

Abstract
Background
Organs are programmed to acquire a particular size during development, but the regulatory mechanisms that dictate when dividing progenitor cells should permanently exit the cell cycle and stop producing additional daughter cells are poorly understood. In differentiated tissues, tumor suppressor genes maintain a constant cell number and intact tissue architecture by controlling proliferation, apoptosis and cell dispersal. Here we report a similar role for two tumor suppressor genes, the Zac1 zinc finger transcription factor and that encoding the cytokine TGFβII, in the developing retina.
Results
Using loss and gain-of-function approaches, we show that Zac1 is an essential negative regulator of retinal size. Zac1 mutants develop hypercellular retinae due to increased progenitor cell proliferation and reduced apoptosis at late developmental stages. Consequently, supernumerary rod photoreceptors and amacrine cells are generated, the latter of which form an ectopic cellular layer, while other retinal cells are present in their normal number and location. Strikingly, Zac1 functions as a direct negative regulator of a rod fate, while acting cell non-autonomously to modulate amacrine cell number. We implicate TGFβII, another tumor suppressor and cytokine, as a Zac1-dependent amacrine cell negative feedback signal. TGFβII and phospho-Smad2/3, its downstream effector, are expressed at reduced levels in Zac1 mutant retinae, and exogenous TGFβII relieves the mutant amacrine cell phenotype. Moreover, treatment of wild-type retinae with a soluble TGFβ inhibitor and TGFβ receptor II (TGFβRII) conditional mutants generate excess amacrine cells, phenocopying the Zac1 mutant phenotype.
Conclusion
We show here that Zac1 has an essential role in cell number control during retinal development, akin to its role in tumor surveillance in mature tissues. Furthermore, we demonstrate that Zac1 employs a novel cell non-autonomous strategy to regulate amacrine cell number, acting in cooperation with a second tumor suppressor gene, TGFβII, through a negative feedback pathway. This raises the intriguing possibility that tumorigenicity may also be associated with the loss of feedback inhibition in mature tissues.
Background
Tissues and organs are genetically programmed to achieve their optimal, mature size, defined by total cell number and individual cellular dimensions. Several regulatory strategies are employed to control cell number, including: direct negative regulators, which inhibit alternative cell fates but permit (or instruct) a primary fate; negative feedback pathways, acting as cell sensors that halt the continued genesis of specific cell types once a feedback signal reaches threshold levels; and cell counting mechanisms, whereby the number of times a progenitor divides before differentiating is genetically determined [1,2]. In the vertebrate retina, negative feedback pathways are used recurrently for cell number control. The retina is composed of one glial and six neuronal cell types that are present in stereotyped proportions in each vertebrate species [3-5]. Based on lineage tracing, all retinal cell types are derived from multipotent progenitor cells [6-11], although distinct cell lineages likely also exist [1,12]. In mouse, retinal ganglion cells (RGCs), horizontal cells, cone photoreceptors and amacrine cells are primarily generated during the second half of the embryonic period, while rod photoreceptor, bipolar and Müller glial cell production ends on postnatal days (P) 5–6 in the central retina [3]. Differentiated RGCs, amacrine cells and cones secrete signals negatively regulating production of additional cells of that type [13-16]. However, only signals limiting production of RGCs have been identified, including Sonic hedgehog (Shh) and growth and differentiation factor-11 (GDF11) [17]. GDF11, a transforming growth factor (TGF)β family member, has similar autoregulatory functions in other tissues, including the olfactory epithelium [18] and pancreas [19], while a related molecule, GDF8 (myostatin), negatively regulates skeletal muscle mass [20], suggesting a common role for these cytokines in cell number control.
We identified Zac1 (zinc finger protein that regulates apoptosis and cell cycle arrest or pleiomorphic adenoma gene-like 1 (Plag-l1)) [21] in a screen designed to isolate genes involved in neural fate specification [22]. Zac1 encodes a seven-C2H2 zinc finger protein that acts as a transcriptional activator or repressor [21]. Zac1 is a known tumor suppressor gene and is frequently lost in multiple carcinomas [21]. Zac1 is also maternally repressed through genomic imprinting, a mode of epigenetic control common to many genes regulating embryonic growth. Recently, a Zac1 null mutation was shown to lead to intrauterine growth restriction, consistent with the kinship theory that paternally expressed genes are growth promoting [23]. However, growth retardation was not expected if Zac1 has tumor suppressor properties, promoting cell cycle exit and apoptosis [21,24]. We therefore examined Zac1 function at the cellular level, focusing on the developing retina, where it is robustly expressed [25]. Notably, in our initial cross-species studies in Xenopus, murine Zac1 unexpectedly promoted proliferation [26]. Herein we describe intra-species loss- and gain-of-function assays in mouse that in contrast reveal tumor suppressor-like properties for Zac1 in the retina. Zac1 is required to induce cell cycle exit and apoptosis at late developmental stages, with Zac1 mutant retinae becoming hypercellular, containing supernumary rod photoreceptors and amacrine cells. Strikingly, Zac1 negatively regulates rod and amacrine cell numbers through distinct autonomous and cell non-autonomous (TGFβII-mediated) inhibitory mechanisms, respectively.
Results
Biphasic expression of Zac1 in retinal progenitors and postmitotic cells
We identified Zac1 in a subtractive screen designed to identify regulators of neuronal fate specification [22]. In an initial expression survey, we noted high Zac1 expression in the developing retina [25]. A detailed spatiotemporal characterization from embryonic day (E) 10.5 through P0 revealed high levels of Zac1 transcripts (Figure 1a–d) and protein (Figure 1f–i) in the outer neuroblast layer (onbl), where proliferating progenitors reside, and not in the inner neuroblast layer (inbl) of postmitotic cells that, prior to P0, primarily includes RGCs and amacrine cells (Additional data file 1 (a–c)). Confirming Zac1 expression in dividing cells, a large number of Zac1+ cells incorporated the S-phase label bromodeoxyuridine (BrdU) after a 30 minute pulse at E15.5 (Additional data file 1 (d–f)). Notably, Zac1 expression declined in central, more mature retinal progenitors by P0 (Figure 1d, i).
Figure 1.

Biphasic Zac1 expression in the retina. Zac1 (a-e) transcript and protein (f-j) distribution from E10.5 to P7. Arrowheads in (d,g,i) mark limits of higher expression domains. (k-o) Identification of Zac1+ P7 retinal cells. Co-labeling with Zac1 (red) and CRALBP (green (k,k')), calbindin (green (l,l')), Pax6 (green (m,m')), PKC (green (n,n')) and Brn3a (green (o,o')). High magnification images of boxed areas are shown in (k'-o'). Arrowheads mark double+ cells. Of 2,154 Zac1+ cells analyzed, 1,238 CRALBP/Zac1 double+ Müller glia; 29 calbindin/Zac1 double+ horizontal cells (based also on morphology), 480 Pax6/Zac1 double+ amacrine cells (in the INL) and 407 Brn3a/Zac1 double+ RGCs were identified. GCL, ganglion cell layer; inbl, inner neuroblast layer; INL, inner nuclear layer; le, lens; lv, lens vesicle; onbl, outer neuroblast layer; ONL, outer nuclear layer; ov, optic vesicle.
At P2 (not shown), P7 (Figure 1e, j) and P21 (Additional data file 1 (g,h)), Zac1 transcripts and protein were detected in scattered postmitotic cells in the inner nuclear layer (INL) and RGC layer (GCL; Figure 1k–o). Double immunolabeling with cell type-specific markers at P7 revealed Zac1 expression in CRALBP+ Müller glia (64.1% ± 6.26% Zac1+cells; n = 3 retinae; Figure 1k,k'), syntaxin+ (not shown) and Pax6+ amacrine cells (17.5% ± 3.6%; Figure 1m,m'), Brn3a+ RGCs (17.2% ± 5.0%; Figure 1o,o') and calbindin+ horizontal cells (1.2% ± 0.7%; Figure 1l,l'). Zac1 was not detected in protein kinase C (PKC)-expressing bipolar cells (Figure 1n,n') or in rod and cone photoreceptors in the outer nuclear layer (ONL).
Zac1 is thus expressed biphasically in the retina, initially in dividing retinal progenitors and later in Müller glia, RGCs, amacrine and horizontal cells.
Zac1 mutants develop hypercellular retinae containing an ectopic cellular layer
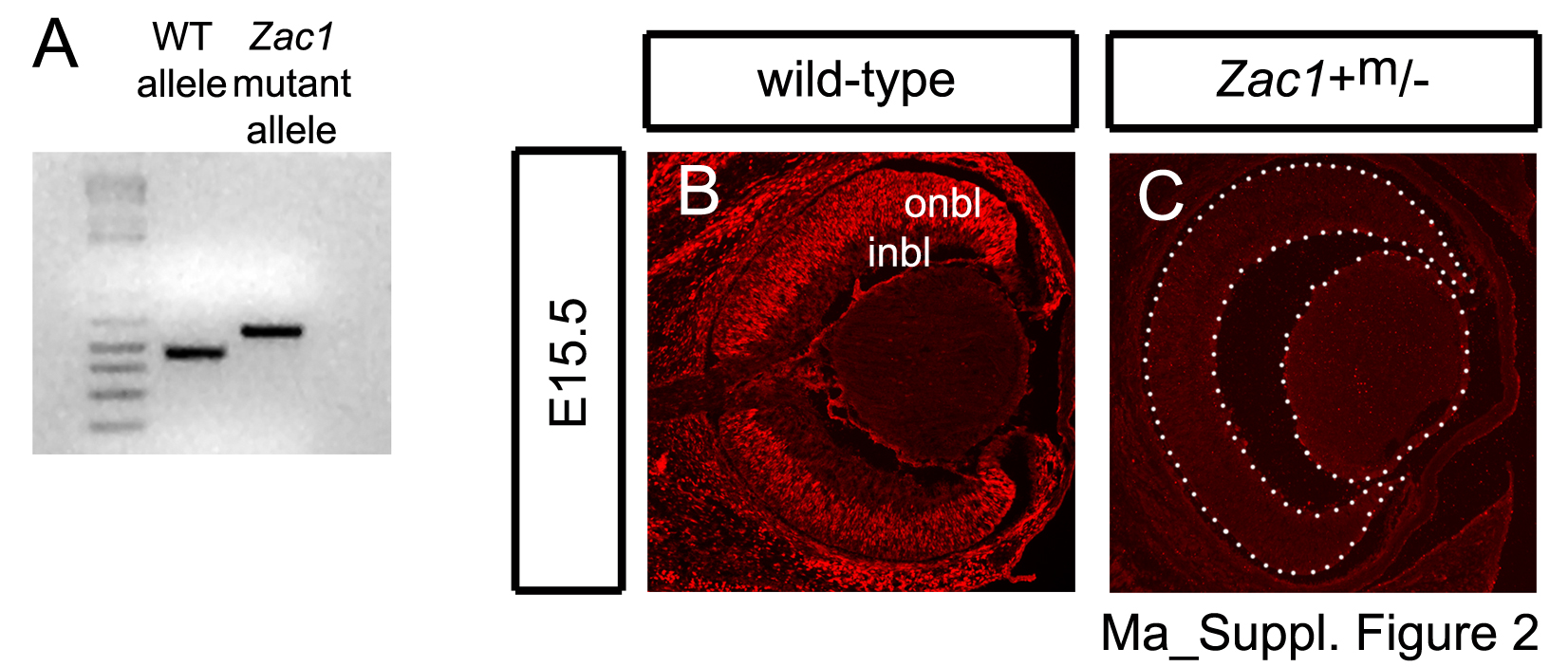
To investigate the in vivo requirement for Zac1, we analyzed embryos with a Zac1 null allele [23]. Because Zac1 is maternally imprinted, Zac1+m/- heterozygotes inheriting a wild-type allele from their mother are effectively mutant for Zac1. Indeed, imprinting occurs in the gametes, and complete methylation of Zac1 is achieved in 96.8% of mature oocytes [27]. Accordingly, Zac1+m/- retinae were devoid of Zac1 immunolabeling (Additional data file 2) and were thus considered equivalent to null mutants throughout this study.
By P3, 80% of Zac1+m/- pups die, with 50% dying within the first 24 hours [23]. To ensure we did not analyze surviving pups with unknown compensatory mechanisms, we studied only Zac1+m/- embryos collected prenatally, when Mendelian ratios of mutants were obtained. To circumvent the problem of retinogenesis not being complete until about P5–6 in the central retina [3], we cultured E18.5 retinae as explants for eight days in vitro (DIV), recapitulating the normal histogenic process [28]. Subsequent phenotypic analyses then focused on the central retina, adjacent to the transected optic nerve, where differentiation was complete. Strikingly, most Zac1+m/- explants (55%; n = 27/49) were thicker than their littermate controls, developing a distinct, ectopic cellular layer (ECL) between the INL and GCL (Figure 2b,d,f,f',h,h'; Additional data file 3). Wild-type (Figure 2a,c,e,e',g,g') and remaining Zac1+m/- retinae (not shown) acquired a normal trilaminar structure.
Figure 2.

