

Abstract
Initially described as a modulator of embryogenesis for a number of organ systems, Wnt1 has recently been linked to the development of several neurodegenerative disorders, none being of greater significance than Alzheimer's disease. We therefore examined the ability of Wnt1 to oversee vital pathways responsible for cell survival during β-amyloid (Aβ1-42)exposure. Here we show that Wnt1 is critical for protection in the SH-SY5Y neuronal cell line against genomic DNA degradation, membrane phosphatidylserine (PS) exposure, and microglial activation, since these neuroprotective attributes of Wnt1 are lost during gene silencing of Wnt1 protein expression. Intimately tied to Wnt1 protection is the presence and activation of Akt1. Pharmacological inhibition of the PI 3-K pathway or gene silencing of Akt1 expression can abrogate the protective capacity of Wnt1. Closely aligned with Wnt1 and Akt1 are the integrated canonical pathways of synthase kinase-3β (GSK-3β) and β-catenin. Through Akt1 dependent pathways, Wnt1 phosphorylates GSK-3β and maintains β-catenin integrity to insure its translocation from the cytoplasm to the nucleus to block apoptosis. Our work outlines a highly novel role for Wnt1 and its integration with Akt1, GSK-3β, and β-catenin to foster neuronal cell survival and repress inflammatory microglial activation that can identify new avenues of therapy against neurodegenerative disorders.
Keywords: Akt, apoptosis, β-catenin, glycogen synthase kinase-3β, inflammation, microglia, wingless, Wnt
1. Introduction
Derived from Drosophila Wingless and the mouse Int-1 genes, Wnt proteins are a family of cysteine-rich glycosylated proteins that function through a canonical pathway targeting β-catenin or through non-canonical β-catenin independent pathways. Members of the Wnt family control the proliferation and differentiation of a variety of cell types that can range from progenitor stem cells to mature organ systems [1, 2]. More recently, the Wnt family, and in particular Wnt1 signaling, has been closely tied to the control of apoptotic cellular injury. Loss of Wnt1 expression [3, 4] leads to apoptosis while the presence of Wnt1 can promote cell survival during insults such as serum deprivation [5]. In the nervous system, altered Wnt1 signaling may incite functional cognitive impairment [6], lead to retinal degeneration [7], and in experimental models, affect amyloid precursor protein metabolism [8] and neurofibrillary pathology [9].
Given the potential impact of Wnt1 upon neurodegeneration, investigations that elucidate the ability of Wnt1 signaling to alter β-amyloid (Aβ1-42) toxicity may shed new light for novel treatments for Alzheimer's disease [10, 11]. One attractive pathway that may work in concert with Wnt1 to maintain cell survival and block inflammatory microglial activation involves protein kinase B, or Akt [12]. Of the three known Akt family members (Akt1, Akt2, and Akt3), Akt1 is highly expressed in brain. During cell injuries that include amyloid toxicity [13-15], Akt1 prevents apoptotic DNA degradation and membrane phosphatidylserine (PS) exposure that is responsible for microglial cell activation [16-19]. Neither Wnt1 or Akt1 function in isolation to foster cell survival, but these proteins can block critical glycogen synthase kinase-3β (GSK-3β) activity and promote β-catenin integrity with nuclear translocation to enhance cell survival [1, 4, 20, 21].
We demonstrate that Wnt1 has a novel capacity to modulate both apoptotic DNA degradation and membrane PS externalization during Aβ1-42toxicity. As a result, Wnt1 is a significant determinant not only of neuronal cell survival, but also of primary microglial activation and proliferation, since gene silencing of Wnt1 protein expression abrogates the ability of Wnt1 to enhance cell survival. Neuroprotection by Wnt1 also is closely integrated with the activation of Akt1 and the cellular substrates of GSK-3β and β-catenin that require the inhibitory phosphorylation of GSK-3β activity and the intracellular trafficking of β-catenin from the cytoplasm to the nucleus.
2. Materials and methods
2.1 Human neuroblastoma SH-SY5Y cell culture and differentiation
Human adrenergic neuroblastoma SH-SY5Y cells were purchased from ATCC (American Type Culture Collection) and maintained in regular Eagle’s Minimum Essential alpha medium (MEM) (Invitrogen, Carlsbad, CA), supplemented with 10% heat-inactivated fetal bovine serum, 1 mM pyruvate, 1.5 g/L sodium bicarbonate, 100 IU/ml penicillin, 100 μg/ml streptomycin at 37°C in 95%/5% (v/v) mixture of humidified atmospheric air and CO2. Cell suspension was prepared at a density of 3-4× 104 (24 well plate) or 1-1.5× 105 (35 mm2 Petri dish). When confluent at 50-60%, cells were differentiated by MEM growth medium containing 10 μM all-trans retinoic acid (Sigma, St. Louis, MO) for 48 h. Experiments were initiated until cells grew to 60% - 70% confluence between passages 4-10 after differentiation.
2.2 Experimental treatments
β-amyloid (Aβ1-42) treatment: Aβ1-42 (US Peptide, Rancho Cucamonga, CA) was dissolved in PBS at a concentration of 100 μM. To allow for Aβ aggregation, Aβ was incubated at 37°C for a 7 day period and then directly applied to neuronal cell cultures per the experimental protocols.
2.3 Assessment of cell survival
SH-SY5Y injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours following treatment with Aβ1-42 per our previous protocols [13]. The mean survival was determined by counting eight randomly selected non-overlapping fields with each containing approximately 10-20 cells (viable + non-viable). Each experiment was replicated 6 times independently with different cultures.
2.4 Assessment of DNA fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay [17, 22]. Briefly, SH-SY5Y cells were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3’-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI) followed by streptavidin-peroxidase and visualized with 3,3’-diaminobenzidine (Vector Laboratories, Burlingame, CA).
2.5 Assessment of membrane phosphatidylserine (PS) residue externalization
Phosphatidylserine (PS) exposure was assessed through the established use of annexin V. Per our prior protocols [16, 17], a 30 μg/ml stock solution of annexin V conjugated to phycoerythrin (PE) (R&D Systems, Minneapolis, MN) was diluted to 3 μg/ml in warmed calcium containing binding buffer (10 mmol/L Hepes, pH 7.5, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2). Plates were incubated with 500 μl of diluted annexin V for 10 minutes. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm.
2.6 Expression of Wnt1, phosphorylated Akt1, phosphorylated β-catenin, and phosphorylated glycogen synthase kinase
Cells were homogenized and following protein determination, each sample (50 μg/lane) was then subjected to 7.5% (GSK-3β, β-catenin) or 12.5% (Wnt1) SDS-polyacrylamide gel electrophoresis. After transfer, the membranes were incubated with a rabbit polyclonal antibody against Wnt1 (1: 200, Santa Cruz Biotechnologies, Santa Cruz, CA), a mouse monoclonal antibody against phosphorylated Akt1 (p-Akt1, Ser473, 1:1000, Active Motif, Carlsbad, CA), a rabbit antibody against phosphorylated-β-catenin (1:1000) (p-β-catenin, Ser33/37 Thr41, Cell Signaling, Beverly, MA), or a rabbit antibody against phosphorylated GSK-3β (p-GSK-3β, Ser9). Following washing, the membranes were incubated with a horseradish peroxidase (HRP) conjugated secondary antibody (goat anti-mouse IgG, 1:2000 (p-Akt1) or goat anti-rabbit IgG, 1:15000 (p-β-catenin, p-GSK-3β). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, NJ).
2.7 Stable transfection of Wnt1 and (myr-Akt1) cDNA constructs into SH-SY5Y cells
Stable SH-SY5Y clones overexpressing Wnt1 were generated by transfecting cells with Wnt1 cDNA construct under the control of a CMV promoter with HA-tagged Wnt1 cDNA (6.75 kb) inserted into the Xba I site of pUSEamp (+) vector (Upstate Biotechnology, Lake Placid, NY). For the myristoylated (active) form of Akt1 (myr-Akt1), stable SH-SY5Y clones were generated by transfecting the cells with a cDNA construct under the control of a CMV promoter with cDNA (6.89 kb) containing sequences corresponding to amino acids 1-11 of avian c-rsc at the 5’ end and a Myc-His tag at the 3’end of the mouse Akt1 open reading frame (Upstate Biotechnology, Lake Placid, NY). Cells were seeded into 35 mm dishes at a concentration of 1-1.5× 105 and transfected with 1 μg cDNA by using lipofection with Lipofectamine reagent (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer. Subsequent selection of the transfectants was performed with 400 μg/ml geneticin (Gibco BRL, Gaithersburg, MD) 48 h later. Stable clones were identified, collected, and expanded over a 3-4 week course with transfection efficiency equal to approximately 98% (n= 20). Individual clones were evaluated independently and characterized for Wnt1 or myr-Akt1 expression on Western analysis and by immunocytochemistry detection with tagged fluorescent hemagglutinin (HA-Tag) (for Wnt1, anti-HA rabbit antibody, 1:1000, Upstate Biotechnology, Lake Placid, NY) or Myc Tag (for Akt1, anti-Myc rabbit antibody, 1:1000, Upstate Biotechnology, Lake Placid, NY) conjugated to biotinylated anti-rabbit IgG (1:50) and fluorescein avidin (1:50) (Vector Laboratories, Burlingame, CA).
