells. Inhibition probably occurred at the reception step, as all of the colicins tested use the same receptor, BtuB, of which there are 200 to 400 molecules per cell. Overproduction of BtuB impaired the ability of ColE2 to inhibit colicins A and E1. Under these conditions, BtuB became a major component of the outer membrane and there were at least 7 × 104 OmpF molecules per cell. The limiting step for colicin importation under these conditions is not receptor binding but translocation across the periplasm, which requires the Tol machinery. E. coli cells contain approximately 1,000 translocation sites (14). So, it was not surprising that 1,600 ColE2 molecules was required to block translocation of group A colicins. Colicin A translocation was not prevented by 1,600 ColE2S37A molecules, which did not interact with Tol machinery. Control experiments with an inactive form of ColE2 demonstrate that the DNase activity of this colicin did not damage the cells in a way that prevents K+ efflux induced by group A pore-forming colicins. Moreover, wild-type and mutated ColE2 did not prevent induction of K+ efflux by ColB, a group B pore-forming colicin.
The major finding of this paper is not that ColE2 blocked reception and translocation of group A colicins but that ColE2 blocked these even when it was introduced 30 min before the addition of pore-forming colicins. Thus, ColE2 remained associated with its receptor and the Tol machinery for at least 30 min. Although the exact amount of time required for complete import of nuclease colicins is unknown, previous reports suggest that nuclease colicins enter the cytoplasm from 3.5 to 20 min after their addition to cells (15, 21, 34). These findings demonstrate that ColE2 releases neither its receptor nor its import machinery when its nuclease domain enters the cytoplasm. Thus, only the C-terminal domain of ColE2 crosses the inner membrane. It is unknown if the nuclease domain is cleaved before entering the cytoplasm, as suggested previously for ColD and ColE7 (11, 30), or remains attached to the other domains. I show that if the nuclease domain is cleaved, the other two domains remain associated with BtuB and the Tol proteins. Colicin structure may explain the ability of colicin to span the whole envelope. Colicins Ia (a pore-forming colicin) and E3 (a nuclease colicin) appear to have an unusually elongated Y shape. The two arms of the Y shape correspond to the N-terminal domain and the C-terminal domain of colicins, whereas the stem corresponds to the central domain. The two arms are long enough to allow the C-terminal domain and the N-terminal domain to interact with the inner membrane, while the central domain remains attached to the outer membrane (32, 36).
It is difficult to evaluate the time required for DNase colicins to kill sensitive cells, because there is no rapid and sensitive assay to measure DNA damage. Nevertheless, previous reports (based on trypsin accessibility, SOS promoter-lux fusion reporter assay, and cell-killing activity) indicate this process takes from 3.5 to 20 min (2, 27, 34). This is more than enough time for pore-forming colicins to be completely imported. As mentioned previously, ColA required only 30 s at 37°C to form voltage-gated channels in the inner membrane of E. coli. Our findings suggest that this time difference was not due to receptor binding and translocation of colicin. Incubation with ColE2 for only 2 s inhibited half of ColA reception, and incubation with ColE2 for 20 s inhibited half of ColA translocation. Thus, 20 s was a sufficient amount of time for ColE2 to interact with its import machinery. This finding suggests that nuclease colicins accumulate delay during translocation of their C-terminal domains from the outer membrane to the cytoplasm. There are two possible reasons for this. First, the nuclease domain must enter the cytoplasm, whereas the pore-forming domain inserts into the inner membrane. Second, in contrast to pore-forming colicins, nuclease colicins are released as heterodimeric complexes with their cognate immunity protein which must be removed during the translocation process. However, it has been demonstrated previously that the amounts of time required for complete translocation of nuclease colicin E9—fixed or not to its immunity protein—were identical, suggesting that release of the immunity protein did not delay colicin translocation (34). Moreover, the nuclease domain entered the cell very quickly (34). Based on these results and those presented in this paper, the amounts of time required for complete translocation of pore-forming and nuclease colicins are probably similar, and any differences were likely due the assays used to measure these times.
Trypsin accessibility experiments have been used extensively to assess the amount of time available for this protease to rescue cells from colicin action. Addition of trypsin as soon as 1 min and up to 5 min after the onset of colicin A-induced K+ efflux has been shown to arrest the efflux (4). It has been reported that after ColA is digested by trypsin, it is released from its receptor and its import machinery (14). The same experiments performed with ColE9 suggested that half of the DNase domains crossed at least the outer membrane within 3.5 min of addition to the cells. Here, I show that 60 s was required for the receptor domain of ColE2 to become inaccessible to trypsin digestion. Indeed, when trypsin was added to sensitive cells 13 s after ColE2 was introduced, ColE2 still inhibited 50% of the incoming colicin A (Fig. 4), suggesting that half of the receptor was still occupied by the ColE2 central domain. ColE2 binds its receptor between 13 s and 3.5 min, with its nuclease domain accessible to the external medium and trypsin digestion. So, once digested, ColE2 was not released from its receptor.