Zac1+m/- retinae develop an ectopic amacrine cell layer and supernumerary rod photoreceptors. (a-h) E18.5→8DIV retinal explants. DAPI-stained (a) wild-type and (b) Zac1+m/- explants. Rhodopsin expression in (c) wild-type and (d) Zac1+m/-+ ECL retinae. (e,e',f,f') Pax6 and (g,g',h,h') syntaxin expression in amacrine cells in wild-type (e,e',g,g') and Zac1+m/-+ECL (f,f',h,h') retinae. Asterisks mark the ECL. The duplicated IPL is labeled by ipl' in (h'). Blue is DAPI counterstain. (i) Average of the absolute number of DAPI+ nuclei/layer in a standard counting field in wild-type (black bar; total DAPI+ nuclei counted in 30 fields; ONL: 23,700; INL: 9,870; GCL: 1,776), Zac1+m/- without an ECL (grey bar; total DAPI+ nuclei counted in 9 fields; ONL: 6,615; INL: 2,826; GCL: 498 nuclei) and Zac1+m/-+ECL (white bar; total DAPI+ nuclei counted in 27 fields; ONL: 26,175; INL: 11,968; GCL: 1,674). (j) Percentage of each retinal cell type based on total cell counts in wild-type (black bar; HC: 56 calbindin+/7,183 DAPI+; AC: 1,832 Pax6+/11,696 DAPI+; BP: 819 Chx10+/9,302 DAPI+; MG: 1,003 p27+/18,465 DAPI+ nuclei; 537 CRALBP+/9,169 DAPI+), Zac1+m/- without an ECL (grey bar; HC: 64 calbindin+/12,960 DAPI+; AC: 1,558 Pax6+/10,304 DAPI+; BP: 1,077 Chx10+/10,171 DAPI+; MG: 430 p27+/9,966 DAPI+; 332 CRALBP+/6,773 DAPI+) and Zac1+m/-+ECL retinae (white bar; HC: 11 calbindin+/1,924 DAPI+; AC: 2,068 Pax6+/11,302 DAPI+; BP: 646 Chx10+/9,157 DAPI+; MG: 395 p27+/9,921 DAPI+; 240 CRALBP+/3,319 DAPI+). AC, amacrine cell; BP, bipolar cell; HC, horizontal cell; MG, Müller glia.
An ECL may develop due to an overall increase in retinal cell number and/or aberrant cellular migration. To determine if Zac1+m/- retinae were hypercellular, DAPI-labeled nuclei were counted. In ECL-containing Zac1+m/- explants (hereafter designated Zac1+m/-+ECL), there was a 1.34-fold increase in the number of INL cells (p < 0.001; 442.9 ± 17.9 cells/field; n = 9 retinae) compared to wild-type controls (329.5 ± 22.0 cells/field; n = 10) or non-ECL containing mutants (henceforth simply designated Zac1+m/-; 314.0 ± 22.1 cells/field; n = 3; Figure 2i). Strikingly, Zac1+m/-+ECL retinae also exhibited a 1.23-fold increase in ONL cells (p < 0.01; 969.4 ± 46.1 cells/field; n = 9) compared to wild-type controls (790.3 ± 40.7 cells/field; n = 10) or Zac1+m/- (735 ± 106.7 cells/field; n = 3; Figure 2i). In contrast, cellular contents of the GCL were comparable in wild-type (59.2 ± 3.1 cells/field; n = 10), Zac1+m/-+ECL (62.0 ± 4.3 cells/field; n = 9) and Zac1+m/- (55.3 ± 1.8 cells/field; n = 3) explants. Zac1 is, therefore, an essential negative regulator of retinal cell number and is also required to orchestrate appropriate cellular migration.
The Zac1+m/- ECL is composed of supernumerary amacrine cells
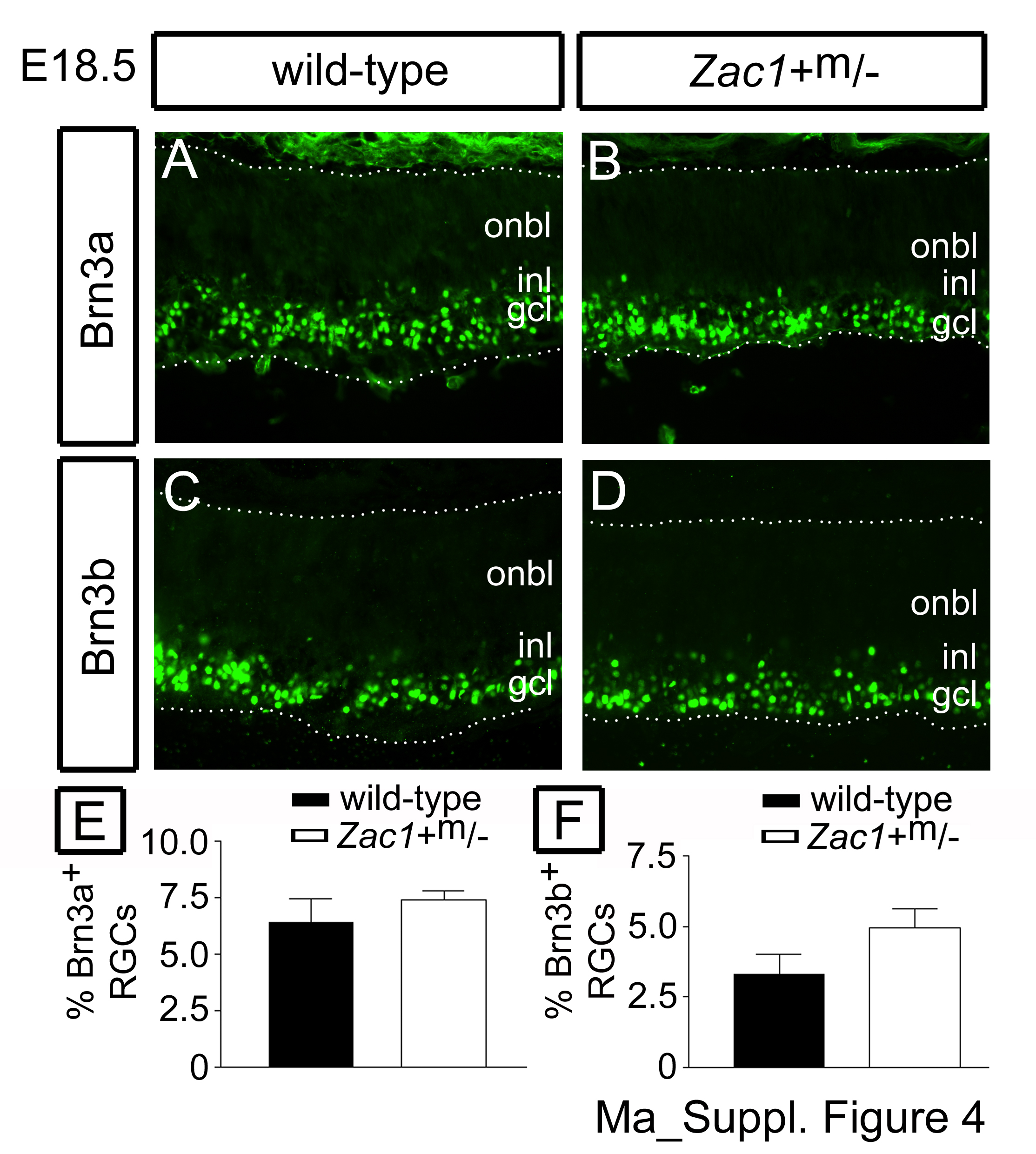
To identify the expanded cell population(s) in Zac1+m/-+ECL retinae, E18.5→8DIV explants were immunostained with cell type-specific markers. Strikingly, almost all cells in the Zac1+m/- ECL expressed the homeodomain transcription factor Pax6 (Figure 2f,f'), which was also expressed by amacrine cells in the INL and GCL in wild-type (Figure 2e,e',f,f') and Zac1+m/- (data not shown) E18.5→8DIV explants. Although Pax6 labels both amacrine cells and RGCs [29], RGCs rapidly undergo apoptosis following optic nerve transection (in explants [30]), allowing us to assign an amacrine cell identity to ECL cells. Accordingly, no other RGC markers (Brn3a/3b, Thy1.2; not shown) were detected in the ECL or GCL of wild-type or Zac1+m/-+ECL explants. Moreover, RGC differentiation is essentially complete by E18.5, and at this stage, equivalent numbers of RGCs were labeled by Brn3a (p = 0.95) and Brn3b (p = 0.23) in wild-type and Zac1+m/- retinae, indicating Zac1 does not regulate RGC number (n = 3 for each; total n = 6; Additional data file 4). Furthermore, syntaxin, which labels amacrine cell membranes and processes in the inner plexiform layer (IPL; Figure 2g,g'), marked duplicated and disorganized synaptic plexi (IPL/IPL') in Zac1+m/-+ECL explants (Figure 2h,h'). Finally, amacrine cell subtype markers, including Bhlhb5, calbindin, GABA and the GlyT1 glycine transporter, were all expressed in Zac1+m/- ECL (Additional data file 3 (k–p)).
Quantitation of Pax6+ nuclei in E18.5→8DIV explants revealed a 1.31-fold increase (p < 0.01) in the percentage of amacrine cells in Zac1+m/-+ECL retinae, while Zac1+m/-explants contained wild-type proportions of these interneurons (wild type: 15.1 ± 0.5%; n = 4; Zac1+m/-: 15.2 ± 1.3%; n = 3; Zac1+m/-+ECL: 19.8 ± 1.3%; n = 3; Figure 2j). In contrast, all other INL cell types were present at equivalent ratios in wild-type and Zac1+m/-+/-ECL retinae, including bipolar cells (Chx10+; wild type: 8.7 ± 0.6%; n = 4; Zac1+m/-: 10.5 ± 0.9%; n = 3; Zac1+m/-+ECL: 7.2 ± 1.3%; n = 3), Müller glia (CRALBP+: wild type: 5.2 ± 0.3%; n = 4; Zac1+m/-: 4.3 ± 0.2%; n = 3; Zac1+m/-+ECL: 3.9 ± 0.5%; n = 3; p27Kip1+: wild type: 5.9 ± 0.5%; n = 4; Zac1+m/-: 5.1 ± 0.7%; n = 3; Zac1+m/-+ECL: 5.8 ± 0.1%; n = 3) and horizontal cells (calbindin+; identified also by morphology and apical location; wild type: 1.1 ± 0.3%; n = 3; Zac1+m/-: 0.8 ± 0.2%; n = 4; Zac1+m/-+ECL: 0.1 ± 0.1%; n = 3; Figure 2j; Additional data file 3 (m,n)).
Cones normally comprise only 3% of the murine photoreceptor pool [31]. In Zac1+m/-+ECL and wild-type retinae, similar numbers of cones were labeled with peanut agglutinin (PNA; p = 0.26; wild type: 50.4 ± 1.2cells/field; n = 3; Zac1+m/-+ECL: 64.6 ± 10.1cells/field; n = 3) and s-opsin (p = 0.70; wild type: 44.22 ± 8.91cells/field; n = 3; Zac1+m/-+ECL: 39.75 ± 6.66cells/field; n = 4; Additional data file 3 (g–j)). Instead, the vast majority of ONL cells in wild-type and Zac1+m/-+ECL explants expressed the rod-specific markers rhodopsin (Figure 2c, d) and Nr2e3 (not shown), indicating that the rod pool is expanded in Zac1+m/-+ECL retinae. Zac1 therefore ensures appropriate numbers of rod photoreceptors and amacrine cells are generated during development.
Retinal progenitors divide ectopically in Zac1 mutants late in retinogenesis
The hypercellularity of Zac1+m/- retinae could arise due to additional rounds of cell division and/or a reduction in apoptosis. To determine if cell cycle exit was perturbed, S-phase progenitors were BrdU pulse-labeled 30 minutes prior to sacrifice. During embryogenesis (E13.5-E18.5) and in E18.5→2DIV explants, BrdU-labeling indices were similar in wild-type and Zac1+m/- retinae (Figure 3e). In contrast, in E18.5→4DIV Zac1+m/- explants, BrdU incorporation was elevated 2.1-fold over wild type (p < 0.002; Zac1+m/-: 3.4 ± 0.4%; n = 10; wild type: 1.6 ± 0.3%; n = 7; Figure 3a,b,e), although an ECL was not yet distinguishable. Notably, BrdU-labeling indices were variable in individual Zac1+m/- retinae, with about 50% of the mutant explants well above wild-type values (Figure 3f), a phenotypic distribution corresponding well with the proportion of mutant explants that later developed a hypercellular phenotype (see above). Furthermore, in 6DIV explants, when cell division had ceased in wild-type central retinae, BrdU uptake persisted in some mutants (p < 0.05; 0.5 ± 0.01%; n = 4/11; Figure 3c–e). As an independent cell cycle parameter, cyclin D1 (CcnD1) expressing cells were also elevated 1.48-fold (p < 0.001) over wild type (11.7 ± 0.2%; n = 4) in approximately half of the 4DIV Zac1+m/- explants (with phenotype: 17.3 ± 0.6%; n = 4/9; without phenotype: 12.0 ± 0.7%; n = 5/9; Figure 3g–j). Cell proliferation was thus specifically elevated at late stages of retinogenesis in Zac1 mutants.
Figure 3.