2.8 Gene silencing of Wnt1 and Akt1 with small interfering RNA (siRNA)
Cells were plated into 35 mm dishes or 24-well plates. To silence Akt1 gene expression, commercial reagents using the SMARTpool Akt1 siRNA kit (Upstate, Lake Placid, NY) were used. To silence Wnt1 gene expression, the following sequences were synthesized using Silencer® siRNA (Ambion, Austin, Texas): the Wnt1 target sequence 5’-AAGACCTGCTGGATGCGGCTG-3’, the siRNA sense strand 5’-GACCUGCUGGAUGCGGCUGTT-3’, and the antisense strand 5’-CAGCCGCAUCCAGCAGGUCTT-3’. Transfection of siRNA duplexes were performed with Oligofectamine reagent according to manufacturer guidelines (Invitrogen, Carlsbad, CA). Experimental assays were performed 72 hours post-transfection. For each siRNA assay, negative controls contain multiple siRNAs including the target siRNA and positive controls are absent of the target siRNA.
2.9 Microglia cell cultures, assessment of microglial activation and proliferation
Microglia were obtained from the cerebral cortex of E-19 Sprague-Dawley rat pups [23]. Briefly, cerebral cortex cells were mechanically dissociated, seeded in 75 cm2 plastic flasks at a density of 8.5×106 cells per flask, and maintained with Dulbecco's Modified Eagle Medium (DMEM/F-12) (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (ICN, Aurora, OH). Microglia were purified from mixed cultures with reciprocal shaking at 180 rpm for 15 hour and then re-seeded at 105 cells/ml for cell adhesion of 3 hour duration to yield an almost pure preparation of microglia (>98%). Microglial cells were identified by α-naphthyl acetate esterase, OX-42, and isolectin B4 from griffonia simplicifolia (Sigma, St. Louis, MO). The cells did not stain for glial fibrillary acidic protein (GFAP) (Sigma, St. Louis, MO).
Microglia were conditioned for 3 hour by media from either wildtype or SH-SY5Y cells overexpressing Wnt1 24 hour following Aβ exposure. Proliferating cell nuclear antigen (PCNA) staining for microglial activation [24] and bromodeoxyuridine (BrdU) staining for microglial proliferation [25] was performed with anti-mouse monoclonal antibody PCNA (1:100) or BrdU (1:100) conjugated with biotinylated anti-mouse IgG (1:50) and visualized through fluorescein avidin (1:50) for PCNA and Texas Red streptavidin (Vector laboratories, Burlingame, CA) for BrdU. BrdU (10 μM) and fluorodexyuridine (1μM) (Sigma, St. Louis, MO) were applied 1 hour prior to the time of fixation.
2.10 β-catenin immunocytochemistry
For immunocytochemical staining of β-catenin, SH-SY5Y cells were fixed with 4% paraformaldehyde and permeabilized using 0.2% Triton X-100. Cells were then incubated with rabbit anti-β-catenin 1 (1:100, Cell Signaling Technology, Beverly, MA) over night at 4°C and then with biotinylated anti-rabbit IgG (1:50, Vector laboratories) for 2 hours followed by Texas Red streptavidin (1:50, Vector laboratories) for 1 hour. Cells are washed in PBS and then stained with DAPI (Sigma, St. Louis, MO) for nuclear identification. Protein for β-catenin was imaged with fluorescence at the wavelengths of 565 nm (red) and 400 nm (DAPI nuclear staining).
2.11 Statistical analysis
For each experiment, the mean and standard error were determined. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) from 6 replicate experiments with the post-hoc Dunnett's test. Statistical significance was considered at p<0.05.
3. Results
3.1 Exposure to Aβ1-42 results in progressive cellular injury
Aβ1-42 was applied to neuronal cultures in a series of concentration (1, 5, 10, 20, and 50 μM) and cell survival was assessed 24 hours later by trypan blue dye exclusion. As shown in Fig. 1A, cell survival was significantly reduced from 93 ± 3% in untreated control cells to 75 ±4%, 55 ± 4% (P<0.01), 40 ± 5% (P<0.01), and 33 ± 6% (P<0.01) during exposure to Aβ1-42 in the serial concentrations of 5 μM, 10 μM, 20 μM, and 50 μM respectively. The Aβ1-42 concentration of 1 μM was not toxic. Since Aβ1-42 concentrations of 10 μM and 20 μM provided robust cell injury models and resulted in a large decrease in neuronal survival ranging from approximately 30-50% (30-50% SH-SY5Y cell loss), these concentrations of Aβ1-42were employed for the subsequent experimental paradigms to allow observation of potential changes in cell survival that would be statistically significant.
Fig. 1.
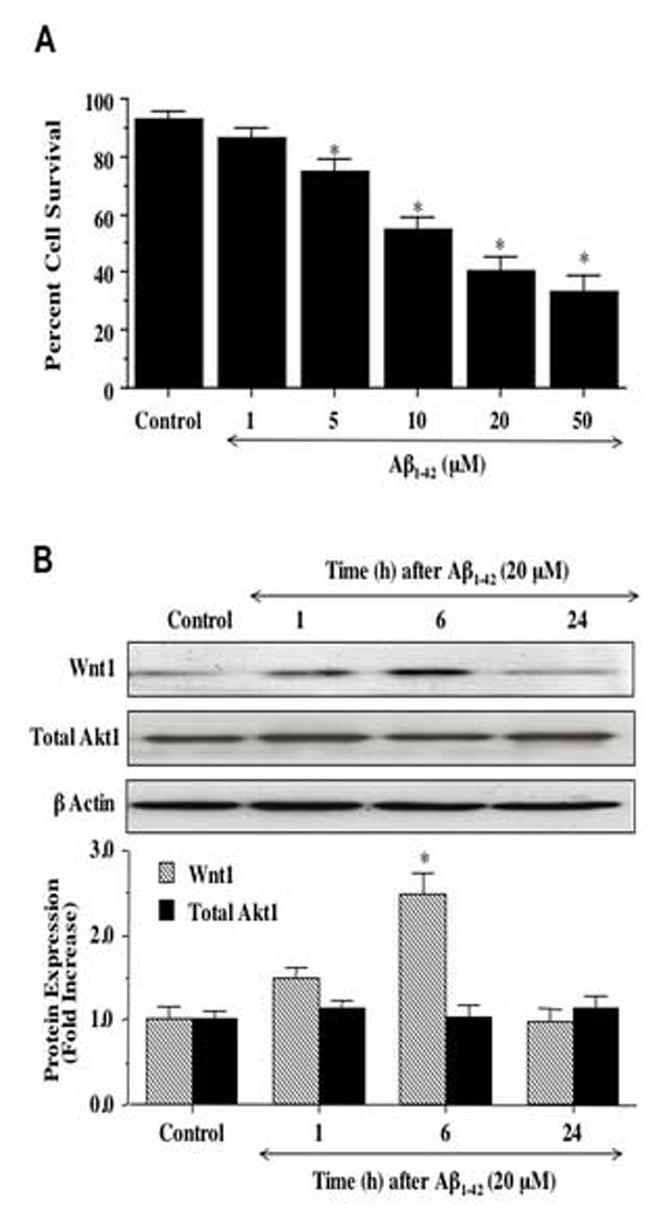
Exposure to Aβ1-42 leads to progressive neuronal injury and is accompanied by an initial increase in expression of Wnt1. (A) Neuronal survival was progressively decreased with increased concentration of Aβ1-42 (*P<0.01 vs. untreated control). (B) Exposure to Aβ1-42resulted in increased Wnt1 expression at 1 hour (h) and significantly at 6 hours (h) following Aβ1-42application, but loss of Wnt1 by 24 hours (h) with expression levels comparable to untreated control (*P<0.01 vs. control). Total Akt1 expression was not altered by Aβ1-42 exposure over a 24 hour course. Each data point represents the mean and ±SEM from 6 experiments.
3.2 Wnt1 protein expression is rapidly increased following Aβ1-42 exposure
Western blot assay was performed for Wnt1 protein expression at 1, 6, and 24 hours following exposure to Aβ1-42 (20 μM) (Fig. 1B). Aβ1-42 initially increased the expression of Wnt1 at 1 hour when compared to control, but a significant increase in Wnt1 expression was observed within 6 hours following Aβ1-42exposure. After 6 hours post Aβ1-42 exposure, expression of Wnt1 returned to untreated control cell levels (Fig. 1B). Total Akt1 expression was not altered by Aβ1-42 exposure over a 24 hour course.
3.3 Wnt1 prevents cellular injury and apoptotic genomic DNA degradation during Aβ1-42 exposure
To examine the ability of Wnt1 to prevent cell injury during Aβ1-42 exposure, SH-SY5Y cells were exposed to the Aβ1-42 (5, 10, 20 μM) and cell survival was assessed 24 hours later by the trypan blue dye exclusion method (Figs. 2A, and 2B). In Fig. 2A, untreated wildtype control cells are without trypan blue staining, but exposure to Aβ1-42 leads to cellular membrane breakdown and trypan blue staining of cells. In wildtype and vector transfected cells exposed to Aβ1-42, cell survival was significantly reduced following 5, 10, and 20 μM Aβ1-42 treatment (P<0.01). In contrast, cells with stable Wnt1 overexpression exposed to Aβ1-42 were with significantly reduced trypan blue staining and had increased survival, illustrating that Wnt1 was capable of preventing Aβ1-42 toxicity (Figs. 2B).
Fig. 2.