In conclusion, I show that ColE2 released neither its receptor nor the Tol machinery with entry of its nuclease domain into the cytoplasm, disproving the hypothesis that the entire colicin is imported. The kinetics of reception and translocation of ColE2 and ColA were similar. However, in contrast to ColA, ColE2 was not released from its receptor after digestion by trypsin.
Acknowledgments
I gratefully acknowledge J. Sturgis and R. Lloubès for advice and discussions. I thank R. Lloubès, P. Chames, and J. Sturgis for comments on the manuscript.
Footnotes
Published ahead of print on 6 April 2007.
REFERENCES
- 1.Baty, D., M. Frenette, R. Lloubès, V. Géli, S. P Howard, F. Pattus, and C. Lazdunski. 1988. Functional domain of colicin A. Mol. Microbiol. 2:807-811. [DOI] [PubMed] [Google Scholar]
- 2.Bénédétti, H., M. Frenette, D. Baty, R. Lloubès, V. Géli, and C. Lazdunski. 1989. Comparison of the uptake system for the entry of various BtuB group colicins into Escherichia coli. J. Gen. Microbiol. 135:3413-3420. [DOI] [PubMed] [Google Scholar]
- 3.Benedetti, H., M. Frenette, D. Baty, M. Knibiehler, F. Pattus, and C. Lazdunski. 1991. Individual domains of colicins confer specificity in colicin uptake, in pore-properties and in immunity requirement. J. Mol. Biol. 217:429-439. [DOI] [PubMed] [Google Scholar]
- 4.Bénédétti, H., R. Lloubès, C. Lazdunski, and L. Letellier. 1992. Colicin A unfolds during its translocation in Escherichia coli cells and spans the whole cell envelope when its pore has formed. EMBO J. 11:441-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boulanger, P., and L. Letellier. 1988. Characterization on ion channels involved in the penetration of phage T4 DNA into Escherichia coli cells. J. Biol. Chem. 263:9767-9775. [PubMed] [Google Scholar]
- 6.Bourdineaud, J. P., P. Boulanger, C. Lazdunski, and L. Letellier. 1990. In vivo properties of colicin A: channel activity is voltage dependent but translocation may be voltage independent. Proc. Natl. Acad. Sci. USA 87:1037-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun, V., H. Pilsl, and P. Gross. 1994. Colicins: structures, modes of action, transfer through membranes and evolution. Arch. Microbiol. 161:199-206. [DOI] [PubMed] [Google Scholar]
- 8.Cavard, D., and C. Lazdunski. 1981. Involvement of BtuB and OmpF proteins in binding and uptake of colicin A. FEMS Microbiol. Lett. 12:311-316. [Google Scholar]
- 9.Davies, J. K., and P. Reeves. 1975. Genetics of resistance to colicins in Escherichia coli K-12: cross-resistance among colicins of group A. J. Bacteriol. 123:102-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies, J. K., and P. Reeves. 1975. Genetics of resistance to colicins in Escherichia coli K-12: cross-resistance among colicins of group B. J. Bacteriol. 123:96-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Zamaroczy, M., L. Mora, A. Lecuyer, V. Geli, and R. H. Buckingham. 2001. Cleavage of colicin D is necessary for cell killing and requires the inner membrane peptidase LepB. Mol. Cell 8:159-168. [DOI] [PubMed] [Google Scholar]
- 12.Duché, D., D. Baty, M. Chartier, and L. Letellier. 1994. Unfolding of colicin A during its translocation through the Escherichia coli envelope as demonstrated by disulfide bond engineering. J. Biol. Chem. 269:24820-24825. [PubMed] [Google Scholar]
- 13.Duché, D., J. Izard, J.-M. González-Mañas, M. W. Parker, M. Crest, M. Chartier, and D. Baty. 1996. Membrane topology of the colicin A pore-forming domain analyzed by disulfide bond engineering. J. Biol. Chem. 271:15401-15406. [DOI] [PubMed] [Google Scholar]
- 14.Duché, D., L. Letellier, V. Géli, H. Bénédétti, and D. Baty. 1995. Quantification of group A colicin import sites. J. Bacteriol. 177:4935-4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duché, D., A. Frenkian, V. Prima, and R. Lloubès. 2006. Release of immunity protein requires functional endonuclease colicin import machinery. J. Bacteriol. 188:8593-8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El Ghachi, M., A. Bouhss, H. Barreteau, T. Touze, G. Auger, D. Blanot, and D. Mengin-Lecreulx. 2006. Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem. 281:22761-22772. [DOI] [PubMed] [Google Scholar]
- 17.Garinot-Schneider, C., C. N. Penfold, G. R. Moore, C. Kleanthous, and R. James. 1997. Identification of residues in the putative TolA box which are essential for the toxicity of the endonuclease toxin colicin E9. Microbiology 143:2931-2938. [DOI] [PubMed] [Google Scholar]