Loss of Zac1 results in increased proliferation and reduced apoptosis at a late stage of retinogenesis. (a-d) BrdU labeling (red) of E18.5 wild-type and Zac1+m/- explants cultured 4DIV (a,b) or 6DIV (c,d). Arrowheads in (d) mark ectopic proliferating cells. (e) Percentage of BrdU+ nuclei in wild-type (black bar; E13.5: 2,824 BrdU+/8,235 DAPI+; E16.5: 2,234 BrdU+/10,663 DAPI+; E18.5: 2,859 BrdU+/27,380 DAPI+; E18.5→2DIV: 4,371 BrdU+/54,554 DAPI+; E18.5→4DIV: 988 BrdU+/55,300 DAPI+; E18.5→6DIV: 0 in 9 fields) and Zac1+m/- retinae (grey bars; E13.5: 3,555 BrdU+/10,413 DAPI+; E16.5: 3,369 BrdU+/15,707 DAPI+; E18.5: 2,212 BrdU+/17,642 DAPI+; E18.5→2DIV: 3,298 BrdU+/35,085 DAPI+; E18.5→4DIV: 3,474 BrdU+/97,499 DAPI+; E18.5→6DIV: 54 BrdU+/11,618 DAPI+). (f) BrdU-labeling indices of individual wild-type (squares) and Zac1+m/- (triangles) E18.5→4DIV retinal explants. (g,h) E18.5→4DIV wild-type (g) and Zac1+m/- (h) retinal explants labeled with CcnD1 (red). (i) Percentage of Ccdn1+ cells in wild-type (black bar; 2,480 CcnD1+/21,329 DAPI+) and Zac1+m/- without aberrant proliferation (grey bar; 3,156 CcnD1+/26,328 DAPI+) and with a proliferative phenotype (w/φ; white bar; 3,266 CcnD1+/18,709 DAPI+) at 4DIV. (j) Ccnd1-labeling indices of individual wild-type (squares) and Zac1+m/- (triangles) E18.5→4DIV retinal explants. (k,l) E18.5→4DIV wild-type (k) and Zac1+m/- (l) retinal explants labeled with pHH3 (red). (m) Apical (Ap) to basal (Ba) ratio of pHH3+ cells in wild-type (black bar; 808 ap:791 ba pHH3+) and Zac1+m/- without (grey bar; 971 ap:796 ba pHH3+) and with (w/φ; white bar; 1,012 ap:480 ba pHH3+) a proliferative phenotype at 4DIV. (n) Ap:Ba ratios of pHH3+ cells in individual wild-type (squares) and Zac1+m/- (triangles) E18.5→4DIV retinal explants. (o-p) Active caspase-3 (Ac-3) expression (red) in wild-type and Zac1+m/- E18.5→4DIV explants. Blue is DAPI counterstain. (q) Percentage of apoptotic cells in the total population of wild-type (black bars; E18.5: 71 ac-3+/18,341 DAPI+; E18.5→2DIV: 532 ac-3+/14,995 DAPI+; E18.5→4DIV: 1,266 ac-3+/27,321 DAPI+; E18.5→8DIV: 294 ac-3+/10,209 DAPI+) and Zac1+m/- (white bars; E18.5: 67 ac-3+/13,768 DAPI+; E18.5→2DIV: 457 ac-3+/13,195 DAPI+; E18.5→4DIV: 488 ac-3+/24,077 DAPI+; E18.5→8DIV: 212 ac-3+/14,377 DAPI+) retinae. (r) Distribution of individual wild-type (squares) and Zac1+m/- (triangles) ac-3-labeling indices at 4DIV.
Ectopic division could occur if progenitors cycled more extensively and/or committed precursors failed to exit the cell cycle. Retinal progenitors are defined by cell cycle-dependent, interkinetic nuclear movements, with G2/M-phase, phospho-histoneH3 (pHH3)-expressing nuclei lining the apical surface (Figure 3k,l), while S-phase nuclei lie more basal in the onbl [32] (Additional data file 1 (e)). This contrasts to committed precursors that migrate towards the vitreal (basal) surface of the inbl to initiate formation of the mature retinal layers. We thus used mitotic position to distinguish proliferating progenitors (apical mitoses) versus precursors (basal mitoses) [33]. In Zac1+m/- retinae, the proportion of pHH3-labeled nuclei was biased towards apical compartments in many Zac1+m/- 4DIV explants (apical to basal ratio: wild type: 1.02 ± 0.07; n = 10; Zac1+m/-+phenotype: 2.30 ± 0.35; n = 3/8; Zac1+m/-: 1.19 ± 0.10; n = 5/8; Figure 3k–e), consistent with an increase in progenitor and not precursor cell divisions. Accordingly, most Pax6+ amacrine precursors did not incorporate BrdU after a 30 minute exposure in wild-type or Zac1+m/- 4DIV explants (Figure 4a,a',b,b'; Additional data file 5 (j)). Similarly, double labeling with Math3, an amacrine and bipolar precursor marker, revealed very few Math3/BrdU double+ cells in wild-type and Zac1+m/- explants (Figure 4c,c',d,d'). Therefore, retinal progenitor cells and not committed precursors are dependent on Zac1 for cell cycle exit.
Figure 4.

Amacrine cell genesis is elevated postnatally in Zac1+m/- retinae. (a-d) E18.5→4DIV wild-type (a,a',c,c') and Zac1+m/- (b,b',d,d') explants co-labeled with BrdU (red, S-phase) and Pax6 (green; amacrine cells (a,a',b,b') or Math3 (green; amacrine and bipolar precursors in INL (c,c',d,d'). (e,f) E18.5→4DIV explants labeled with Pax6 alone (red). (g,g',h,h') Birthdating of E18.5→8DIV wild-type (g,g') and Zac1+m/- (h,h') retinal explants exposed to BrdU (green) at 2DIV and co-labeled with anti-Pax6 (red). BrdU/Pax6 double+ cells are marked by arrowheads in (g',h'), which are high magnification images of boxed areas in (g,h), respectively. (i) Percentage of BrdU/Pax6 double+ nuclei (amacrine cells born at time of BrdU exposure) in wild-type (black bar; BrdU at 1DIV: 562 BrdU/Pax6 double+/2,385 Pax6+; 2DIV: 527 BrdU/Pax6 double+/6,022 Pax6+; 4DIV: 77 BrdU/Pax6 double+/1,496 Pax6+; all counts in 8DIV explants) and Zac1+m/- explants without an ECL (grey bar; BrdU at 1DIV: 1,307 BrdU/Pax6 double+/4,084 Pax6+; 2DIV: 527 BrdU/Pax6 double+/4,926 Pax6+; 4DIV: 75 BrdU/Pax6 double+/1,660 Pax6+) and Zac1+m/-+ECL explants (white bar; BrdU at 1DIV: 2,126 BrdU/Pax6 double+/6,107 Pax6+; 2DIV: 883 BrdU/Pax6 double+/4,386 Pax6+; 4DIV: 335 BrdU/Pax6 double+/3,587 Pax6+). (j) Model of amacrine cell genesis in wild-type (red line) versus Zac1+m/-+ECL (blue line) retinae.
Apoptosis is reduced during late developmental stages in Zac1-deficient retinae
Compensatory mechanisms exist in the retina to ensure that cellular content remains constant, with excess proliferation often balanced by an increase in apoptosis [34,35]. Given that Zac1 induces apoptosis when misexpressed in cell lines [24], we tested if it were also required for the normal program of cell death in the retina, using activated-caspase-3 (ac-3), a downstream effector and early marker of commitment to the cell death pathway [36]. During embryonic retinal development, apoptosis peaks during the optic cup stage (E10–E11) in the presumptive retinal pigmented epithelium (rpe) and optic stalk and again between E15.5–E17.5, primarily in retinal cells adjacent to the optic nerve head [37-40]. We analyzed ac-3 staining in wild-type (n = 6) and Zac1 mutant retinae (n = 6) at E10.5 and E15.5 but did not observe more than a few apoptotic cells per retinal section in either genotype (Additional data file 5 (a-d)). Similarly, at E18.5 (p = 0.14; wild type: 0.4 ± 0.02%; n = 3; Zac1+m/-: 0.5 ± 0.03%; n = 3) and in E18.5→2DIV explants (p = 0.93; wild type: 3.5 ± 0.4%; n = 3; Zac1+m/-: 3.5 ± 0.2%; n = 3), comparable levels of apoptosis were observed in both genotypes (Figure 3q). In contrast, after 4 and 8DIV, there were 3.48-fold (p < 0.01; wild type: 4.5 ± 1.0%; n = 7; Zac1+m/-: 1.3 ± .0.2%; n = 5/6) and 2.02-fold (p < 0.05; wild type: 2.9 ± 0.2%; n = 3; Zac1+m/-: 1.4 ± 0.3%; n = 4) reductions, respectively, in the number of ac-3+ retinal cells in Zac1+m/- explants (Figure 3o–r).
The reduction in cell death in Zac1+m/- explants could contribute to the increase in amacrine and rod cell numbers. However, the number of ac-3/Pax6-double+ amacrine cells was similar in E18.5→4DIV explants from both genotypes (p = 0.15; wild type: 1.6 ± 0.4%; n = 3; Zac1+m/-: 0.9 ± 0.1%; n = 3; Additional data file 5 (e–i)). In contrast, there was a 1.82-fold reduction in ac-3+ ONL photoreceptors in Zac1+m/- E18.5→8DIV explants (p < 0.05; wild type: 2.9 ± 0.2%; n = 3; Zac1+m/-: 1.60 ± 0.4%; n = 4). Zac1 deficiency therefore perturbs pro-apoptotic pathways that adjust cell numbers at late stages of retinogenesis, likely contributing to the increase in rod cell number.
Zac1 is a direct negative regulator of proliferation and rod differentiation
To test if Zac1 was a direct negative regulator of amacrine and rod cell fates, we established a gain-of-function assay, electroporating retinal explants with a pCIG2 vector, containing an internal ribosome entry site (IRES) 2-enhanced green fluorescent protein (EGFP) cassette, or a pCIG2-Zac1 vector, expressing both EGFP and Zac1 (Figure 5). E15.5 and P0 retinal explants misexpressing Zac1 were BrdU-pulse labeled 24 hours post-electroporation, revealing 1.96-fold and 2.49-fold reductions, respectively, in the number of BrdU/EGFP-double+ cells compared to controls at E15.5 (p < 0.05; pCIG2: 15.4 ± 1.6%; n = 3; Zac1: 7.9 ± 0.8%; n = 3; Figure 5g) and P0 (p < 0.05; pCIG2: 6.3 ± 0.3%; n = 3; Zac1: 2.5 ± 1.0%; n = 3; Figure 5a–d,g). Zac1 therefore promotes cell cycle exit and/or increases cell cycle length in the murine retina. In contrast, Zac1 misexpression did not increase the number of ac-3+ cells compared to controls 24 hour post-electroporation at P0 (p = 0.2; pCIG2: 3.1 ± 0.6%; n = 3; Zac1: 5.2 ± 1.3%; n = 3; Figure 5e,f,h), indicating that Zac1 is not sufficient to induce retinal apoptosis.
Figure 5.

Zac1 inhibits cell division and rod fate specification. (a-f) P0 retinae electroporated with pCIG2 control (a-c,e) or pCIG2-Zac1 (d,f) cultured 1DIV. GFP+ electroporated cells (green (a-f)) labeled with anti-BrdU (red (c,d)) and anti-ac-3 (red (e,f)). Blue in (b) is DAPI counterstain. (g) Percentage of GFP+ cells that incorporated BrdU after electroporation of pCIG2 (black bar; E15.5→1DIV: 88 BrdU/GFP double+/542 GFP+; P0→1DIV: 124 BrdU/GFP double+/1,784 GFP+) and pCIG2-Zac1 (white bar; E15.5→1DIV: 24 BrdU/GFP double+/290 GFP+; P0→1DIV: 14 BrdU/GFP double+/816 GFP+). (h) Percentage of GFP+ cells that expressed ac-3 in P0→1DIV retinae electroporated with pCIG2 (black bar; 157 ac-3/GFP double+/5,402 GFP+) and pCIG2-Zac1 (white bar; 97 ac-3/GFP double+/1,808 GFP+). (i-r) P0→8DIV retinae electroporated with pCIG2 (i-k,m,o,q) or pCIG2-Zac1 (l,n,p,r). GFP+ electorporated cells (green (i-r)) co-labeled with Pax6 (red; amacrine cells (j-l)), rhodopsin (red; rods (m,n)), Chx10 (red; bipolar (o,p)) and p27Kip1 (red; Müller glia (q,r)). (s) Percentage of GFP+ cells expressing cell-type specific markers post-electroporation of pCIG2 (black bar; 290 Pax6/GFP double+/3,939 GFP+; 81 syntaxin/GFP double+/552 GFP+; 955 rhodopsin/GFP double+/1,751 GFP+; 384 Nr2e3/GFP double+/1,261 GFP+; 279 Chx10/GFP double+/3,146 GFP+; 520 p27/GFP double+/3,846 GFP+) or pCIG2-Zac1 (white bar; 140 Pax6/GFP double+/1,284 GFP+; 83 syntaxin/GFP double+/376 GFP+; 56 rhodopsin/GFP double+/356 GFP+; 131 Nr2e3/GFP double+/816 GFP+; 263 Chx10/GFP double+/1,455 GFP+; 541 p27/GFP double+/1,888 GFP+). Arrowheads mark double+ cells. le, lens; Rho, Rhodopsin; Syn, Syntaxin.
To determine if Zac1 was a direct, negative regulator of rod and/or amacrine fates, we examined the molecular phenotype of retinal cells electroporated at P0 and cultured 8DIV. No differences were observed in the ratio of GFP+ cells that became Pax6+ amacrine cells after electroporation of pCIG2 (p = 0.73; 11.7 ± 3.4%; n = 6; Figure 5i–,k,s) versus pCIG2-Zac1 (10.2 ± 2.3%; n = 6; Figure 5l,s). Similarly, misexpression of Zac1 at E15.5 and E17.5, during the peak of amacrine cell genesis, did not affect amacrine cell number (Additional data file 6). In contrast, Zac1 misexpression at P0 resulted in a 4.49-fold reduction in rhodopsin+ rods (p < 0.01; pCIG2: 52.3 ± 4.5%; n = 3; Zac1: 11.6 ± 5.4%; n = 3; Figure 5m,n,s) and a 2.43-fold reduction in Nr2e3-labeled rods (p < 0.05; pCIG2: 30.8 ± 3.6%; n = 3; Zac1: 12.7 ± 3.1%; n = 3; Figure 5s). Zac1-misexpressing progenitors instead preferentially differentiated into Chx10+ bipolar cells (1.72-fold increase; p < 0.05; pCIG2: 11.1 ± 1.8%; n = 6; Zac1: 19.1 ± 2.2%; n = 6) and p27Kip1+ Müller glia (1.95-fold increase; p < 0.05; pCIG2: 14.4 ± 2.2%; n = 6; Zac1: 28.1 ± 4.5%; n = 6), cells types normally generated along with rods postnatally (Figure 5o–s). Zac1 is thus a potent inhibitor of a rod fate but does not directly suppress amacrine cell genesis.