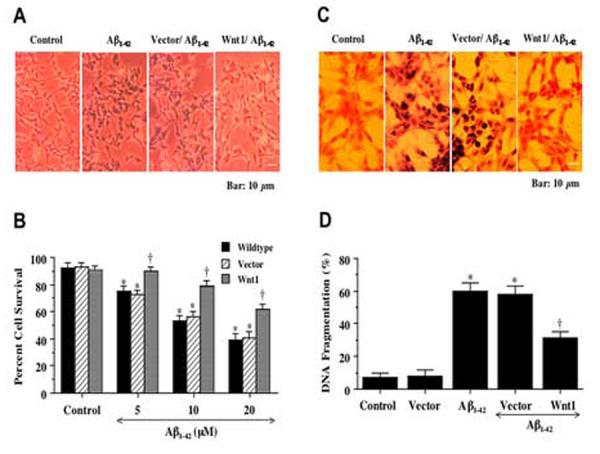
Wnt1 overexpression enhances cell survival and blocks nuclear DNA degradation during Aβ1-42 exposure. In A, representative images illustrate that overexpression of Wnt1 significantly prevents cell injury 24 hours after administration of Aβ1-42(20 μM) when compared to Aβ1-42(20 μM) exposure alone. Vector transfected cells that lack Wnt1 cDNA resulted in cell injury and trypan blue during Aβ1-42(20 μM) exposure. In B, significant increased cell survival is present in cells with stable Wnt1 overexpression during Aβ1-42(5 μM, 10 μM, and 20 μM) (*P<0.01 vs. Control; †P<0.01 vs. Wildtype at corresponding point of concentration). In C, representative images illustrate that Wnt1 overexpression significantly prevents apoptotic DNA fragmentation with TUNEL stain. Vector transfected cells that lack Wnt1 cDNA also lead to DNA fragmentation during Aβ1-42(20 μM) exposure. In D, a significant reduction in apoptotic DNA fragmentation with Wnt1 overexpression 24 hours after Aβ1-42 (20 μM) using the TUNEL assay is present (*P<0.01 vs. Control or Vector; †P<0.01 vs. Aβ1-42alone or Aβ1-42 with vector). In all cases, Control = untreated cells and Vector = transfection without Wnt1 cDNA. For B and D, each data point represents the mean and ±SEM from 6 experiments.
In Figs. 2C and 2D, SH-SY5Y cells were exposed to Aβ1-42and 24 hours later cell apoptosis was assessed by DNA fragmentation using TUNEL. Untreated wildtype control cells are without DNA fragmentation. In Fig. 2C, wildtype cells are without evidence of nuclear DNA damage, but exposure to Aβ1-42 results in significant nuclear DNA damage with TUNEL labeling. In contrast, cells with stable Wnt1 overexpression exposed to Aβ1-42 are absent of significant DNA degradation when compared to wildtype cells and vector transfected cells (Figs. 2C and 2D). Data for all stable expressing clones with a transfection efficiency equal to or greater than 98% (highest expressing clones resulting from selection methods) were combined since no significant differences in cell injury or DNA fragmentation were present among the individual clones with stable Wnt1 overexpression.
3.4 Wnt1 blocks apoptotic membrane phosphatidylserine (PS) exposure in SH-SY5Y cells and subsequently prevents microglial proliferation and activation
Apoptotic neuronal cell injury is a dynamic process that consists of both genomic DNA degradation as well as early membrane PS exposure [10]. Subsequent externalization of membrane PS residues in neurons undergoing apoptosis can result in inflammatory cell activation and identify these cells for phagocytic disposal [26-28]. We therefore examined the ability of Wnt1 to modulate externalization of membrane PS exposure in SH-SY5Y cells during Aβ1-42 exposure. In Fig. 3A, SH-SY5Y cells were exposed to the Aβ1-42 (20 μM) and 24 hours later cell membrane PS exposure was determined by annexin V. Untreated wildtype control cells were without PS externalization. In SH-SY5Y wildtype cells (Aβ1-42) and vector transfected cells exposed to Aβ1-42 (Vector/Aβ1-42) there was significant membrane PS exposure. In contrast, cells with stable Wnt1 overexpression exposed to Aβ1-42 (Wnt1/Aβ1-42) had minimal PS exposure (Fig. 3A). Quantification illustrated an increase in membrane PS exposure in SH-SY5Y wildtype cells (Aβ1-42) to 63 ± 4% and vector transfected cells exposed to Aβ1-42(Vector/Aβ1-42) to 62 ± 5% at 24 hours after Aβ1-42 exposure when compared to untreated control cultures of 12 ± 3%. Cells with stable Wnt1 overexpression (Wnt1/Aβ1-42) demonstrated a significant reduction in membrane PS exposure to 27 ± 4% at 24 hours after exposure to Aβ1-42.
Fig. 3.
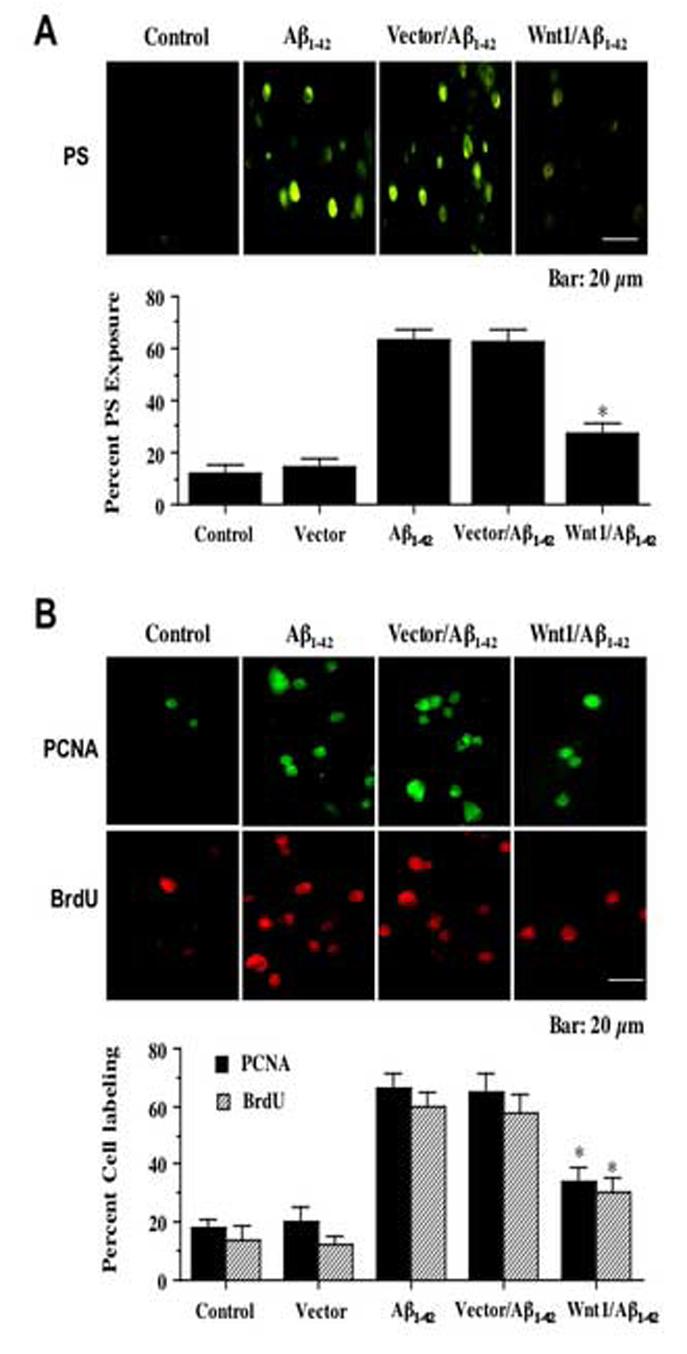
Wnt1 controls membrane phosphatidylserine (PS) exposure in SH-SY5Y cells and subsequent microglial proliferation and activation. In A, Wnt1 overexpression (Wnt1/Aβ1-42) in during Aβ1-42 application (Aβ1-42) significantly prevents membrane PS externalization in SH-SY5Y cells (*P<0.01 vs. Aβ1-42alone or Aβ1-42 with vector). Vector transfected SH-SY5Y cells (Vector/Aβ1-42) did not reduce PS exposure following Aβ1-42application. In B, representative images and quantitative analysis illustrate that PCNA and BrdU expression was significantly increased in microglia exposed to media from Aβ1-42 treated wildtype SH-SY5Y cells (Aβ1-42), but minimal PCNA expression and BrdU uptake occur following exposure to media from SH-SY5Y cells overexpressing Wnt1 (Wnt1/Aβ1-42) (*P<0.01 vs. Aβ1-42alone or Aβ1-42 with vector). Media from vector transfected SH-SY5Y cells following Aβ1-42 application (Vector/Aβ1-42) did not reduce PCNA expression or BrdU uptake in primary microglia. In all cases, Control = untreated cells and Vector = transfection without Wnt1 cDNA. Each data point represents the mean and ±SEM from 6 experiments.
Since Wnt1 can prevent the externalization of membrane PS residues on SH-SY5Y cells which are critical for microglial activation and proliferation [17, 18, 26], we next investigated whether Wnt1 fostered inflammatory cell protection through the prevention of microglial activation and proliferation. In Fig. 3B, representative microglial cultures illustrate a marked induction of microglial activation and proliferation during treatment with Aβ1-42 exposed media from SH-SY5Y wildtype cells (Aβ1-42) as evidenced by significant PCNA expression [24] and BrdU uptake [25] when compared to microglia treated with media not exposed to Aβ1-42 (Control). Minimal activation (PCNA) or proliferation (BRDU) of microglia is present during treatment with media from SH-SY5Y cells overexpressing Wnt1 (Wnt1/Aβ1-42) which are also absent of membrane PS exposure (Fig. 3A). In Fig. 3B, quantification of PCNA and BrdU labeling revealed that a significant expression in PCNA (66 ± 5%, p<0.01) and BrdU (60 ± 5%, p<0.01) was present in microglia cultures following the application of Aβ1-42 exposed media (Aβ1-42) when compared to untreated control cultures (18 ± 3%, PCNA; 14 ± 5%, BrdU). Media from vector transfected SH-SY5Y cells following Aβ1-42 application (Vector/Aβ1-42) also resulted in significant PCNA expression and BrdU uptake in primary microglia. Yet, application of media from SH-SY5Y cells overexpressing Wnt1 during Aβ1-42 exposure (Wnt1/Aβ1-42) prevented microglial activation with reduced PCNA (34 ± 5%) expression and reduced BrdU (30 ± 5%) uptake.