- 18.Gudmundsdottir, A., P. E. Bell, M. D. Lundrigan, C. Bradbeer, and R. J. Kadner. 1989. Point mutations in a conserved region (TonB box) of Escherichia coli outer membrane protein BtuB affect vitamin B12 transport. J. Bacteriol. 171:6526-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Housden, N. G., S. R. Loftus, G. R. Moore, R. James, and C. Kleanthous. 2005. Cell entry mechanism of enzymatic bacterial colicins: porin recruitment and the thermodynamics of receptor binding. Proc. Natl. Acad. Sci. USA 102:13849-13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.James, R., C. N. Penfold, G. R. Moore, and C. Kleanthous. 2002. Killing of E. coli cells by E group nuclease colicins. Biochimie 84:381-389. [DOI] [PubMed] [Google Scholar]
- 21.Krone, W. J. A., P. de Vries, G. Koningstein, A. J. R. de Jonge, F. K. de Graaf, and B. Oudega. 1986. Uptake of cloacin DF13 by susceptible cells: removal of immunity protein and fragmentation of cloacin molecules. J. Bacteriol. 166:260-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurisu, G., S. D. Zakharov, M. V. Zhalnina, S. Bano, V. Y. Eroukova, T. I. Rokitskaya, Y. N. Antonenko, M. C. Wiener, and W. A. Cramer. 2003. The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat. Struct. Biol. 10:948-954. [DOI] [PubMed] [Google Scholar]
- 23.Lakey, J. H., and S. L. Slatin. 2001. Pore-forming colicins and their relatives. Curr. Top. Microbiol. Immunol. 257:131-161. [DOI] [PubMed] [Google Scholar]
- 24.Lloubès, R., E. Cascales, A. Walburger, E. Bouveret, C. Lazdunski, A. Bernadac, and L. Journet. 2001. The Tol-Pal proteins of the Escherichia coli cell envelope: an energized system required for outer membrane integrity? Res. Microbiol. 152:523-529. [DOI] [PubMed] [Google Scholar]
- 25.Mosbahi, K., C. Lemaitre, A. H. Keeble, H. Mobasheri, B. Morel, R. James, G. R. Moore, E. J. Lea, and C. Kleanthous. 2002. The cytotoxic domain of colicin E9 is a channel-forming endonuclease. Nat. Struct. Biol. 9:476-484. [DOI] [PubMed] [Google Scholar]
- 26.Pressler, B., V. Braun, B. Wittmann-Liebold, and R. Benz. 1986. Structural and functional properties of colicin B. J. Biol. Chem. 261:2654-2659. [PubMed] [Google Scholar]
- 27.Reynolds, B. L., and P. R. Reeves. 1969. Kinetics of absorption of colicin CA42-E2 and reversal of its bactericidal activity. J. Bacteriol. 100:301-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaller, K., and M. Nomura. 1976. Colicin E2 is DNA endonuclease. Proc. Natl. Acad. Sci. USA 73:3989-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz, S. A., and D. R. Helinski. 1971. Purification and characterization of colicin E1. J. Biol. Chem. 246:6318-6327. [PubMed] [Google Scholar]
- 30.Shi, Z., K. F. Chak, and H. S. Yuan. 2005. Identification of an essential cleavage site in ColE7 required for import and killing of cells. J. Biol. Chem. 280:24663-24668. [DOI] [PubMed] [Google Scholar]
- 31.Slatin, S. L., A. Nardi, K. S. Jakes, D. Baty, and D. Duché. 2002. Translocation of a functional protein by a voltage-dependent ion channel. Proc. Natl. Acad. Sci. USA 99:1286-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soelaiman, S., K. Jakes, N. Wu, C. Li, and M. Shoham. 2001. Crystal structure of colicin E3: implications for cell entry and ribosome inactivation. Mol. Cell 8:1053-1062. [DOI] [PubMed] [Google Scholar]
- 33.Sun, T.-P., and R. E. Webster. 1986. fii, a bacterial locus required for filamentous phage infection and its relation to colicin-tolerant tolA and tolB. J. Bacteriol. 165:107-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vankemmelbeke, M., B. Healy, G. R. Moore, C. Kleanthous, C. N. Penfold, and R. James. 2005. Rapid detection of colicin E9-induced DNA damage using Escherichia coli cells carrying SOS promoter-lux fusions. J. Bacteriol. 187:4900-4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallis, R., A. Reilly, K. Barnes, C. Abell, D. G. Campbell, G. R. Moore, R. James, and C. Kleanthous. 1994. Tandem overproduction and characterization of the nuclease domain of colicin E9 and its cognate inhibitor protein Im9. Eur. J. Biochem. 220:447-454. [DOI] [PubMed] [Google Scholar]
- 36.Wiener, M., D. Freymann, P. Ghosh, and R. M. Stroud. 1997. Crystal structure of colicin Ia. Nature 385:461-464. [DOI] [PubMed] [Google Scholar]