Elevated amacrine cell genesis continues for a prolonged period in Zac1 mutants
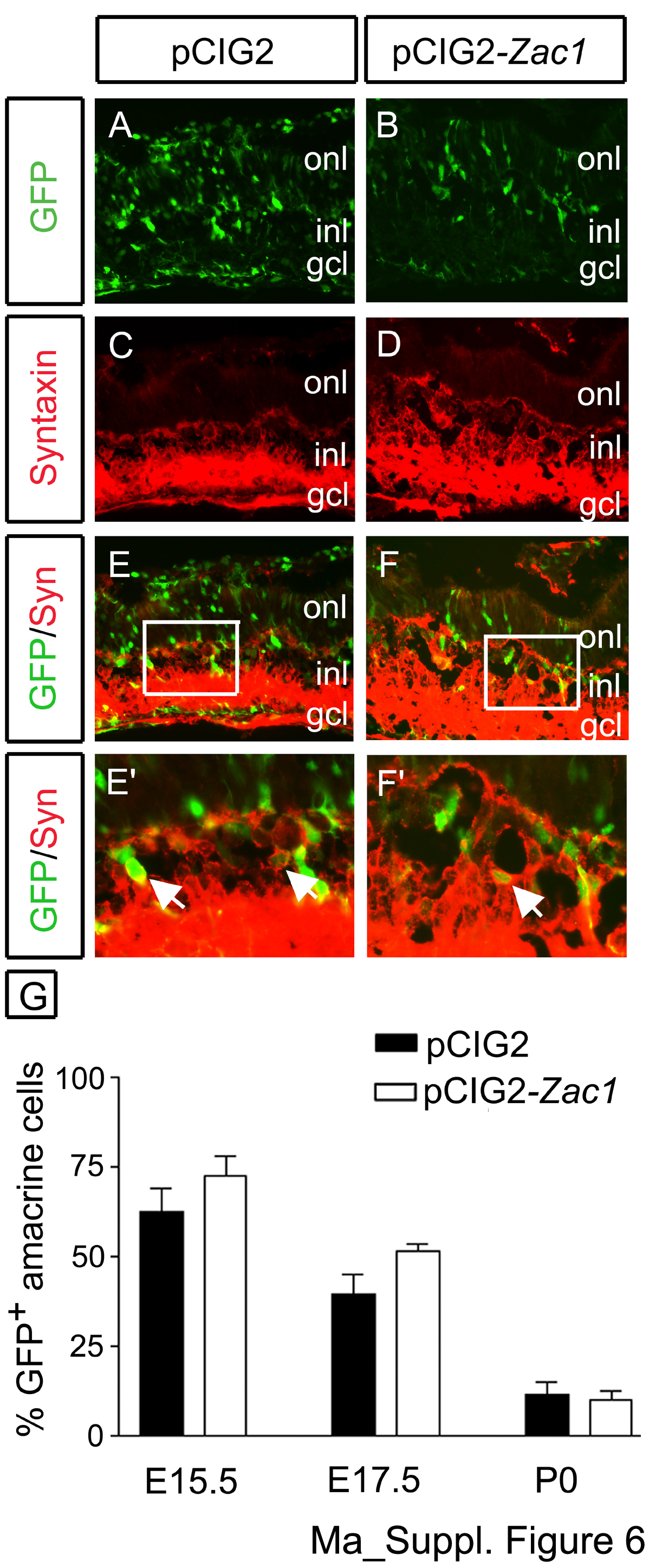
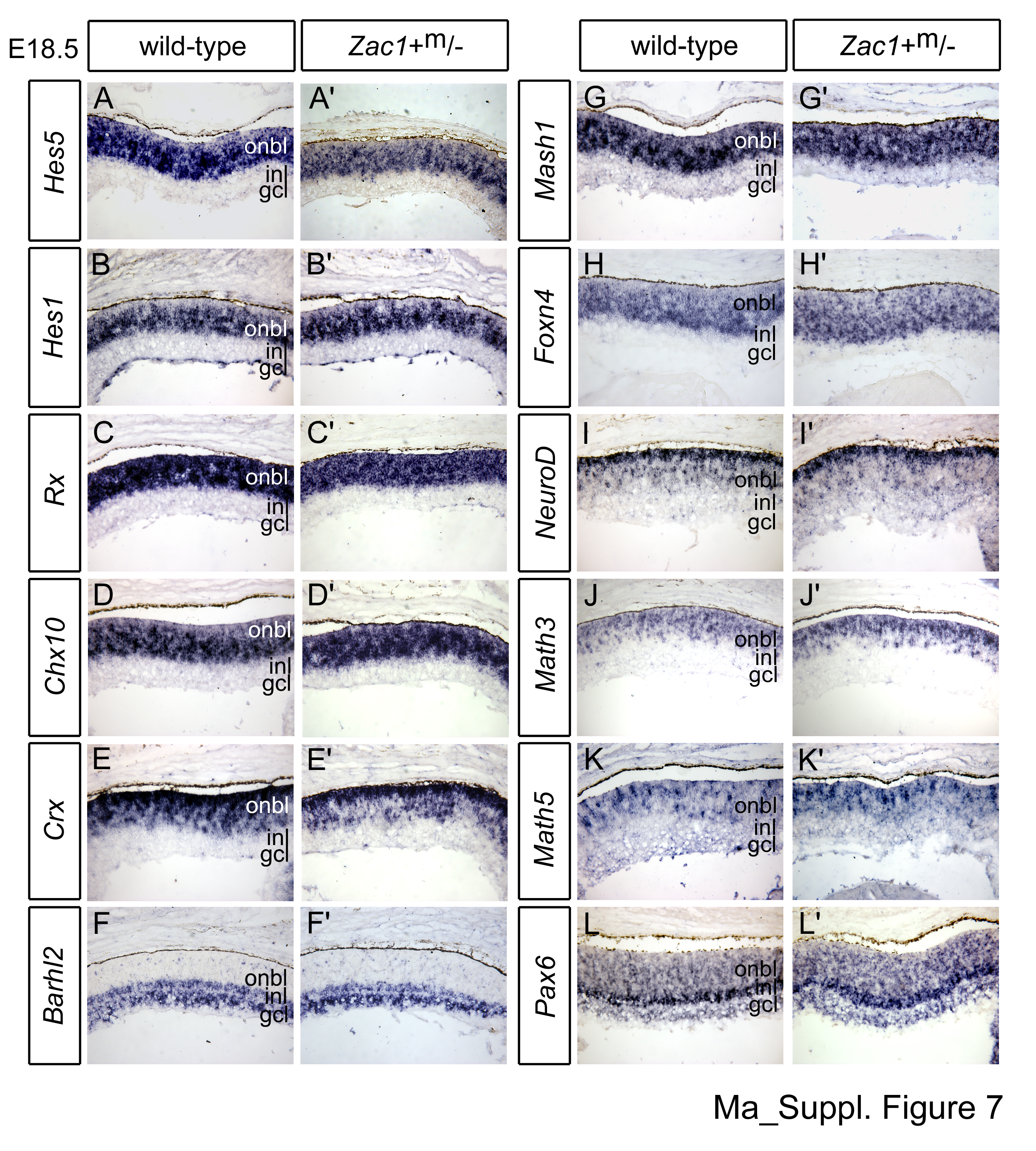
To understand how Zac1 controls amacrine cell numbers, we next determined when ectopic amacrine cells first appeared in Zac1+m/- retinae. In mouse, amacrine cell genesis normally peaks at E15.5, tapering off before birth [3] (Figure 4j). At E18.5, genes involved in amacrine fate specification/differentiation, including Math3, Foxn4, NeuroD, Pax6 and Barhl2 [41-43], were expressed in an indistinguishable manner in wild-type and Zac1+m/- retinae, as were several other genes involved in the specification of all other cell types (Additional data file 7). Cell fate specification was thus grossly normal in E18.5 Zac1+m/- retinae. In contrast, in E18.5→4DIV Zac1+m/- explants, Pax6 (Figure 4a,a',b,b',e,f), Six3, Barhl2 and Math3 (Additional data file 8 (i–n)) expression increased, suggesting the amacrine cell population expanded during early postnatal stages in Zac1+m/- retinae.
To verify that amacrine genesis increased postnatally in Zac1+m/- retinae, we performed birthdating. E18.5 retinal explants were labeled with BrdU after 1, 2 and 4DIV and then cultivated for 8DIV (Figure 4g,g',h,h'). More BrdU+/Pax6+ amacrine cells were born at 1DIV (1.76-fold increase; p < 0.05; wild type: 22.7 ± 3.4%; n = 3; Zac1+m/-+ECL: 39.9 ± 3.8%; n = 4; Zac1+m/-: 29.8 ± 3.0%; n = 3), 2DIV (2.34-fold increase; p < 0.05; wild type: 8.6 ± 1.3%; n = 7; Zac1+m/-+ECL: 20.0 ± 4.4%; n = 4; Zac1+m/-: 10.1 ± 2.8%; n = 6) and 4DIV (5.42-fold increase; p < 0.05; wild type: 1.7 ± 1.0%; n = 6; Zac1+m/-+ECL: 9.2 ± 2.4%; n = 3; Zac1+m/-: 4.3 ± 0.4%; n = 2; Figure 4g,g',h,h',i) in Zac1+m/-+ECL explants compared to wild type, confirming that the period of amacrine cell genesis was prolonged.
Negative feedback signals are deficient in Zac1+m/- amacrine cells
Our data suggested that the 'stop' or negative feedback signals that normally limit amacrine cell production later in development [13,14] were deficient in Zac1+m/- retinae (Figure 4j). To thus test if Zac1 was an essential component of the amacrine cell negative feedback loop, we performed aggregation assays. Dissociated E14.5 wild-type retinal cells pre-labeled with BrdU were either cultured alone as intact pellets or in pellet aggregations with a 20-fold excess of dissociated E18.5 wild-type or Zac1+m/- retinal cells, the latter populations serving as a source of amacrine cell feedback signals (Figure 6a). After 8DIV, pellets were dissociated and Pax6+/BrdU+ amacrine cells derived from E14.5 progenitors were quantified (Figure 6b–J). Of the E14.5 cells cultured alone, 39.6 ± 3.4% (n = 9; 3 independent experiments) of BrdU+cells became Pax6+ amacrine cells (Figure 6b–d,k). Consistent with feedback signals being emitted from differentiating, E18.5 wild-type cells, in co-cultures, amacrine cell development from the E14.5-labeled cohort was reduced 1.40-fold (p < 0.01; 27.9 ± 2.0%; n = 10; Figure 6e–g,k). In contrast, amacrine cell development from the E14.5 cohort was restored to normal levels (compared to E14.5 cells alone) in mixed aggregates containing E18.5 Zac1+m/- cells (p < 0.05, 37.7 ± 2.4%; n = 21), indicative of impaired negative feedback (Figure 6h–k). Zac1 is thus required in postnatal retinal cells to negatively regulate amacrine cell genesis.
Figure 6.

Zac1+m/- retinae lose amacrine cell feedback inhibition. (a) Schematic of aggregation assay protocol. (b-j) Immunolabeling of dissociated cell pellets with Pax6 (red (c,d,f,g,i,j)), BrdU (green (b,d,e,g,h,j)) and merged image with DAPI (blue (d,g,j)). E14.5 progenitors cultured alone (b-d) or with E18.5 wild-type (e-g) or Zac1+m/- (h-j) retinal cells. Arrowheads mark Pax6/BrdU double+ nuclei (d,g,j). (k) Percentage of BrdU+ E14.5 cells that differentiated into Pax6+ amacrine cells when cultured alone (black bar; 1,085 BrdU/Pax6 double+/2,892 BrdU+) or with E18.5 wild-type (grey bar; 853 BrdU/Pax6 double+/3,215 BrdU+) or Zac1+m/- (white bar; 2,559 BrdU/Pax6 double+/7,196 BrdU+) retinal cells. n indicates number of individual retinal aggregates quantified.
TGFβ signaling inhibits amacrine cell genesis in the retina
The cell non-autonomous requirement for Zac1 as a negative regulator of amacrine cell production implied that this transcription factor must regulate the expression of an unknown secreted signal. We focused on TGFβ cytokines, given their role in feedback control in other systems. Specifically, we studied TGFβII, which regulates cell cycle exit at late stages of rat retinogenesis [44], corresponding to the period when Zac1+m/- cells proliferated aberrantly. In E18.5 > 4DIV explants, the cognate receptors, TGFβRI and TGFβRII, were expressed at low levels in dividing, Ccnd1+ progenitors (Figure 7a,b,d,e) and at higher levels in Pax6+ amacrine cells (Figure 7c,f). TGFβII was similarly expressed in Pax6+ amacrine cells in the GCL and INL (Figure 7g–i) and at lower levels in Ccnd1+ progenitors in the onbl (not shown). Thus, TGFβII signaling could correspond to the amacrine cell stop signal.
Figure 7.

Zac1 regulates TGFβII signaling in the retina. (a-f) Co-expression of TGFβRI (green (a-c)) and TGFβRII (green (d-f)) with Ccnd1 (red, proliferating progenitors (b,e)) and Pax6 (red, amacrine cells (c,f)) in E18.5 > 4DIV wild-type retinal explants. (g-l) TGFβII expression in E18.5→4DIV wild-type (green (g-i)) and Zac1+m/- (green (j-l)) retinal explants co-labeled with Pax6 (red, amacrine cells (i,l)). Arrowheads mark double+ cells. Asterisk in (j) marks reduction in onbl/INL expression. (m,n) Expression of pSmad2/3 in E18.5→4DIV wild-type (m) and Zac1+m/- (n) retinal explants. (o) Western blot analysis of TGFβII, pSmad2/3, total Smad2/3, and β-actin. Asterisks in (o) indicate mutants with reduced expression of TGFβII or pSmad2/3. (p,q) Quantitation of expression levels normalized to β-actin via densitometry for TGFβII (p) and pSmad2/3 (q).