3.5 Gene silencing of Wnt1 eliminates neuroprotection, leads to apoptotic injury, and results in microglial activation and proliferation
To assess whether Wnt1 was necessary for the protection of neuronal cells during Aβ1-42 exposure, we examined the effects of siRNA gene silencing of Wnt1 to specifically knockdown Wnt1 expression. In SH-SY5Y cells with stable overexpression of Wnt1, siRNA for Wnt1 was co-transfected into these cells and the expression of Wnt1 was observed through Western blot analysis (Fig. 4A). For Wnt1 overexpression, a vector control was used and for gene silencing of Wnt1, a mock control that represents the use of transfection reagents for gene silencing without siRNA specific for Wnt1 was employed. In Fig. 4A, transfection of Wnt1 leads to significant protein expression of Wnt1 that is not present in untreated wildtype control cells or in vector transfected cells. In contrast, co-transfection of Wnt1 siRNA in cells with stable overexpression of Wnt1 significantly blocks Wnt1 expression. Over expression of Wnt1 is not affected in mock controls.
Fig. 4.

Expression of Wnt1 is blocked during siRNA gene silencing. In A, stable overexpression of Wnt1 demonstrates expression of the Wnt1 protein, but cells with co-transfection of Wnt1 overexpression and siRNA Wnt1 (+siRNA) illustrate loss of Wnt1 expression during siRNA gene silencing (*P<0.01 vs. control, vector, or Wnt1/+siRNA). For Wnt1 overexpression, a vector control was used and for gene silencing of Wnt1, a mock control (-siRNA) that represents the use of transfection reagents for gene silencing without siRNA specific for Wnt1 was employed. Each data point represents the mean and ±SEM from 6 experiments. In B, tagged fluorescent hemagglutinin (HA-Tag) of Wnt1 overexpression in SH-SY5Y cells demonstrates the specificity and utility for siRNA gene silencing. Top panel illustrates SH-SY5Y cells acquired through transmitted light and bottom panel represents Wnt1 overexpression tracked with HA-Tag labeling. Wnt1 expression occurs in SH-SY5Y cells with stable overexpression of Wnt1, but is not observed in non-transfected control cells or cells with co-transfection of Wnt1 siRNA and stable overexpression of Wnt1.
For the technique of siRNA transfection, we independently assessed the ability to specifically silence Wnt1 overexpression by immunocytochemistry detection with Wnt1 tagged fluorescent hemagglutinin (HA-Tag). In Fig. 4B, Wnt1 overexpression is tracked with HA-Tag labeling in SH-SY5Y cells and demonstrates specificity and utility for siRNA gene silencing. Wnt1 expression occurs in SH-SY5Y cells with stable overexpression of Wnt1, but is not observed in non-transfected control cells or cells with co-transfection of Wnt1 siRNA and stable overexpression of Wnt1.
In Fig. 5A, we examined cell survival following Aβ1-42exposure in wildtype SH-SY5Y cells and in cells with stable Wnt1 overexpression with and without co-transfection of siRNA for Wnt1. In wildtype cells, Aβ1-42exposure (20 μM) significantly decreased cell survival assessed by trypan blue dye exclusion to 38 ± 4% (p<0.01) (Aβ1-42) when compared to untreated cell survival of 93 ± 3% (Untreated). Stable Wnt1 overexpression in SH-SY5Y cells (Wnt1/Aβ1-42) significantly increased survival to 63 ± 3% during Aβ1-42exposure. Mock co-transfection without Wnt1 siRNA (-siRNA/Aβ1-42) did not alter the neuroprotective capacity of Wnt1 during Aβ1-42administration, but co-transfection with Wnt1 siRNA and Wnt1 overexpression (+siRNA/Aβ1-42) eliminated protection by Wnt1 (Fig. 5A). In addition, transfection with Wnt1 siRNA alone prior to Aβ1-42exposure (20 μM) enhanced cell injury to approximately 20% cell survival when compared to Aβ1-42exposure alone with approximately 38% cell survival (data not shown).
Fig. 5.
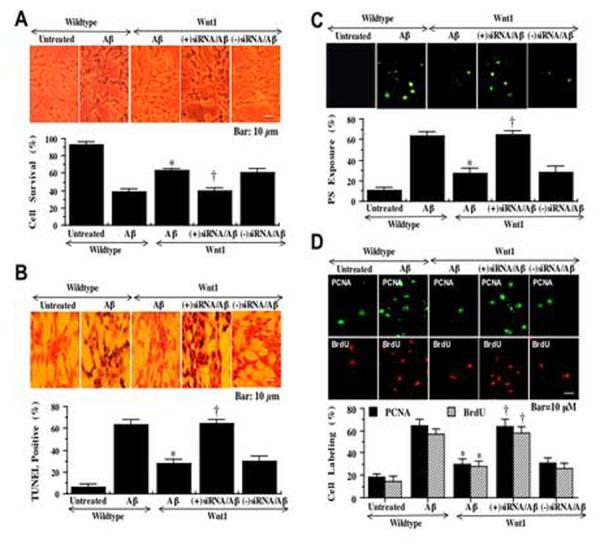
Gene silencing of Wnt1 results in apoptotic cell injury and microglial activation and proliferation. In A, Wnt1 overexpression significantly increases cell survival (Wnt1/Aβ). Gene silencing of Wnt1 with co-transfection of Wnt1 overexpression and Wnt1 siRNA during Aβ1-42(20 μM) application (Wnt1/(+)siRNA/Aβ) blocks Wnt1 from increasing cell survival (increased trypan blue uptake) during Aβ1-42exposure. Mock co-transfection without Wnt1 siRNA (Wnt1/(-) siRNA/Aβ) did not affect protection by Wnt1 overexpression during Aβ1-42administration (*P<0.01 vs. Aβ1-42alone; †P<0.01 vs. Aβ1-42 alone or mock siRNA). In B, Wnt1 overexpression significantly prevents DNA fragmentation (Wnt1/Aβ), but co-transfection of Wnt1 overexpression and Wnt1 siRNA during Aβ1-42(20 μM) application (Wnt1/(+)siRNA/Aβ) eliminates the ability of Wnt1 to block DNA fragmentation during Aβ1-42exposure. Mock co-transfection without Wnt1 siRNA (Wnt1/(-) siRNA/Aβ) did not affect alter DNA fragmentation by Wnt1 overexpression during Aβ1-42administration (*P<0.01 vs. Aβ1-42alone; †P<0.01 vs. Aβ1-42 alone or mock siRNA). In C, Wnt1 overexpression significantly reduces membrane PS externalization (Wnt1/Aβ). However, co-transfection of Wnt1 overexpression and Wnt1 siRNA during Aβ1-42(20 μM) application (Wnt1/(+)siRNA/Aβ) blocks the ability of Wnt1 to prevent PS exposure during Aβ1-42administration. Mock co-transfection without Wnt1 siRNA (Wnt1/(-) siRNA/Aβ) did not change PS exposure by Wnt1 overexpression during Aβ1-42administration (*P<0.01 vs. Aβ1-42alone; †P<0.01 vs. Aβ1-42 alone or mock siRNA). In D, media from Aβ1-42(20 μM) application alone (Aβ) leads to both PCNA expression and BrdU uptake in representative images and quantitative analysis in primary microglial cells. Wnt1 overexpression significantly reduces PCNA expression and BrdU uptake (Wnt1/Aβ). Yet, co-transfection of Wnt1 overexpression and Wnt1 siRNA during Aβ1-42(20 μM) application (Wnt1/(+)siRNA/Aβ) blocks the ability of Wnt1 to inhibit PCNA expression and BrdU uptake. Mock co-transfection without Wnt1 siRNA (Wnt1/(-) siRNA/Aβ) did not alter the ability of Wnt1 overexpression to prevent microglial PCNA expression or BrdU uptake (*P<0.01 vs. Aβ1-42alone; †P<0.01 vs. Aβ1-42 alone or mock siRNA). In all cases, each data point represents the mean and ±SEM from 6 experiments.
Similar to our observations with cell survival, protection against genomic DNA degradation was dependent upon Wnt1 expression (Fig. 5B). In wildtype cells, Aβ1-42exposure (20 μM) significantly increased DNA fragmentation per TUNEL assessment to 63 ± 5% (p<0.01) (Aβ1-42) when compared to untreated control TUNEL label of 6 ± 3% (Untreated). In contrast, cells with stable Wnt1 overexpression (Wnt1/Aβ1-42) were protected against DNA fragmentation, but the ability of Wnt1 overexpression to prevent DNA fragmentation following Aβ1-42exposure was lost during co-transfection with Wnt1 siRNA (+siRNA/Aβ1-42) (Fig. 5B). Mock co-transfection without Wnt1 siRNA (-siRNA/Aβ1-42) did not affect the ability of Wnt1 to prevent DNA fragmentation.