In Zac1 mutants, a notable reduction in TGFβII expression was observed in onbl progenitors and in Pax6+ amacrine cells in the INL, while GCL levels were similar to wild type (Figure 7j–l). An overall reduction in TGFβII levels was confirmed by western blot, demonstrating that the 25 kDa isoform (note: labile 12 kDa mature form not detected) was reduced in most (n = 8/12) Zac1+m/- explants (p < 0.05; signal normalized to β-actin; wild type: 1.5 ± 0.04; n = 4; Zac1+m/-: 0.9 ± 0.2; n = 3/4; Figure 7o,p). To confirm that TGFβ signaling was indeed reduced in Zac1+m/- retinae, we examined expression of the downstream effector, pSmad 2/3. In E18.5→4 DIV wild-type explants, pSmad2/3 was expressed at diffuse levels throughout the retinae, but at significantly higher levels in the GCL and the basal half of the INL, where differentiated amacrine cells reside, as well as at lower levels in dividing progenitor cells in the onbl (Figure 7m). In contrast, pSmad2/3 levels were decreased in the INL and onbl progenitors in Zac1+m/- explants (Figure 7n). Accordingly, western blot analysis revealed a significant reduction in pSmad2/3 protein levels in Zac1+m/- versus wild-type E18.5 > 4 DIV explants when normalized to β-actin (p < 0.01; n = 6/8 mutants analyzed), while total Smad2/3 protein levels were similar in both genotypes (n = 4 for each genotype; Figure 7o,q). TGFβ signaling was thus attenuated in Zac1+m/-retinae.
To determine if reduced TGFβ signaling results in amacrine cell expansion, conditional TGFβRII mutants were analyzed. Mice heterozygous (Figure 8a,c,e,g) or homozygous (Figure 8b,d,f,h,i,j) for a floxed mutant allele of TGFβRII (hereafter referred to as flTGFβRII; [45]) were crossed with mice carrying a R26R reporter and a GLAST::CreERT2 knock-in allele [46]. GLAST was expressed in the embryonic retina (Figure 8a, inset) and, accordingly, tamoxifen administered at E16 specifically induced CreERT2 recombinase activity in the E18.5 retina as evidenced by R26R reporter expression (that is, X-Gal staining in tamoxifen injected (Figure 8b) and not un-injected control (Figure 8a) retinae). Accordingly, expression of TGFβRII was reduced in E18.5 flTGFβRII-/- (Figure 8d) compared to flTGFβRII+/- retinae (Figure 8c). An overt expansion of the amacrine cell layer, as labeled by Pax6 (Figure 8e,f) and syntaxin (Figure 8g–j), was also evident in tamoxifen-induced E18.5 flTGFβRII-/- mutant retinae (Figure 8f,h and Figure 8i,j show different mutants) compared to wild-type controls (Figure 8e,g).
Figure 8.

TGFβII negatively regulates amacrine cell genesis. (a,b) X-gal staining of E18.5 GLAST::CreERT2+/-;R26R reporter+ transgenic without (a) and with (b) administration of tamoxifen at E16. Inset in (a) shows GLAST immunostaining of E15.5 retina. (c-j) Analysis of TGFβRII+/-;GLAST::CreERT2+/-;R26R+ (c,e,g) and TGFβRII-/-;GLAST::CreERT2+/-;R26R+ (d,f,h,i,j) retinae immunostained with anti-TGFβRII (c,d), Pax6 (e,f) and syntaxin (g-j). Arrowheads in (i) mark ectopic amacrine cell clusters. TGFβRII-/- retinae in panels (d-h) and (i,j) are from two different mutant embryos. (k,l) E18.5 > 8DIV retinal explants cultured with vehicle control (k) or TGFβRII-Fc (l) and labeled with anti-Pax6. (m) Percentage of Pax6+ amacrine cells/field in vehicle control (black bar; 2,148 Pax6+ in 10 fields) and TGFβRII-Fc treated (white bar; 4,966 Pax6+ in 15 fields) retinal explants. (n,o) E18.5→8DIV wild-type (n) or Zac1+m/- (o) retinal explants cultured with rTGFβII. Asterisk in (o) indicates ECL formation in Zac1+m/- retinae even in the presence of rTGFβII. (p) Percentage of amacrine cells in wild-type explants (black bar; vehicle control: 1,761 Pax6+/3,353 DAPI+; rTGFβII: 2,605 Pax6+/4,301 DAPI+ in INL+GCL) and Zac1+m/-+ECL explants (white bar; vehicle control: 2,232 Pax6+/3,328 DAPI+; rTGFβII: 3,243 Pax6+/5,282 DAPI+ in INL+GCL).
While the analysis of TGFβRII mutants supported a role for this signaling pathway in regulating amacrine cell number, we were precluded from analyzing the effects of mutating TGFβRII at postnatal stages as the mutants unexpectedly died at early postnatal stages. We therefore used a complementary pharmacological approach to mimic the late reduction in TGFβ signaling observed in Zac1 mutant retinae. The pharmacological inhibition of TGFβII in the early postnatal rat retina increases proliferation and cell number [44], but specific effects on amacrine cell genesis were not analyzed. In accordance with experiments in rat [44], addition of 0.5 μg/ml soluble TGFβRII-Fc receptor to E18.5→8DIV retinal explants resulted in a 1.55-fold increase in INL/GCL cell number compared to vehicle controls (p < 0.01; control: 387.1 ± 35.0 cells/field; n = 3; TGFβRII-Fc: 601.6 ± 62.1 cells/field; n = 3; Figure 8k,l), while 0.1 μg/ml had no effect (not shown). Moreover, the inhibition of TGFβII signaling resulted in a 1.50-fold increase in the absolute number of amacrine cells (p < 0.01; vehicle control: 220.2 ± 5.9 cells/field; n = 3; TGFβRII-Fc: 331.2 ± 15.3 cells/field; n = 3; Figure 8k–m). These results are consistent with a requirement for TGFβ signaling to negatively regulate amacrine cell number during development.
Next, to show that attenuation of TGFβ signaling underlies amacrine cell expansion in Zac1+m/- retinae, we performed a rescue experiment. Recombinant TGFβII (or vehicle control) was added to wild-type and Zac1+m/- E18.5→8DIV explants. In control explants, the percentage of Pax6+ amacrine cells was elevated 1.38-fold in Zac1+m/-+ECL versus wild-type explants (p < 0.01; wild type: 53.6 ± 4.2% INL/GCL cells; n = 4; Zac1+m/-+ECL; 68.7 ± 4.8% INL/ECL/GCL cells; n = 3; Figure 8p). In contrast, following exposure to TGFβII for 8DIV, the percentage of amacrine cells was equivalent in wild-type and Zac1+m/-+ECL explants (wild type: 60.4 ± 2.5% INL/GCL cells; n = 3; Zac1+m/-+ECL: 61.5 ± 2.9% INL/ECL/GCL cells; n = 3; Figure 8n–p). Strikingly, however, an ECL still formed in TGFβII-treated Zac1+m/- explants (Figure 8o), suggesting that an alternative, non-TGFβ-mediated pathway underlies amacrine cell migration defects. This is also consistent with the inability of TGFβRII-Fc to induce an ECL (Figure 8l). These studies implicate attenuated TGFβII signaling in amacrine cell expansion in Zac1+m/- retinae.
Discussion
The development of a functional retina requires that appropriate numbers of each cell type be generated. Hence, the molecular events that guide cell fate specification and differentiation must be tightly coordinated with those that govern cell number control. Here we demonstrate that the Zac1 tumor suppressor is an essential negative regulator of retinal size, controlling the absolute number of rod photoreceptors and amacrine cells generated during development. Strikingly, Zac1 regulates rod and amacrine cell genesis through distinct cell autonomous and non autonomous mechanisms, respectively (Figure 9). While Zac1 is a direct negative regulator of a rod photoreceptor fate, it regulates amacrine cell genesis by controlling the expression of TGFβII, which serves as an amacrine cell negative feedback signal. Zac1 and TGFβII thus join a growing list of tumor suppressor genes with established roles in retinogenesis (for example, Rb, p53, p27Kip1 [33,35,40,44,47-52]), but are the first tumor surveillance molecules shown to control neuronal number through a negative feedback or 'cell sensing' mechanism.
Figure 9.

Model of Zac1 function in the retina. Zac1 negatively regulates amacrine cell number cell non-autonomously, controlling TGFβII expression, which inhibits amacrine cell genesis at threshold levels (negative feedback). In contrast, Zac1 negatively regulates rod number cell-autonomously. Zac1 negatively regulates (either directly or indirectly) the expression of genes involved in the specification/differentiation of an amacrine cell (Pax6, Six3, Foxn4, Math3, NeuroD, Bhlhb5, Barhl2) and rod cell (Rb, Otx2, Crx, Nr2e3, Nrl) fate by controlling the decision by retinal progenitor cells to differentiate along these lineages. AC, amacrine cell; APC, amacrine precursor cell; PPC, photoreceptor precursor cell; RPC, retinal progenitor cell.
Zac1 promotes cell cycle exit and apoptosis in the developing retina
The widespread expression of Zac1 in dividing progenitor cells in the retina (this study) and throughout the developing neural tube [25,53-55] suggested that it would have an early role in neural development. Unexpectedly, we found that in the murine retina, Zac1 function is restricted to the early postnatal period. While we cannot rule out the possibility that Zac1 functions redundantly with other factors to regulate early events in retinal development, we would predict that the tumor suppressor-like properties of Zac1 would have to be actively suppressed during early retinal development as most cells that express Zac1 at these stages continue to divide for some time. Indeed, we show here that Zac1 is required to promote cell cycle exit only at late stages of retinogenesis, a context dependency that is also characteristic of other tumor suppressor genes and oncogenes [56]. Specifically, we show that, in Zac1 mutants, retinal progenitor cells divide excessively, similar to p27Kip1 mutants [35,52] and in contrast to Rb mutants, where committed precursors instead fail to exit the cell cycle [33,47,48]. Our demonstration that cyclin D1 expression is upregulated in Zac1+m/- retinae provides some insight into the molecular mechanisms underlying Zac1-mediated control of the cell cycle. However, several observations make it unlikely that Zac1 functions directly through p27Kip1 or the related cyclin dependent kinase (CDK) inhibitor (CDKI) p57Kip2 to regulate cell cycle exit. Firstly, p27Kip1 is not required in a temporally restricted manner in the retina, and p57Kip2 is only required at early stages of retinal development [35,52,57], which contrasts with the late temporal requirement for Zac1. Furthermore, expression of the Kip family CDKIs was not altered in Zac1 mutants, and while there was an increase in p27Kip1 expression following Zac1 misexpression, it was specific to Müller glia, where this CDKI is normally expressed, and not observed in other cell types. Moreover, a previous cell culture study reported that Zac1 promoted cell cycle exit independently of Kip-family CDKIs or other classic cell cycle regulators such as Rb [24].
Zac1 functions as a direct negative regulator of rod cell fate
The requirement for Zac1 to promote cell cycle exit and apoptosis at late stages of retinal development likely contributes to the formation of hypercellular retinae in mutants, but does not explain why rod photoreceptors and amacrine cells are the only two cell types that are specifically expanded. Strikingly, misexpression of Zac1 robustly inhibited rod differentiation, implicating Zac1 as a bona fide negative regulator of this cell fate. Accordingly, Zac1 expression declines in progenitor cells at P0 when rod photoreceptor genesis begins to peak. Zac1 is also not expressed in differentiated ONL photoreceptors. However, we cannot rule out the possibility that cell non-autonomous mechanisms may also underlie the expansion of the rod pool in Zac1+m/- retinae. Indeed, we found that the generation of excess rods is directly linked to the formation of an ECL, both occuring in the same approximately 55% of Zac1+m/- retinae. Notably, we implicated attenuated TGFβ signaling [58], a proapoptotic pathway, in the amacrine cell expansion. However, reduced TGFβ signaling may also underlie the decreased apoptosis we observed in Zac1+m/- ONLs, consequently contributing to the expansion of the rod pool.
Zac1 misexpression also increased bipolar and Müller glial production in our gain-of-function assays, but rather than proposing that Zac1 is instructive for these fates, we favor the interpretation that progenitor cells prevented from adopting a rod fate instead acquire later-born fates by default. Accordingly, in Zac1+m/- retinae, we did not observe compensatory decreases in bipolar and Müller glial cell number. Nevertheless, in Xenopus, murine Zac1 also promoted Müller glial as well as RGC genesis, suggesting it might be instructive for a glial identity in different vertebrate species [26]. However, there are numerous examples whereby misexpression of a murine gene in Xenopus specifies distinct cell fates compared to misexpression in a mouse model (for example, Mash1 promotes a rod fate when misexpressed in mouse and a bipolar fate in Xenopus [59,60]. Moreover, in a previous study we showed that murine Zac1 unexpectedly promoted proliferation in Xenopus retina [26], in sharp contrast to its ability to promote cell cycle exit in the murine retina (this study) and cell lines in vitro [21,24]. To simplify our model of Zac1 retinal function, we therefore consider results obtained in mouse and Xenopus as independent systems where gene function may differ substantively.
Zac1 regulates amacrine cell production cell non-autonomously
Previous studies based on ablation of mature amacrine cells [14] and aggregation of early progenitors with post-mitotic retinal cells [13] demonstrated that amacrine cell number is regulated by negative feedback, but the molecular mechanisms were unknown. Using similar aggregation assays, we showed that Zac1 is required in postnatal retinal cells to limit the number of amacrine cells generated [14]. With the exception of rods, numbers of all other retinal cells were not grossly perturbed in Zac1 mutants. The loss of amacrine cell negative feedback therefore does not affect later-born cell types, consistent with previous cell aggregation experiments [1,13]. We thus propose a model whereby initial reductions in amacrine cell genesis, beginning at E16 in wild-type retinae, occurs when progenitors switch to the next competence window to make later-born rods, bipolar cells and Müller glia, an event that is Zac1-independent. This would be followed early postnatally by Zac1/TGFβII-regulated feedback inhibition serving as the final signal to halt amacrine cell genesis (Figure 9).
Feedback pathways exist in diverse biological systems, including the counting factor in Dictyostelium, which dictates group size [2], Drosophila miRNA9a, which regulates sensory organ precursor number by downregulating Senseless expression [61], and the well established role of feedback signals in regulating cell number in vertebrate liver, pancreas, olfactory epithelium and retina [2]. Feedback pathways operate by secreting limiting amounts of extrinsic signals that must reach threshold levels to signal cessation of cell genesis [2]. Our data support the idea Zac1 acts in post-mitotic amacrine cells during the postnatal period to regulate TGFβII expression, which in turn suppresses amacrine cell genesis. However, our analysis of TGFβRII mutants also indicates that deleting TGFβ signaling earlier in development (that is, from E16 to E18.5), during the peak period of amacrine cell genesis, can also influence amacrine cell genesis. Invoking a threshold model for TGFβII could help explain why defects in cell cycle exit and expansion of the amacrine cell population were not completely penetrant phenotypes in Zac1 mutants. Indeed, developmental processes are known to be highly sensitive to levels of signaling molecules, and stochastic differences in signaling often account for phenotypic variability [62]. Moreover, abrogation of the feedback pathway regulating sense organ production in Drosophila, through deletion of miR-9a, similarly results in variable expressivity and penetrance of neuronal overproduction [61].
Notably, amacrine cell migration defects and the subsequent formation of an ECL were independent of attenuated TGFβ signaling in Zac1 mutant retinae. While we attribute the generation of an ECL to the mutation of Zac1, it remains a possibility that ECL formation requires both this genetic deletion as well as the loss of RGCs that occurs in retinal explant cultures, a possibility we cannot directly address given that Zac1 mutants die at birth. Another possibility is that Zac1 directly regulates cell migration by controlling the expression of cell adhesion genes, an idea based on a meta-analysis of microarray data in which several extracellular matrix molecules that could potentially modulate cell adhesion/migration were found to be co-regulated with Zac1 [23]. The underlying cause of ECL formation is the subject of current investigations.
Conclusion
Here we demonstrate that Zac1 is an essential negative regulator of retinal size, controlling the absolute number of rod and amacrine cells generated during development. Strikingly, while Zac1 acts as a direct negative regulator of a rod fate, it negatively regulates amacrine cell genesis via TGFβII-mediated negative feedback inhibition. Zac1 and TGFβII are thus the first tumor surveillance molecules shown to control neuronal number through a negative feedback, 'cell sensing' mechanism. In summary, Zac1 regulates cell number and migration in the developing retina, highly reminiscent of its function in the prevention of tumor formation, suggesting that similar cellular and molecular mechanisms may underlie these processes.
Materials and methods
Animals and genotyping
For embryo staging, the day of the vaginal plug was considered E0.5. Generation of the Zac1 mutant allele was previously described [23]. The Zac1 mutant allele was maintained on a C57BL/6 background. Zac1+m/- heterozygous embryos were generated by crossing Zac1+/- heterozygous males to C57BL/6 females. Primers for PCR genotyping (35 cycles; 94° for 1 minute, 60° for 1 minute, and 72° for 1 minute) of Zac1 were: wild type 5': AGTGACTCCCCACCTTCTTTCTG; wild type 3': CTTGCCACATTTTTGACAGCG; mutant 5': TGACCGCTTCCTCGTGCTTTAC; mutant 3': CCCCCCAGAATAGAATGACACC. Genotyping of GLAST::CreERT2 and R26R reporter mice were previously described [46]. The floxed TGFβRII allele was previously reported [45] and was genotyped by PCR (38 cycles; 95° for 30 s, 62° for 30 s, and 72° for 40 s) with: primer 1 5': TGG GGATAGAGGTAGAAAGACATA-3'; primer 2 5': TATGGACTGGCT TTTGTATTC. To induce deletion of the TGFβRII gene, 3 mg of tamoxifen was administered by oral gavage at E16.0 as previously described [46].
RNA in situ hybridization
For RNA in situ hybridization, tissue preparation and experimental procedures were followed as previously described [25]. Briefly, tissue was fixed in 4% paraformaldehyde (PFA)/1X-phosphate buffered saline (PBS) overnight at 4°C, cryopreserved in 20%sucrose/1X PBS overnight at 4°C and embedded in Cryomatrix™ (Anatomical Pathology USA [Pittsburgh, PA, USA]). Digoxygenin (dig)-labelled probes were generated using a dig-UTP labeling mix and T3, T7 or SP6 RNA polymerases according to the manufacturer's instructions (Roche [Laval, QC, Canada]). Mouse probes included Zac1 [25], Hes1, Hes5, Mash1 [63], Ngn2 [64], Math3 [65], Math5 [60], NeuroD [66], Pax6 [67], Rx [68], Crx [69], Chx10 [70]. Foxn4 [42] and Barhl2 [71], Six3 [41] and s-opsin [72].
Immunohistochemistry and histochemistry
For immunohistochemistry, fixation in 4% PFA/1 × PBS was shortened to 1–2 h at 4°C. Primary antibodies were incubated on slides overnight at 4°C or 1 h at room temperature. The following primary antibodies were used: rabbit active-caspase 3 (1/500; Promega [Madison, WI, USA]), mouse Brn3a (1/500; Chemicon [Temecula, CA, USA]), goat anti-Brn3b (1/250; Santa Cruz [Santa Cruz, CA, USA]), mouse anti-BrdU (5-bromo-2'-deoxyuridine, 1/500; Roche), rat-anti-BrdU (1/10; Oxford Biotech [now Antibodies by Design, Raleigh, NC, USA]), rabbit anti-calbindin (1/1,000; SWANT [Bellinzona, Switzerland]), mouse anti-cyclinD1 (1/100; Santa Cruz), rabbit anti-Chx10 (1/50; Rod McInnes), mouse anti-CRALBP (1/5,000; Jack Saari), rabbit anti-GFP (1/500, Chemicon), goat anti-Math3 (1/100, Santa Cruz), mouse anti-neurofilament 200 (1/500; NF200, Sigma [Oakville, ON, Canada]), rabbit anti-Nr2e3 (1/100; Chemicon), rabbit anti-Pax6 (1/500; Babco [Richmond, CA, USA]]), mouse anti-Pax6 (1/4, Developmental Studies Hybridoma Bank [Iowa City, IA, USA]), rabbit anti-p27Kip1 (1/500; NeoMarker Lab Vision, [Freemont, CA, USA] ]), mouse anti-protein kinase C (PKC; 1/500; Sigma), mouse anti-rhodopsin (1/500; Chemicon), mouse anti-syntaxin (1/2,000; Sigma), rabbit anti-TGFβII (1/100; Santa Cruz), rabbit anti-TGFβRI (1/100; Santa Cruz), rabbit anti-TGFβRII (1/100; Santa Cruz), rabbit anti-phospho-Smad2/3 (1/100; Santa Cruz), guinea pig anti-GLAST (1/8,000; Chemicon) and rabbit anti-Zac1 (1/1,000 [24]). Primary antibodies were washed 3 times in PBS with 0.1% triton X-100 (PBT) and detected using secondary antibodies conjugated with Cy3- (1/500; Jackson ImmunoResearch Laboratories, Inc. [West Grove, PA, USA]) or Alexa488 (1/500; Molecular Probes [Invitrogen, Eugene, OR, USA]). Secondary antibodies were diluted in PBT and left on the slides for 1 h prior to 3–10 minute washes with PBT. Note that the TSA™ Tyramide-Fluorescein Immunostaining Kit (NEL701, Perkin-Elmer [Shelton, CT, USA]) was used to amplify anti-TGFβII, TGFβRI, TGFβRII and phospho-Smad2/3 immunostaining as per the manufacturer's instructions. Peanut Agglutinin (PNA) staining was carried out using a 1:200 dilution of the PNA lectin incubated at 37°C for 30 minutes. Sections were then stained for five minutes with DAPI, washed an additional three times with PBS, and mounted with AquaPolymount. β-Galactosidase activity was detected using X-gal as a substrate as previously described [73].
BrdU labeling
To label S-phase progenitors, pregnant females were injected intraperitoneally with 100 μg/g body weight BrdU (Sigma) 30 minutes prior to sacrifice. For birthdating studies, BrdU was added to the culture media at a final concentration of 10 μM. Embryos were processed for anti-BrdU staining as above except for the addition of a pretreatment with 2N HCl for 30 minutes at 37°C. BrdU immunolabeling after RNA in situ hybridization was carried out using 3,3'-diaminobenzidine (DAB) as a substrate using the Vectastain kit (Vector Laboratories Inc. [Burlingame, CA, USA]).
Western blotting
Retinae were lysed for 15 minutes on ice in RIPA buffer (1% SDS, 1% sodium deoxycholate, 0.1% Nonidet P-40 in 50 mM Tris-HCl (pH 7.6)/150 mM NaCl) plus protease (Complete inhibitor tablet, Roche) and phosphatase (5 mM NaF and 1 mM orthovanadate) inhibitors. Cell lysates were cleared and protein concentrations determined via Bradford analysis. Cell free extract (25 μg) was loaded per lane on a 12% (Smad/phospho-Smad) or 15% (TGFβII) SDS-PAGE gel. Protein was then transferred to PVDF membrane at 80 V for 1 h. Membranes were blocked in 5% skim milk powder or 5% bovine serum albumin (for phospho-Smad) in tris-buffered saline with 0.1% tween 20 (TBST) and then incubated with anti-phospho-Smad2/3 (1/200; Santa Cruz), Smad2/3 (1/200; Santa Cruz), TGFβII (1/200; Santa Cruz) or anti-β-actin (1/5,000, AbCam [Cambridge, MA, USA]) overnight at 4°C. Membranes were washed three times for ten minutes each prior to incubation in horse radish peroxidase (HRP)-conjugated secondary antibodies and development with ECL (Roche).
Retinal explants
Retinae were dissected and grown as explants as previously described [74]. Briefly, the retinal pigmented epithelium (RPE) and lens were removed from dissected eyes, and the retina was flattened and cultured GCL-up on a Nucleopore Track-Etch membrane (13 mm; Whatman [Maldstone, England]) in explant media (50% MEM, 25% Hanks Solution, 25% horse serum, 6.75 mg/ml glucose, 200 μM L-glutamine, 2.5 mM HEPES) at 37°C in 5% CO2. The TGFβRII-Fc soluble receptor inhibitor (R&D systems [Burlington, ON, Canada]) was added at 0.5 μg/ml dissolved in PBS (vehicle control) every second day as described [44]. Recombinant TGFβII (R&D systems) was added to explants every second day at 1 ng/ml.
Aggregation assays
Retinae were dissected, dissociated into single cell suspensions and cultured as aggregates essentially as described [13,75]. Briefly, E14.5 wild-type retinae were dissociated in trypsin (10 min/37°C) and triturated in DMEM/10% fetal calf serum with 100 μl DNAseI (2 mg/ml). Dissociated progenitors were labeled in media with 10 μM BrdU for 1 h. BrdU was washed out and cells were resuspended in culture media at 5 × 105 cells/ml. For co-cultures, 100 μl (5 × 104 cells) of labeled E14.5 progenitors were added to a 20-fold excess (1 × 106 cells) of dissociated E18.5 wild-type or Zac1 mutant cells. Aggregated cells were pelleted by centrifuging for 8 minutes at 2,200 rpm and pellets were transferred after 1 h onto Nucleopore membranes and cultured 8DIV. Pellets were then dissociated and plated on poly-D-lysine-coated slides for immunostaining.
Retinal electroporation
For misexpression, full-length Zac1 cDNA [26] was cloned into a pCIG2 expression vector containing a CMV-enhancer/chicken β-actin promoter and IRES-EGFP cassette (gift from Franck Polleux) [76]. For electroporation, eyes were dissected and the RPE removed prior to immersion in 10 μl DNA (3 μg/μl) on a 3% agarose gel plug. Platinum electrodes were placed on either side of the eye (E15.5, 4 mm spacing; and E18.5, 5 mm spacing) and seven 20 ms pulses of 25 V were applied. Electroporated retinae were then cultured as explants.
Cell counts and statistical analysis
Immunoreactive cells were counted in sections adjacent to the optic nerve or site of optic nerve transection in explants. In all experiments, cells were counted from a minimum of three embryos (or explants) and three sections per embryo (or explant). The total number of individual retinae analyzed per experiment (n values) is presented in the results section and the total number of cells counted per experiment is presented in the figure legends. All quantification was done from photomicrographs representing a 0.33 mm × 0.25 mm counting field. Statistical variation was determined using the standard error of the mean (SEM). Statistical significance was calculated using a Student's t-test, individually comparing experimental bars against wild-type or control counts.
Additional data files
The following additional data are available with the online version of this paper. Additional data file 1 is a figure showing that Zac1 is expressed in dividing progenitors at embryonic stages and differentiated cells at postnatal stages. Additional data file 2 is a figure showing Zac1 genotyping and verification of maternal imprinting in the embryonic retina. Additional data file 3 is a figure showing that equivalent numbers of bipolar cells, Müller glia, horizontal cells and cone photoreceptors develop in wild-type and Zac1 mutant retinal explants, while the number of amacrine cells increased in Zac1 mutant retinae. Additional data file 4 is a figure showing that RGC differentiation is unperturbed in Zac1-deficient retinae at E18.5. Additional data file 5 is a figure showing that amacrine cell precursors do not undergo more apoptosis or divide ectopically in Zac1 mutant retinae. Additional data file 6 is a figure showing that misexpression of Zac1 in the retina does not affect amacrine cells genesis. Additional data file 7 is a figure showing that the molecular profile of Zac1-deficient retinal progenitors is unperturbed at E18.5. Additional data file 8 is a figure showing that amacrine cell marker expression domains are expanded in E18.5 Zac1 mutant retinal explants cultured 4 DIV.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
LM carried out the vast majority of the experiments with technical assistance from NK. RC carried out western blot analysis, apoptosis studies and TGFβRII conditional knock-out analysis. AV generated Zac1 knock-out mice in the laboratory of LJ, who also provided Zac1 antiserum and comments on the manuscript. DC generated TGFβRII conditional knock-out embryos in the laboratory of MG. SMF provided intellectual input and comments on the manuscript. The experiments were primarily designed by LM and CS.