Since gene silencing of Wnt1 protein expression could prevent the ability of Wnt1 to protect against cell injury and DNA fragmentation during Aβ1-42administration, we next investigated whether removal of Wnt1 protein expression could affect the capacity of Wnt1 to modulate early apoptotic membrane PS exposure and subsequent microglial activation and proliferation. In Fig. 5C, Aβ1-42administration (20 μM) significantly increased membrane PS exposure in wildtype cells to 63 ± 4% (p<0.01) (Aβ1-42) when compared to untreated control survival of 12 ± 3% (Untreated). Yet, stable Wnt1 overexpression (Wnt1/Aβ1-42) significantly decreased membrane PS exposure to 27 ± 5% during Aβ1-42application, but co-transfection with Wnt1 siRNA (+siRNA/Aβ1-42) prevented Wnt1 overexpression from maintaining membrane PS asymmetry and blocking PS externalization (Fig. 5C). These observations correlated with the demonstration that expression of PCNA (65 ± 5%, p<0.01) and BrdU (57 ± 5%, p<0.01) uptake was significantly increased in microglia cultures following the application of Aβ1-42 exposed media (Aβ1-42) (Fig. 5D). Yet, reduction in PCNA expression and reduced BrdU uptake application of media from cells with overexpression of Wnt1 during Aβ1-42 exposure (Wnt1/Aβ1-42) was reversed by administration of media from cells with stable overexpression of Wnt1 and co-transfection with Wnt1 siRNA (+siRNA/Aβ1-42). Mock co-transfection without Wnt1 siRNA (-siRNA/Aβ1-42) did not affect the ability of Wnt1 overexpression to prevent microglial activation or proliferation (Fig. 5D).
3.6 Neuronal protection by Wnt1 requires the activity of the PI 3-K pathway
Western blot assay was performed for phosphorylated Akt1 (p-Akt1) (activated form of Akt1) (Fig. 6A) 12 hours following Aβ1-42 administration. For Akt1 phosphorylation, Ser473 was examined since phosphorylation of Ser473 is considered to be a critical component for the complete activation of Akt1 [29]. In Fig. 6A, stable Wnt1 overexpression, either in untreated cells or in the presence of Aβ1-42 (20 μM), elevated p-Akt1 expression to a greater extent than Aβ1-42 exposure alone. This increased expression of p-Akt1 was blocked by the inhibitors of PI 3-K. The inhibitor wortmannin (500 nM) forms a covalent link with the lysine residue of PI 3-K [30] and the inhibitor LY294002 (10 μM) reversibly competes for ATP binding [31] (Fig. 6A). In conjunction with p-Akt1 expression, Wnt1 overexpression was assessed by Western analysis.
Fig. 6.
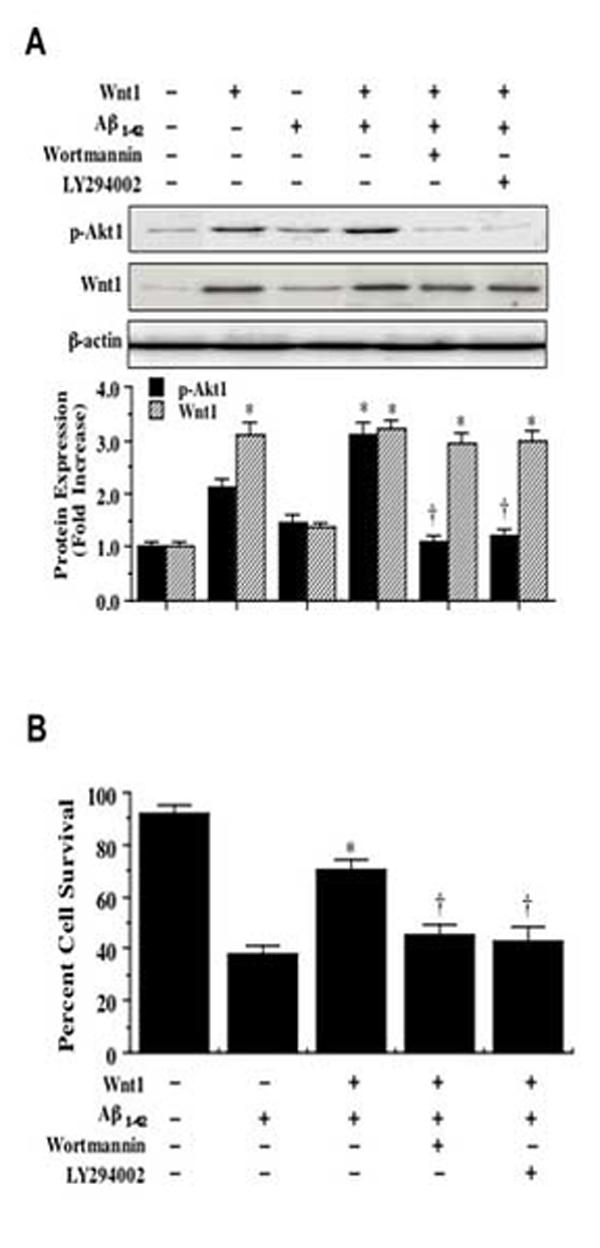
Wnt1 employs the PI 3-K pathway for neuroprotection. In A, phosphorylated (active) Akt1 (p-Akt1) expression is increased 12 hours following application of Aβ1-42 (20 μM), but decreased by a 1 hour pretreatment of the PI 3-K inhibitor wortmannin (0.5 μM) or LY294002 (10 μM). Wnt1 overexpression also was assessed by Western analysis in conjunction with p-Akt1 expression (*P<0.01 vs. Aβ1-42; †P<0.01 vs. Wnt1 alone or with Aβ1-42). In B, the PI 3-K inhibitors wortmannin (0.5 μM) and LY294002 (10 μM) significantly decreased cell survival in Wnt1 transfected cells (*P<0.01 vs. Aβ1-42; †P<0.01 vs. Wnt1 with Aβ1-42). Control =untreated cultures of wildtype cells. Each data point represents the mean and ±SEM from 6 experiments.
In Fig. 6B, SH-SY5Y cells with stable Wnt1 overexpression following Aβ1-42 exposure (20 μM) increased neuronal survival from 38 ± 3% to 70 ± 4%. Yet, application of wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) at concentrations that block activation of Akt1 activation (Fig. 6A) significantly reduced the ability of Wnt1 to protect against Aβ1-42 injury, suggesting that Wnt1 requires activation of the PI 3-K and Akt to exert neuronal protection against Aβ1-42.
3.7 Gene silencing of Akt1 eliminates the capacity of Wnt1 to prevent apoptosis
Since wortmannin and LY294002 function at the level of PI 3-K to modulate Akt1 activity, we next examined whether direct elimination of Akt1 expression through the specific gene silencing of Akt1 with siRNA could also alter Wnt1 neuroprotection. SH-SY5Y cells were transfected with Akt1 siRNA alone or during the co-transfection with stable Wnt1 overexpression. In Fig. 7A, Aβ1-42 application (20 μM) significantly increased trypan blue staining. In addition, quantitative analysis demonstrated that Aβ1-42 lead to significant loss of membrane integrity with trypan blue staining increasing from 3 ± 2% to 64 ± 4% (p<0.01) (Fig. 7B). Loss of cell membrane integrity as demonstrated by the trypan blue dye exclusion assay was accompanied by increased DNA fragmentation with TUNEL staining during Aβ1-42 exposure (20 μM) (Fig. 7A) and TUNEL labeling increased from 3 ± 2% to 66 ± 4% (p<0.01) following Aβ1-42. As expected per our prior results, Wnt1 overexpression significantly prevented cell injury assessed by trypan blue staining and cell apoptosis assessed by TUNEL labeling (Figs. 7A and 7B), but this protection was lost with gene silencing of Akt1 with the increase of trypan blue staining to 60 ± 5% (p<0.01) and TUNEL labeling to 61 ± 5% (p<0.01) during Aβ1-42 exposure (Figs. 7A and 7B), illustrating that the presence and activation of Akt1 is an essential component for Wnt1 to prevent neuronal cell injury and genomic DNA degradation. Transfection with siRNA for Akt1 alone was not toxic to SH-SY5Y cells (data not shown).
Fig. 7.