Supplementary Material
Zac1 is expressed in dividing progenitors at embryonic stages and differentiated cells at postnatal stages. (a-c) E15.5 retinae co-immunolabeled with anti-Zac1 (red, a,c) and anti-syntaxin (green, b) and merged image (c). (d-f) E15.5 retinae co-immunolabeled with anti-Zac1 (red, d) and anti-BrdU (green, e) and merged image (f). (g,h) Expression of Zac1 transcripts (g) and protein (h) in P21 retinae.
{kind=link}
Zac1 genotyping and verification of maternal imprinting in the embryonic retina. (a) PCR genotyping of wild-type and Zac1 mutant alleles. (b,c) Zac1 immunostaining of E15.5 wild-type and Zac1+m/- mutant retinae revealed a loss of expression in heterozygous embryos carrying a maternal wild-type allele.
{kind=link}
Equivalent numbers of bipolar cells, Müller glia, horizontal cells and cone photoreceptors develop in wild-type and Zac1 mutant retinal explants, while the number of amacrine cells increased in Zac1 mutant retinae. E18.5 wild-type (a,c,e,g,i,k,m,o,q) and Zac1-deficient (b,d,f,h,j,l,n,p,r) retinae were cultured 8DIV and labelled with Chx10 (red, a,b) for bipolar cells, p27Kip1 (red, c,d) and CRALBP (red, e,f) for Müller glia, peanut agglutinin (PNA, green, g,h) and s-opsin (i,j) for cones, Bhlhb5 (green, k,l) for GABAergic amacrine cells, calbindin for horizontal cell bodies (red, with processes in outer plexiform layer; m,n) and AII amacrine cells (deeper in INL; m,n), and GABA (red, o,p) and GlyT1 (red, q,r) for amacrine cell subtypes. Explants were counterstained with DAPI (blue).
{kind=link}
RGC differentiation is unperturbed in Zac1-deficient retinae at E18.5. Brn3a (a,b) and Brn3b (c,d) immunolabeling of RGCs in wild-type (a,c) and Zac1 mutant (b,d) retinae at E18.5. Quantitation of Brn3a (e) and Brn3b (f) expressing cells revealed equivalent numbers of RGCs in wild-type (n = 3 retinae; black bar) and Zac1 mutant (n = 3 retinae; white bar) retinae. Brn3a (p = 0.95; wild-type: 6.4 ± 1.0% retinal cells; 774 Brn3a+/12138 DAPI+; Zac1 mutant: 7.4 ± 0.4%; 743 Brn3a+/10123 DAPI+) and Brn3b (p = 0.23; wild-type: 3.3 ± 0.7%; 269 Brn3b+/6813 DAPI+; Zac1 mutant: 5.0 ± 0.7%; 393 Brn3b+/7818 DAPI+).
{kind=link}
Amacrine cell precursors do not undergo more apoptosis or divide ectopically in Zac1 mutant retinae. (a-d) E10.5 (a,b) and E15.5 (c,d) retinae immunostained for activated caspase-3 (ac-3) (red) in wild-type (a,c) and Zac1+m/-(b,d) embryos. Inserts in c,d are high magnification images of ac-3+ cells. (e-h) Ac-3 (red)/Pax6 (green) double+ cells label apoptotic amacrine cells in E18.5→4DIV explants. g and h are high magnification images of boxed areas in e and f, respectively. Ac-3+ amacrine cells are marked by arrowheads (g,h). (i) Percentage of Pax6+/ac-3+ apoptotic amacrine cells in wild-type (black bars; 45 ac-3/Pax6 double+/2925 Pax6+) and Zac1+m/- (white bars; 22 ac-3/Pax6 double+/2538 Pax6+) E18.5→4DIV explants. (j) Percentage of BrdU+/Pax6+ dividing amacrine cells in total Pax6+ population in E18.5 explants cultured 1DIV, 2DIV and 4DIV. 1DIV (p = 0.40; wild-type: 1.1 ± 0.3%; n = 3 explants; 12 BrdU/Pax6 double+/1071 Pax6+; Zac1 mutant: 0.7 ± 0.2%; n = 3 explants; 10 BrdU/Pax6 double+/1386 Pax6+), 2DIV (p = 0.76; wild-type: 0.9 ± 0.3%; n = 3 explants; 16 BrdU/Pax6 double+/1698 Pax6+; Zac1 mutant: 0.8 ± 0.3%; n = 3 explants; 16 BrdU/Pax6 double+/1983 Pax6+) and 4DIV (p = 0.44; wild-type: 0.2 ± 0.2%; n = 3 explants; 5 BrdU/Pax6 double+/712 Pax6+; Zac1 mutant: 0.4 ± 0.3%; n = 3 explants; 16 BrdU/Pax6 double+/2307 Pax6 single+). Blue is DAPI counterstain.
{kind=link}
Misexpression of Zac1 in the retina does not affect amacrine cells genesis. (a-f) P0 retinae were electroporated with control pCIG2 (a,c,e) or pCIG2-Zac1 (b,d,f) and cultured 8DIV. Electroporated cells were detected by GFP epifluorescence (green; a,b) and amacrine cells were identified by anti- syntaxin (red; c,d). (e,e',f,f') Merged images show similar numbers of GFP-positive electroporated cells that expressed syntaxin (Syn) after control (e) and Zac1 (f) electroporations. Arrowheads indicate electroporated cells that differentiated into amacrine cells. e' and f' are high magnification images of boxed area in e and f. (g) Quantitation of the percentage of electroporated cells that differentiate into amacrine cells after control pCIG2 (black bar; n = 3) or pCIG2-Zac1 (white bar; n = 3) electroporations. pCIG2 at E15.5: 62.3 ± 6.5%; 519 syntaxin/GFP double+/787 GFP+; Zac1 at E15.5: 72.7 ± 5.4%; 379 syntaxin/GFP double+/680 GFP+; pCIG2 at E17.5: 39.3 ± 5.4%; 897 syntaxin/GFP double+/2520 GFP+; Zac1 at E17.5: 51.3 ± 2.0%; 456 syntaxin/GFP double+/928 GFP+; pCIG2 at P0: 11.7 ± 3.4%; 81 syntaxin/GFP double+/552 GFP+; Zac1 at P0: 10.2 ± 2.3%; 83 syntaxin/GFP double+/376 GFP+.
{kind=link}
Molecular profile of Zac1-deficient retinal progenitors is unperturbed at E18.5. RNA in situ hybridization of E18.5 wild-type (non-prime) and Zac1-deficient (prime) retinae with Hes5 (a,a'), Hes1 (b,b'), Rx (c,c'), Chx10 (d,d'), Crx (e,e'), Barhl2 (f,f'), Mash1 (g,g'), Foxn4 (h,h'), NeuroD (i,i'), Math3 (j,j'), Math5 (k,k') and Pax6 (l,l') probes.
{kind=link}
Amacrine cell marker expression domains are expanded in E18.5 Zac1 mutant retinal explants cultured 4 DIV. Marker expression in E18.5 retinal explants cultured 4 DIV from wild-type (a,c,e,g,i,k,m) and Zac1-deficient (b,d,f,h,j,l,n) embryos. Chx10 transcript (a,b) and Chx10 protein (c,d) distribution in retinal explants. Crx (e,f), Hes1 (g,h),Six3 (i,j), Barhl2 (k,l) and Math3(m,n) expression. Explants processed for Hes1 and Six3 RNA in situ hybridization were also immunolabeled with anti-BrdU (after 30 min exposure) to label dividing cells.
{kind=link}
Acknowledgments
Acknowledgements
We thank C Logan, K Markham, R Wevrick, D Eisenstat, M Vetter, V Wallace, F Polleux, D Zinyk, P Mattar, R Slack, R Kageyama, C Cepko, R McInnes, J Wigle, M Xiang, J Saari, T Doetschmann, Y Wang, J Cross, S Hill, S Rawn, R Dixit and T Glaser for reagents, technical assistance and/or critical reading of the manuscript. CS and SM are Alberta Heritage Foundation for Medical Research (AHFMR) Senior Scholars. This work was supported by CIHR (MOP-44094), March of Dimes (FY05-107) and Networks of Centers of Excellence (NCE; Stem Cell Network) grants to CS, by CNRS and European Commission grants (CT-1999-00602) to LJ and by CIHR grant MOP-14138 to SM. LM and RC were supported by CIHR Training Grant in Genetics, Child Development & Health and LM is a William H Davies Scholar.
Contributor Information
Lin Ma, Email: linma@ucalgary.ca.
Robert Cantrup, Email: r.cantrup@ucalgary.ca.
Annie Varrault, Email: annie.varrault@igf.cnrs.fr.
Dilek Colak, Email: dilek.ertuerk@gsf.de.
Natalia Klenin, Email: nklenin@ucalgary.ca.
Magdalena Götz, Email: magdalena.goetz@gsf.de.
Sarah McFarlane, Email: smcfarla@ucalgary.ca.
Laurent Journot, Email: Laurent.journot@igf.cnrs.fr.
Carol Schuurmans, Email: cschuurm@ucalgary.ca.
References
- Pearson BJ, Doe CQ. Specification of temporal identity in the developing nervous system. Annu Rev Cell Dev Biol. 2004;20:619–647. doi: 10.1146/annurev.cellbio.19.111301.115142. [DOI] [PubMed] [Google Scholar]
- Gomer RH. Not being the wrong size. Nat Rev Mol Cell Biol. 2001;2:48–54. doi: 10.1038/35048058. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- Stiemke MM, Hollyfield JG. Cell birthdays in Xenopus laevis retina. Differentiation. 1995;58:189–193. doi: 10.1046/j.1432-0436.1995.5830189.x. [DOI] [PubMed] [Google Scholar]
- Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proc Natl Acad Sci U S A. 1996;93:589–595. doi: 10.1073/pnas.93.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Cepko CL. A common progenitor for neurons and glia persists in rat retina late in development. Nature. 1987;328:131–136. doi: 10.1038/328131a0. [DOI] [PubMed] [Google Scholar]
- Holt CE, Bertsch TW, Ellis HM, Harris WA. Cellular determination in the Xenopus retina is independent of lineage and birth date. Neuron. 1988;1:15–26. doi: 10.1016/0896-6273(88)90205-X. [DOI] [PubMed] [Google Scholar]
- Wetts R, Fraser SE. Multipotent precursors can give rise to all major cell types of the frog retina. Science. 1988;239:1142–1145. doi: 10.1126/science.2449732. [DOI] [PubMed] [Google Scholar]
- Fekete DM, Perez-Miguelsanz J, Ryder EF, Cepko CL. Clonal analysis in the chicken retina reveals tangential dispersion of clonally related cells. Dev Biol. 1994;166:666–682. doi: 10.1006/dbio.1994.1346. [DOI] [PubMed] [Google Scholar]
- Jensen AM, Raff MC. Continuous observation of multipotential retinal progenitor cells in clonal density culture. Dev Biol. 1997;188:267–279. doi: 10.1006/dbio.1997.8645. [DOI] [PubMed] [Google Scholar]
- Alexiades MR, Cepko CL. Subsets of retinal progenitors display temporally regulated and distinct biases in the fates of their progeny. Development. 1997;124:1119–1131. doi: 10.1242/dev.124.6.1119. [DOI] [PubMed] [Google Scholar]
- Cayouette M, Poggi L, Harris WA. Lineage in the vertebrate retina. Trends Neurosci. 2006;29:563–570. doi: 10.1016/j.tins.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Belliveau MJ, Cepko CL. Extrinsic and intrinsic factors control the genesis of amacrine and cone cells in the rat retina. Development. 1999;126:555–566. doi: 10.1242/dev.126.3.555. [DOI] [PubMed] [Google Scholar]
- Reh TA, Tully T. Regulation of tyrosine hydroxylase-containing amacrine cell number in larval frog retina. Dev Biol. 1986;114:463–469. doi: 10.1016/0012-1606(86)90210-1. [DOI] [PubMed] [Google Scholar]
- Waid DK, McLoon SC. Ganglion cells influence the fate of dividing retinal cells in culture. Development. 1998;125:1059–1066. doi: 10.1242/dev.125.6.1059. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hoyuela M, Barbas JA, Rodriguez-Tebar A. The autoregulation of retinal ganglion cell number. Development. 2001;128:117–124. doi: 10.1242/dev.128.1.117. [DOI] [PubMed] [Google Scholar]
- Kim J, Wu HH, Lander AD, Lyons KM, Matzuk MM, Calof AL. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308:1927–1930. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37:197–207. doi: 10.1016/S0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
- Harmon EB, Apelqvist AA, Smart NG, Gu X, Osborne DH, Kim SK. GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development. 2004;131:6163–6174. doi: 10.1242/dev.01535. [DOI] [PubMed] [Google Scholar]
- Tobin JF, Celeste AJ. Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr Opin Pharmacol. 2005;5:328–332. doi: 10.1016/j.coph.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Abdollahi A. LOT1 (ZAC1/PLAGL1) and its family members: mechanisms and functions. J Cell Physiol. 2007;210:16–25. doi: 10.1002/jcp.20835. [DOI] [PubMed] [Google Scholar]
- Mattar P, Britz O, Johannes C, Nieto M, Ma L, Rebeyka A, Klenin N, Polleux F, Guillemot F, Schuurmans C. A screen for downstream effectors of Neurogenin2 in the embryonic neocortex. Dev Biol. 2004;273:373–389. doi: 10.1016/j.ydbio.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, Pavlidis P, Journot L. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, Journot L. Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. Embo J. 1997;16:2814–2825. doi: 10.1093/emboj/16.10.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam S, Zinyk D, Ma L, Schuurmans C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev Dyn. 2005;234:772–782. doi: 10.1002/dvdy.20577. [DOI] [PubMed] [Google Scholar]
- Ma L, Hocking JC, Hehr CL, Schuurmans C, McFarlane S. Zac1 promotes a Muller glial cell fate and interferes with retinal ganglion cell differentiation in Xenopus retina. Dev Dyn. 2007;236:192–202. doi: 10.1002/dvdy.21002. [DOI] [PubMed] [Google Scholar]
- Hiura H, Obata Y, Komiyama J, Shirai M, Kono T. Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells. 2006;11:353–361. doi: 10.1111/j.1365-2443.2006.00943.x. [DOI] [PubMed] [Google Scholar]
- Caffe AR, Visser H, Jansen HG, Sanyal S. Histotypic differentiation of neonatal mouse retina in organ culture. Curr Eye Res. 1989;8:1083–1092. doi: 10.3109/02713688908997401. [DOI] [PubMed] [Google Scholar]