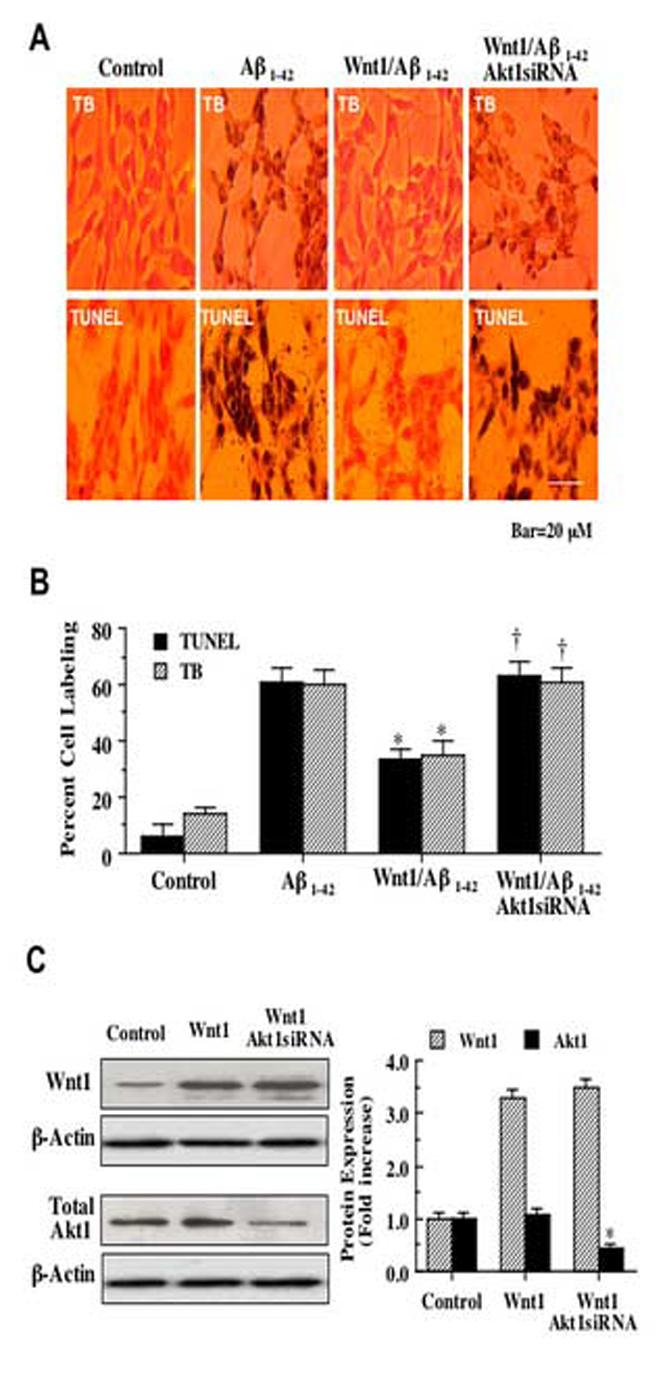
Wnt1 requires Akt1 to block apoptotic cell death during Aβ1-42. In A, overexpression of Wnt1 significantly reduces cell injury with significant reduction in trypan blue (TB) staining and nuclear DNA fragmentation (TUNEL), but co-transfection with Wnt1 overexpression and Akt1 siRNA removes protection by Wnt1 and leads to cell injury with increased trypan blue staining and DNA fragmentation. In B, quantification of results in Aillustrate that gene silencing of Akt1 with siRNA (Wnt1/Aβ1-42Akt1siRNA) significantly prevented the ability of Wnt1 overexpression (Wnt1/Aβ1-42) during Aβ1-42 exposure to block cell injury with decreased trypan blue staining and DNA fragmentation with decreased TUNEL labeling (*P<0.01 vs. Aβ1-42; †P<0.01 vs. Wnt1/Aβ1-42). For B, each data point represents the mean and ±SEM from 6 experiments. In C, the expression of Wnt1 and total Akt1 was observed during Wnt1 overexpression through Western blot analysis. Top panel illustrates that Wnt1 expression is not altered during Akt1 siRNA co-transfection with stable Wnt1 overexpression. In the bottom panel, gene silencing of Akt1 during stable Wnt1 overexpression significantly depresses total Akt1 expression.
The expression of Wnt1 and total Akt1 was observed during Wnt1 overexpression through Western blot analysis (Fig. 7C). Fig. 7C (top panel) illustrates that Wnt1 expression is not altered during Akt1 siRNA co-transfection with stable Wnt1 overexpression. In addition, gene silencing of Akt1 during stable Wnt1 overexpression significantly depresses total Akt1 expression (Fig. 7C, bottom panel).
3.8 Wnt1 phosphorylates glycogen synthase kinase-3β (GSK-3β) through an Akt1 mediated pathway
We investigated the ability of Wnt1 to modulate GSK-3β phosphorylation during Aβ1-42 exposure at the conserved regulatory residue of Ser9 [32]. Using western blot analysis, Wnt1 overexpression either alone or during Aβ1-42 (20 μM) exposure significantly increased the phosphorylation of GSK-3β over 12 hours following application of Aβ1-42 (Fig. 8A). In addition, Wnt1 overexpression led to the phosphorylation of GSK-3β to a significantly greater degree than phosphorylation of GSK-3β by Aβ1-42alone. Yet, the capacity of Wnt1 to phosphorylate GSK-3β was abrogated during co-transfection of stable Wnt1 overexpresssion with Akt1 siRNA, suggesting the Wnt1 inhibition of GSK-3β is intimately tied to the presence and activity of Akt1 (Fig. 8A). Concurrent Western analysis illustrated that total Akt1 expression was significantly decreased by Akt1 siRNA (Fig. 8A).
Fig. 8.
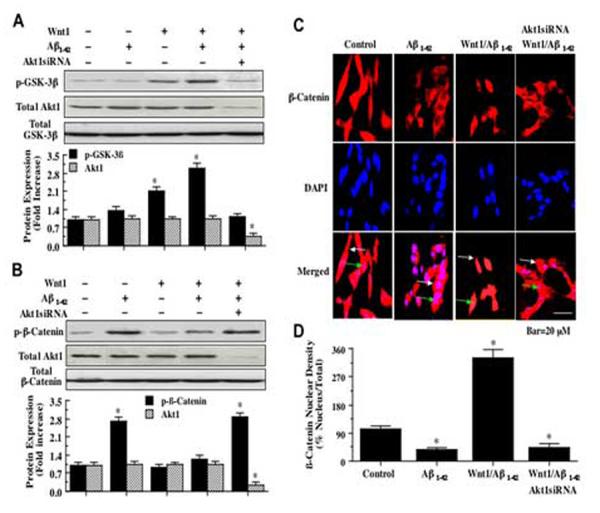
Wnt1 phosphorylates glycogen synthase kinase-3β (GSK-3β) during Aβ1-42 toxicity and maintains the integrity and nuclear translocation of β-catenin through Akt1. In A, phosphorylated GSK-3β (p-GSK-3β) expression is increased during Wnt1 overexpression with or without Aβ1-42 (20 μM) application, but p-GSK-3β expression is blocked during cotransfection with Wnt1 overexpression and Akt1 siRNA (Akt1siRNA) with Aβ1-42 (20 μM) application and p-GSK-3β expression levels match those of untreated control (*P<0.01 vs. Akt1siRNA or Aβ1-42). Concurrent Western analysis illustrated that total Akt1 expression was significantly decreased by Akt1 siRNA. In B, phosphorylated β-catenin (p-β-catenin) expression is significantly increased during Aβ1-42 (20 μM) exposure, but p-β-catenin expression is markedly reduced during Wnt1 overexpression with or without Aβ1-42 (20 μM) application. Loss of Akt1 during co-transfection with Wnt1 overexpression and Akt1 siRNA (Akt1siRNA) with Aβ1-42 (20 μM) application results in a significant increase in p-β-catenin expression. Concurrent Western analysis illustrated that total Akt1 expression was significantly decreased by Akt1 siRNA. In C, green arrows identify the cell nucleus and white arrows identify the cell cytoplasm. Aβ1-42 exposure (20 μM) leads to predominant distribution of β-catenin in the cytoplasm, but Wnt1 overexpression translocates β-catenin to the cell nucleus. Translocation of β-catenin to the cell nucleus is lost during gene silencing of Akt1 with co-transfection of Wnt1 overexpression and Akt1 siRNA (Akt1siRNA) with Aβ1-42 (20 μM) application. In merged images, cells with Aβ1-42 exposure or Akt1 siRNA show decreased β-catenin staining (pink) while Wnt1 overexpression reveals strong β-catenin staining (yellow). In D, Wnt1 overexpression promotes β-catenin translocation from the cytoplasm to the nucleus during Aβ1-42 (20 μM) application, but loss of Akt1 during Akt1 siRNA administration leads to minimal nuclear β-catenin translocation. Intensity of β-catenin nuclear staining was performed using the public domain NIH imaging program (http://rsb.info.nih.gov/nih-image/). Control = untreated cells (*P<0.01 vs. Control). For A, B, and D, each data point represents the mean and ±SEM from 6 experiments.
3.9 Wnt1 relies upon Akt1 for the maintenance of the “anti-apoptotic” protein β-catenin and its intracellular trafficking
Given that Wnt1 depends upon Akt1 for neuronal protection, we examined the ability of both Wnt1 and Akt1 to modulate the downstream pathway of β-catenin. Initially, we investigated the ability of Wnt1 to alter the phosphorylation of β-catenin during Aβ1-42 toxicity. Phosphorylation of β-catenin through the Wnt pathway can occur sequentially with Ser45, Thr41, Ser37, and Ser33, but recent work suggests that phosphorylation at Ser33, Ser37, and Thr41 occurs without the requirement of Ser45 [33]. We therefore examined phosphorylation of β-catenin at Ser33/37 and Thr41. Overexpression of Wnt1 either alone or in conjunction with Aβ1-42 exposure (20 μM) prevented the expression of phosphorylated β-catenin (p-β-catenin) 12 hours following Aβ1-42 administration (Fig. 8B). In contrast, expression of p-β-catenin was increased during Aβ1-42 alone or during co-transfection with stable Wnt1 overexpression and gene silencing of Akt1 during Aβ1-42 application, illustrating that Wnt1 requires Akt1 to prevent the phosphorylation of β-catenin (Fig. 8B). Concurrent Western analysis illustrated that total Akt1 expression was significantly decreased by Akt1 siRNA (Fig. 8B).
Immunofluorescent staining for β-catenin and DAPI nuclear staining were used to follow the subcellular translocation of β-catenin in neurons during Aβ1-42 exposure (20 μM) alone, during Wnt1 overexpression with Aβ1-42, or during cotransfection with Wnt1 overexpression and Akt1 siRNA following Aβ1-42 (Figs. 8C and 8D). With Aβ1-42 (20 μM) exposure alone, significant immunofluorescent staining for β-catenin in the cytoplasm of neurons with minimal nuclear staining is present. This is evident by the ability to detect significant DAPI nuclear staining (pink in color) in cells during merged Aβ1-42 images since prominent β-catenin staining is not present in the nucleus (Figs. 8C). In contrast, with overexpression of Wnt1 during Aβ1-42 exposure, β-catenin is translocated to the nucleus with minimal cytoplasmic staining as shown by the light nuclear regions on merged images of β-catenin and DAPI (Figs. 8C). However, the ability to shuttle β-catenin to the nucleus by Wnt1 during Aβ1-42 application is lost during Akt1 siRNA transfection (Fig. 8C), illustrating that Wnt1 requires Akt1 to allow the translocation of β-catenin from the cell cytoplasm to the nucleus. In Fig. 8D, nuclear β-catenin staining is quantitated that reveals a high proportion of β-catenin in the nucleus during Wnt1 overexpression during Aβ1-42 application, but with minimal β-catenin in the nucleus during cotransfection of Wnt1 and Akt1 siRNA, suggesting that Wnt1 fosters nuclear translocation of β-catenin to allow it to initiate an anti-apoptotic program through an Akt1 mediated pathway.
4. Discussion
The production of β-amyloid peptide aggregates composed of a 39-42 amino acid peptides are considered to be one of the pathological mechanisms that lead to Alzheimer's disease [10]. β-amyloid is cleaved by amyloid precursor protein into products that include a 40-residue peptide and a 42-residue peptide (Aβ1-42), but Aβ1-42 is considered to be the β-amyloid product that most directly leads to Alzheimer's disease and apoptotic injury. The mechanisms that govern Aβ1-42 toxicity are complex in nature and involve apoptotic pathways that affect neuronal cell injury as well as inflammatory microglial activation [10, 11].
We therefore initially examined the effect of Aβ1-42on neuronal survival and demonstrated that Aβ1-42exposure is toxic to SH-SY5Y cells and leads to progressive injury with significant loss of cell survival observed at 5 μM and increasing to approximately 70% cell loss at a concentration of 50 μM. In conjunction with this cell loss during Aβ1-42administration is the rapid and robust increase in Wnt1 protein expression within 1 and 6 hours following Aβ1-42application, but over the course of 24 hours the expression of Wnt1 is lost. Although increases in the expression of Wnt1 have been observed in other scenarios that involve the later stages of wound healing [34] or during the development of hematological cancers [35], our work with the early induction and subsequent loss of Wnt1 during Aβ1-42 toxicity may suggest that endogenous activation of Wnt1 represents an attempt at early reparative process by neurons. Our subsequent work with the stable overexpression of Wnt1 provides further support for such a premise. We show that Wnt1 overexpression prevents cellular injury during increasing concentrations of Aβ1-42 and that Wnt1 is both necessary and sufficient for this neuronal protection. In co-transfection experiments with stable Wnt1 expression and Wnt1 siRNA, cellular protection by Wnt1 against Aβ1-42 application is lost.
Protection by Wnt1 during Aβ1-42 exposure involves the specific prevention of apoptotic genomic DNA fragmentation. Apoptotic neuronal degeneration that involves genomic DNA fragmentation may represent a significant component for neuronal cell loss during Alzheimer's disease [10, 11]. Similar to studies that examined apoptotic induction in other cell systems [3, 4], Wnt1 overexpression in our neuronal cell line model significantly prevented apoptotic nuclear DNA fragmentation during Aβ1-42 administration. In addition, gene silencing of Wnt1 expression abrogated this protection against genomic DNA fragmentation.
Interestingly, the protective capacity of Wnt1 against Aβ1-42 toxicity extends beyond the maintenance of genomic DNA integrity and encompasses the blockade of cellular membrane PS externalization and the inhibition of inflammatory cell activation. Cellular membrane PS exposure can precipitate the subsequent removal of cells in the nervous system by activated microglia [26-28]. We illustrate that exposure to Aβ1-42 results in the significant externalization of PS that can be blocked by Wnt1 overexpression. Furthermore, we show that this unique capacity of Wnt1 to control membrane PS exposure appears to be directly linked to the ability to modulate inflammatory microglial activation and proliferation. Both microglial activation and proliferation as assessed through PCNA expression and BrdU uptake occur during Aβ1-42 administration. In studies that employ media obtained from cells that overexpress Wnt1 during Aβ1-42 exposure, Wnt1 overexpression in neuronal SH-SY5Y cells significantly blunts both the activation and proliferation of primary microglial cells that otherwise would occur with Aβ1-42 exposure alone. In addition, Wnt1 appears to be both necessary and sufficient to control the exposure of membrane PS residues during apoptotic injury with Aβ1-42, since silencing of Wnt1 expression in the presence of Aβ1-42 results in the loss of membrane asymmetry and PS externalization. Our work illustrates that Wnt1 modulates microglial activation and proliferation through membrane PS exposure on neuronal cells and may potentially prevent the shedding of membrane PS residues that is known to occur during apoptosis [28].
Wnt1 appears to work in concert with several pathways to foster protection against apoptotic cell injury, such as nuclear factor-κB, cytochrome c, and caspase 9 [5, 20]. Yet, Wnt1 also may rely upon the central pathways of Akt1 that have been shown to promote cellular integrity and survival during free radical injury [16, 19], oxidative stress [17, 18], tumor invasion [36], cell longevity [37], and amyloid toxicity [13-15]. Evidence for the dependence of Wnt1 on the Akt pathway can be drawn from a variety of cell populations. For example, PC12 cell differentiation that is dependent upon Wnt1 signaling and trophic factor induction is blocked during the repression of Akt activity [38], Wnt1 has been shown in preadipocytes to increase Akt phosphorylation [39], and the Wnt1-induced secreted protein in a fibroblast cell line uses Akt to block apoptotic death [40]. We show that Wnt1 overexpression independently increases the phosphorylation and the activation of Akt1 to a significantly greater degree than Aβ1-42 alone, suggesting that Wnt1 uses Akt1 activation to promote neuronal protection. Additionally, inhibition of the PI 3-K pathway with application of wortmannin (500 nM) or LY294002 (10 μM) at concentrations that prevent activation of Akt1 during Aβ1-42 exposure or co-transfection of cells with Akt1 siRNA to specifically silence Akt1 expression prevents Wnt1 from blocking both cellular membrane injury and apoptotic genomic DNA degradation during Aβ1-42 application.
Both Wnt1 and Akt1 share a common pathway through the phosphorylation of GSK-3β. Inhibition of GSK-3β activity through its phosphorylation can prevent apoptotic cell death and potentially block amyloid toxicity and neurodegenerative disease [10, 41, 42]. We demonstrate that neuronal protection by Wnt1 may require integration of the GSK-3β pathway that involves phosphorylation and inhibition of GSK-3β activity via Akt1. We show that phosphorylation of GSK-3β occurs 12 hours after Aβ1-42 exposure, but that overexpression of Wnt1 significantly increases the phosphorylation of GSK-3β to a greater degree than Aβ1-42alone. In addition, the capacity of Wnt1 to phosphorylate GSK-3β is regulated by Akt1, since co-transfection of Akt1 siRNA with Wnt1 overexpression removes the ability of Wnt1 to phosphorylate GSK-3β.
Cell injury during GSK-3β activation may occur through the phosphorylation of β-catenin that can lead to its ubiquitination and subsequent degradation. As a result, Wnt1 “antiapoptotic” pathways can be dismantled during the phosphorylation of β-catenin since β-catenin cannot translocate into the nucleus and activate the transcription of its target genes that support cell survival and block apoptosis [1, 4, 20, 21]. We therefore investigated the ability of Wnt1 to modulate β-catenin phosphorylation and intracellular trafficking during Aβ1-42 application. We demonstrate that phosphorylation of β-catenin is increased during Aβ1-42 exposure, but overexpression of Wnt1 alone or in conjunction with Aβ1-42 application blocks phosphorylation of β-catenin 12 hours following Aβ1-42 exposure. This ability of Wnt1 to modulation the phosphorylation and integrity of β-catenin is tied to the presence of Akt1, since gene silencing of Akt1 expression during Wnt1 overexpression leads to phosphorylation of β-catenin during Aβ1-42 application. Our work also suggests that blockade of β-catenin phosphorylation by Wnt1 promotes the translocation of β-catenin from the cytoplasm to the nucleus that is mediated through Akt1. Exposure to Aβ1-42 alone maintains β-catenin in the cytoplasm of neurons, but Wnt1 overexpression fosters the translocation of β-catenin from the cytoplasm to the nucleus. Yet, without the presence of Akt1 during the application of Akt1 siRNA, β-catenin remains in the cytoplasm of cells during Wnt1 overexpression and Aβ1-42administration.
5. Conclusions
Effective development of therapeutic entities for neurodegenerative disorders rests upon the discovery of novel systems that can block apoptotic nuclear DNA degradation and membrane PS exposure to avert the phagocytic demise of neurons during microglial cell activation. We demonstrate that Wnt1 is extremely robust in its ability to foster neuronal protection by not only preventing apoptotic DNA degradation and membrane PS exposure, but also by blocking inflammatory microglial activation and proliferation (Fig. 9). Expression of Wnt1 is vital for the maintenance of intrinsic cellular integrity and the prevention of primary microglial activation and proliferation, since these neuroprotective attributes of Wnt1 are lost during the gene silencing of Wnt1 protein expression. Enhanced cell survival and cessation of microglial activation during Aβ1-42exposure by Wnt1 is tightly coupled to the presence and activation of Akt1. Closely aligned with Wnt1 and Akt1 to afford neuronal protection against Aβ1-42are the integrated pathways of GSK-3β and β-catenin. Through Akt1 dependent pathways, Wnt1 phosphorylates GSK-3β and maintains β-catenin integrity to insure its translocation from the cytoplasm to the nucleus. Further insight into the novel pathways required for Wnt1 neuroprotection and inflammatory cell modulation may foster increased understanding for the cellular basis of neurodegenerative disorders as well as the development of effective therapeutics.
Fig. 9.
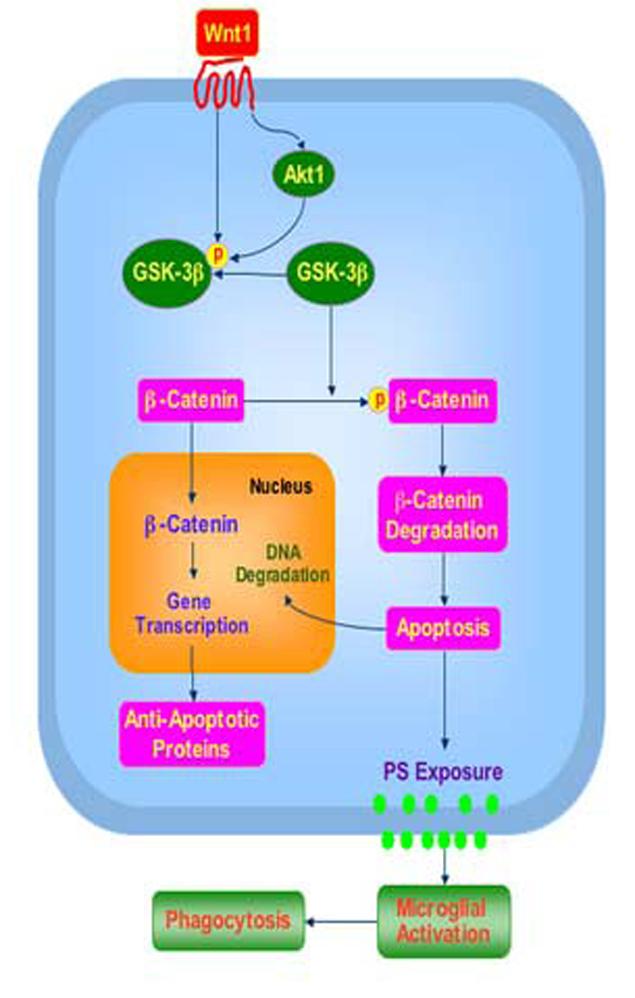
Wnt1 governs intrinsic cellular pathways to modulate apoptotic injury and inflammatory cell activation. Wnt1 fosters cellular survival during Aβ1-42 toxicity through a series of pathways that involve the serine-threonine kinase Akt1 and its downstream substrates of GSK-3β and β-catenin. Targeting these pathways by Wnt1 ultimately provides the novel capacity to both preserve genomic integrity and prevent early apoptotic injury with phosphatidylserine (PS) exposure and subsequent microglial activation.
Acknowledgments
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, Nelson Foundation Award, NIH NIEHS (P30 ES06639), and NIH NINDS/NIA (NS053946).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Li F, Chong ZZ, Maiese K. Histol Histopathol. 2006;21:103. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nelson WJ, Nusse R. Science. 2004;303:1483. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].He B, Reguart N, You L, Mazieres J, Xu Z, Lee AY, Mikami I, McCormick F, Jablons DM. Oncogene. 2005;24:3054. doi: 10.1038/sj.onc.1208511. [DOI] [PubMed] [Google Scholar]
- [4].You L, He B, Uematsu K, Xu Z, Mazieres J, Lee A, McCormick F, Jablons DM. Cancer Res. 2004;64:3474. doi: 10.1158/0008-5472.CAN-04-0115. [DOI] [PubMed] [Google Scholar]
- [5].Bournat JC, Brown AM, Soler AP. J Neurosci Res. 2000;61:21. doi: 10.1002/1097-4547(20000701)61:1<21::AID-JNR3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [6].Miyaoka T, Seno H, Ishino H. Schizophr Res. 1999;38:1. doi: 10.1016/s0920-9964(98)00179-0. [DOI] [PubMed] [Google Scholar]
- [7].Kameya S, Hawes NL, Chang B, Heckenlively JR, Naggert JK, Nishina PM. Hum Mol Genet. 2002;11:1879. doi: 10.1093/hmg/11.16.1879. [DOI] [PubMed] [Google Scholar]
- [8].Morin PJ, Medina M, Semenov M, Brown AM, Kosik KS. Neurobiol Dis. 2004;16:59. doi: 10.1016/j.nbd.2004.01.004. [DOI] [PubMed] [Google Scholar]
- [9].Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Neuron. 2002;34:509. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- [10].Chong ZZ, Li F, Maiese K. Prog Neurobiol. 2005;75:207. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- [11].Colurso GJ, Nilson JE, Vervoort LG. Life Sci. 2003;73:1795. doi: 10.1016/s0024-3205(03)00512-5. [DOI] [PubMed] [Google Scholar]
- [12].Chong ZZ, Li F, Maiese K. Histol Histopathol. 2005;20:299. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chong ZZ, Li F, Maiese K. Curr Neurovasc Res. 2005;2:387. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Du B, Ohmichi M, Takahashi K, Kawagoe J, Ohshima C, Igarashi H, Mori-Abe A, Saitoh M, Ohta T, Ohishi A, Doshida M, Tezuka N, Takahashi T, Kurachi H. J Endocrinol. 2004;183:605. doi: 10.1677/joe.1.05775. [DOI] [PubMed] [Google Scholar]
- [15].Nakagami Y, Nishimura S, Murasugi T, Kubo T, Kaneko I, Meguro M, Marumoto S, Kogen H, Koyama K, Oda T. Eur J Pharmacol. 2002;457:11. doi: 10.1016/s0014-2999(02)02657-2. [DOI] [PubMed] [Google Scholar]
- [16].Chong ZZ, Kang JQ, Maiese K. Exp Cell Res. 2004;296:196. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- [17].Kang JQ, Chong ZZ, Maiese K. Mol Pharmacol. 2003;64:557. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- [18].Kang JQ, Chong ZZ, Maiese K. J Neurosci Res. 2003;74:37. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- [19].Wang H, MacNaughton WK. Cancer Res. 2005;65:8604. doi: 10.1158/0008-5472.CAN-05-1169. [DOI] [PubMed] [Google Scholar]
- [20].Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. J Cell Biol. 2001;152:87. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Terry S, Yang X, Chen MW, Vacherot F, Buttyan R. J Cell Biochem. 2006;99:402. doi: 10.1002/jcb.20983. [DOI] [PubMed] [Google Scholar]
- [22].Chong ZZ, Kang JQ, Maiese K. Circulation. 2002;106:2973. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- [23].Giulian D, Baker TJ. J Neurosci. 1986;6:2163. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Williams K, Schwartz A, Corey S, Orandle M, Kennedy W, Thompson B, Alvarez X, Brown C, Gartner S, Lackner A. Am J Pathol. 2002;161:575. doi: 10.1016/S0002-9440(10)64213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Martinez-Contreras A, Huerta M, Lopez-Perez S, Garcia-Estrada J, Luquin S, Beas Zarate C. J Neurosci Res. 2002;67:200. doi: 10.1002/jnr.10093. [DOI] [PubMed] [Google Scholar]
- [26].Chong ZZ, Kang J, Li F, Maiese K. Curr Neurovasc Res. 2005;2:197. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Maiese K, Li F, Chong ZZ. JAMA. 2005;293:90. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Simak J, Holada K, Vostal JG. BMC Cell Biol. 2002;3:11. doi: 10.1186/1471-2121-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Oncogene. 1998;17:313. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- [30].Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Mol Cell Biol. 1996;16:1722. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vlahos CJ, Matter WF, Hui KY, Brown RF. J Biol Chem. 1994;269:5241. [PubMed] [Google Scholar]
- [32].Eldar-Finkelman H, Argast GM, Foord O, Fischer EH, Krebs EG. Proc Natl Acad Sci U S A. 1996;93:10228. doi: 10.1073/pnas.93.19.10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang Z, Vogelstein B, Kinzler KW. Cancer Res. 2003;63:5234. [PubMed] [Google Scholar]
- [34].Chakravarti S, Wu F, Vij N, Roberts L, Joyce S. Invest Ophthalmol Vis Sci. 2004;45:3475. doi: 10.1167/iovs.04-0343. [DOI] [PubMed] [Google Scholar]
- [35].Simon M, Grandage VL, Linch DC, Khwaja A. Oncogene. 2005;24:2410. doi: 10.1038/sj.onc.1208431. [DOI] [PubMed] [Google Scholar]
- [36].Hardee ME, Rabbani ZN, Arcasoy MO, Kirkpatrick JP, Vujaskovic Z, Dewhirst MW, Blackwell KL. Mol Cancer Ther. 2006;5:356. doi: 10.1158/1535-7163.MCT-05-0196. [DOI] [PubMed] [Google Scholar]
- [37].Li F, Chong ZZ, Maiese K. Curr Med Chem. 2006;13:883. doi: 10.2174/092986706776361058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fukumoto S, Hsieh CM, Maemura K, Layne MD, Yet SF, Lee KH, Matsui T, Rosenzweig A, Taylor WG, Rubin JS, Perrella MA, Lee ME. J Biol Chem. 2001;276:17479. doi: 10.1074/jbc.C000880200. [DOI] [PubMed] [Google Scholar]
- [39].Longo KA, Kennell JA, Ochocinska MJ, Ross SE, Wright WS, MacDougald OA. J Biol Chem. 2002;277:38239. doi: 10.1074/jbc.M206402200. [DOI] [PubMed] [Google Scholar]
- [40].Su F, Overholtzer M, Besser D, Levine AJ. Genes Dev. 2002;16:46. doi: 10.1101/gad.942902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li F, Chong ZZ, Maiese K. Curr Neurovasc Res. 2006;3:187. doi: 10.2174/156720206778018758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Phiel CJ, Wilson CA, Lee VM, Klein PS. Nature. 2003;423:435. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]