- de Melo J, Qiu X, Du G, Cristante L, Eisenstat DD. Dlx1, Dlx2, Pax6, Brn3b, and Chx10 homeobox gene expression defines the retinal ganglion and inner nuclear layers of the developing and adult mouse retina. J Comp Neurol. 2003;461:187–204. doi: 10.1002/cne.10674. [DOI] [PubMed] [Google Scholar]
- de Araujo EG, Linden R. Trophic factors produced by retinal cells increase the survival of retinal ganglion cells in vitro. Eur J Neurosci. 1993;5:1181–1188. doi: 10.1111/j.1460-9568.1993.tb00972.x. [DOI] [PubMed] [Google Scholar]
- Rich KA, Zhan Y, Blanks JC. Migration and synaptogenesis of cone photoreceptors in the developing mouse retina. J Comp Neurol. 1997;388:47–63. doi: 10.1002/(SICI)1096-9861(19971110)388:1<47::AID-CNE4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Pearson RA, Luneborg NL, Becker DL, Mobbs P. Gap junctions modulate interkinetic nuclear movement in retinal progenitor cells. J Neurosci. 2005;25:10803–10814. doi: 10.1523/JNEUROSCI.2312-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Livne-bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–551. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development. 2000;127:3593–3605. doi: 10.1242/dev.127.16.3593. [DOI] [PubMed] [Google Scholar]
- Levine EM, Close J, Fero M, Ostrovsky A, Reh TA. p27(Kip1) regulates cell cycle withdrawal of late multipotent progenitor cells in the mammalian retina. Dev Biol. 2000;219:299–314. doi: 10.1006/dbio.2000.9622. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Hitomi M, van der Wee K, Rothenberg F, Fisher SA, Zucker R, Svoboda KK, Goldsmith EC, Heiskanen KM, Nieminen AL. The pros and cons of apoptosis assays for use in the study of cells, tissues, and organs. Microsc Microanal. 2002;8:375–391. doi: 10.1017/S1431927602010346. [DOI] [PubMed] [Google Scholar]
- Pei YF, Rhodin JA. The prenatal development of the mouse eye. Anat Rec. 1970;168:105–125. doi: 10.1002/ar.1091680109. [DOI] [PubMed] [Google Scholar]
- Silver J, Hughes AF. The role of cell death during morphogenesis of the mammalian eye. J Morphol. 1973;140:159–170. doi: 10.1002/jmor.1051400204. [DOI] [PubMed] [Google Scholar]
- Hero I. Optic fissure closure in the normal cinnamon mouse. An ultrastructural study. Invest Ophthalmol Vis Sci. 1990;31:197–216. [PubMed] [Google Scholar]
- Duenker N. Transforming growth factor-beta (TGF-beta) and programmed cell death in the vertebrate retina. Int Rev Cytol. 2005;245:17–43. doi: 10.1016/S0074-7696(05)45002-0. [DOI] [PubMed] [Google Scholar]
- Inoue T, Hojo M, Bessho Y, Tano Y, Lee JE, Kageyama R. Math3 and NeuroD regulate amacrine cell fate specification in the retina. Development. 2002;129:831–842. doi: 10.1242/dev.129.4.831. [DOI] [PubMed] [Google Scholar]
- Li S, Mo Z, Yang X, Price SM, Shen MM, Xiang M. Foxn4 controls the genesis of amacrine and horizontal cells by retinal progenitors. Neuron. 2004;43:795–807. doi: 10.1016/j.neuron.2004.08.041. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Furukawa T, Lee JE, Cepko CL. NeuroD regulates multiple functions in the developing neural retina in rodent. Development. 1999;126:23–36. doi: 10.1242/dev.126.1.23. [DOI] [PubMed] [Google Scholar]
- Close JL, Gumuscu B, Reh TA. Retinal neurons regulate proliferation of postnatal progenitors and Muller glia in the rat retina via TGF{beta} signaling. Development. 2005;132:3015–3026. doi: 10.1242/dev.01882. [DOI] [PubMed] [Google Scholar]
- Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–568. doi: 10.1182/blood.V100.2.560. [DOI] [PubMed] [Google Scholar]
- Mori T, Tanaka K, Buffo A, Wurst W, Kuhn R, Gotz M. Inducible gene deletion in astroglia and radial glia--a valuable tool for functional and lineage analysis. Glia. 2006;54:21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gray J, Wu L, Leone G, Rowan S, Cepko CL, Zhu X, Craft CM, Dyer MA. Rb regulates proliferation and rod photoreceptor development in the mouse retina. Nat Genet. 2004;36:351–360. doi: 10.1038/ng1318. [DOI] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004;18:1681–1694. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs WB, Walsh GS, Miller FD. Neuronal survival and p73/p63/p53: a family affair. Neuroscientist. 2004;10:443–455. doi: 10.1177/1073858404263456. [DOI] [PubMed] [Google Scholar]
- Li L, Liu F, Ross AH. PTEN regulation of neural development and CNS stem cells. J Cell Biochem. 2003;88:24–28. doi: 10.1002/jcb.10312. [DOI] [PubMed] [Google Scholar]
- Gomes FC, Sousa Vde O, Romao L. Emerging roles for TGF-beta1 in nervous system development. Int J Dev Neurosci. 2005;23:413–424. doi: 10.1016/j.ijdevneu.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J Neurosci. 2001;21:4259–4271. doi: 10.1523/JNEUROSCI.21-12-04259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente T, Auladell C. Expression pattern of Zac1 mouse gene, a new zinc-finger protein that regulates apoptosis and cellular cycle arrest, in both adult brain and along development. Mech Dev. 2001;108:207–211. doi: 10.1016/S0925-4773(01)00492-0. [DOI] [PubMed] [Google Scholar]
- Tsuda T, Markova D, Wang H, Evangelisti L, Pan TC, Chu ML. Zinc finger protein Zac1 is expressed in chondrogenic sites of the mouse. Dev Dyn. 2004;229:340–348. doi: 10.1002/dvdy.10439. [DOI] [PubMed] [Google Scholar]
- Valente T, Junyent F, Auladell C. Zac1 is expressed in progenitor/stem cells of the neuroectoderm and mesoderm during embryogenesis: differential phenotype of the Zac1-expressing cells during development. Dev Dyn. 2005;233:667–679. doi: 10.1002/dvdy.20373. [DOI] [PubMed] [Google Scholar]
- Baker SJ, McKinnon PJ. Tumour-suppressor function in the nervous system. Nat Rev Cancer. 2004;4:184–196. doi: 10.1038/nrc1297. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. The p57Kip2 cyclin kinase inhibitor is expressed by a restricted set of amacrine cells in the rodent retina. J Comp Neurol. 2001;429:601–614. doi: 10.1002/1096-9861(20010122)429:4<601::AID-CNE7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Franke AG, Gubbe C, Beier M, Duenker N. Transforming growth factor-beta and bone morphogenetic proteins: cooperative players in chick and murine programmed retinal cell death. J Comp Neurol. 2006;495:263–278. doi: 10.1002/cne.20869. [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, Tomita K, Inoue T, Kageyama R. Roles of homeobox and bHLH genes in specification of a retinal cell type. Development. 2001;128:1313–1322. doi: 10.1242/dev.128.8.1313. [DOI] [PubMed] [Google Scholar]
- Brown NL, Kanekar S, Vetter ML, Tucker PK, Gemza DL, Glaser T. Math5 encodes a murine basic helix-loop-helix transcription factor expressed during early stages of retinal neurogenesis. Development. 1998;125:4821–4833. doi: 10.1242/dev.125.23.4821. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang F, Lee JA, Gao FB. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes Dev. 2006;20:2793–2805. doi: 10.1101/gad.1466306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea-Gomez A, Vella FD, Shawlot W, Oulad-Abdelghani M, Chazaud C, Meno C, Pfister V, Chen L, Robertson E, Hamada H, Behringer RR, Ang SL. Nodal antagonists in the anterior visceral endoderm prevent the formation of multiple primitive streaks. Dev Cell. 2002;3:745–756. doi: 10.1016/S1534-5807(02)00321-0. [DOI] [PubMed] [Google Scholar]
- Cau E, Gradwohl G, Casarosa S, Kageyama R, Guillemot F. Hes genes regulate sequential stages of neurogenesis in the olfactory epithelium. Development. 2000;127:2323–2332. doi: 10.1242/dev.127.11.2323. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Fode C, Guillemot F. Restricted expression of a novel murine atonal-related bHLH protein in undifferentiated neural precursors. Dev Biol. 1996;180:227–241. doi: 10.1006/dbio.1996.0297. [DOI] [PubMed] [Google Scholar]
- Takebayashi K, Takahashi S, Yokota C, Tsuda H, Nakanishi S, Asashima M, Kageyama R. Conversion of ectoderm into a neural fate by ATH-3, a vertebrate basic helix-loop-helix gene homologous to Drosophila proneural gene atonal. Embo J. 1997;16:384–395. doi: 10.1093/emboj/16.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Hallonet M, Hollemann T, Pieler T, Gruss P. Vax1, a novel homeobox-containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1999;13:3106–3114. doi: 10.1101/gad.13.23.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker P, Laemle L, Munson A, Kanekar S, Oliver ER, Brown N, Schlecht H, Vetter M, Glaser T. The eyeless mouse mutation (ey1) removes an alternative start codon from the Rx/rax homeobox gene. Genesis. 2001;31:43–53. doi: 10.1002/gene.10003. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Morrow EM, Cepko CL. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91:531–541. doi: 10.1016/S0092-8674(00)80439-0. [DOI] [PubMed] [Google Scholar]
- Liu IS, Chen JD, Ploder L, Vidgen D, van der Kooy D, Kalnins VI, McInnes RR. Developmental expression of a novel murine homeobox gene (Chx10): evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron. 1994;13:377–393. doi: 10.1016/0896-6273(94)90354-9. [DOI] [PubMed] [Google Scholar]
- Mo Z, Li S, Yang X, Xiang M. Role of the Barhl2 homeobox gene in the specification of glycinergic amacrine cells. Development. 2004;131:1607–1618. doi: 10.1242/dev.01071. [DOI] [PubMed] [Google Scholar]
- Yaron O, Farhy C, Marquardt T, Applebury M, Ashery-Padan R. Notch1 functions to suppress cone-photoreceptor fate specification in the developing mouse retina. Development. 2006;133:1367–1378. doi: 10.1242/dev.02311. [DOI] [PubMed] [Google Scholar]
- Beddington RS, Morgernstern J, Land H, Hogan A. An in situ transgenic enzyme marker for the midgestation mouse embryo and the visualization of inner cell mass clones during early organogenesis. Development. 1989;106:37–46. doi: 10.1242/dev.106.1.37. [DOI] [PubMed] [Google Scholar]
- Tomita K, Ishibashi M, Nakahara K, Ang SL, Nakanishi S, Guillemot F, Kageyama R. Mammalian hairy and Enhancer of split homolog 1 regulates differentiation of retinal neurons and is essential for eye morphogenesis. Neuron. 1996;16:723–734. doi: 10.1016/S0896-6273(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Jensen AM, Wallace VA. Expression of Sonic hedgehog and its putative role as a precursor cell mitogen in the developing mouse retina. Development. 1997;124:363–371. doi: 10.1242/dev.124.2.363. [DOI] [PubMed] [Google Scholar]
- Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Zac1 is expressed in dividing progenitors at embryonic stages and differentiated cells at postnatal stages. (a-c) E15.5 retinae co-immunolabeled with anti-Zac1 (red, a,c) and anti-syntaxin (green, b) and merged image (c). (d-f) E15.5 retinae co-immunolabeled with anti-Zac1 (red, d) and anti-BrdU (green, e) and merged image (f). (g,h) Expression of Zac1 transcripts (g) and protein (h) in P21 retinae.
Zac1 genotyping and verification of maternal imprinting in the embryonic retina. (a) PCR genotyping of wild-type and Zac1 mutant alleles. (b,c) Zac1 immunostaining of E15.5 wild-type and Zac1+m/- mutant retinae revealed a loss of expression in heterozygous embryos carrying a maternal wild-type allele.
Equivalent numbers of bipolar cells, Müller glia, horizontal cells and cone photoreceptors develop in wild-type and Zac1 mutant retinal explants, while the number of amacrine cells increased in Zac1 mutant retinae. E18.5 wild-type (a,c,e,g,i,k,m,o,q) and Zac1-deficient (b,d,f,h,j,l,n,p,r) retinae were cultured 8DIV and labelled with Chx10 (red, a,b) for bipolar cells, p27Kip1 (red, c,d) and CRALBP (red, e,f) for Müller glia, peanut agglutinin (PNA, green, g,h) and s-opsin (i,j) for cones, Bhlhb5 (green, k,l) for GABAergic amacrine cells, calbindin for horizontal cell bodies (red, with processes in outer plexiform layer; m,n) and AII amacrine cells (deeper in INL; m,n), and GABA (red, o,p) and GlyT1 (red, q,r) for amacrine cell subtypes. Explants were counterstained with DAPI (blue).
RGC differentiation is unperturbed in Zac1-deficient retinae at E18.5. Brn3a (a,b) and Brn3b (c,d) immunolabeling of RGCs in wild-type (a,c) and Zac1 mutant (b,d) retinae at E18.5. Quantitation of Brn3a (e) and Brn3b (f) expressing cells revealed equivalent numbers of RGCs in wild-type (n = 3 retinae; black bar) and Zac1 mutant (n = 3 retinae; white bar) retinae. Brn3a (p = 0.95; wild-type: 6.4 ± 1.0% retinal cells; 774 Brn3a+/12138 DAPI+; Zac1 mutant: 7.4 ± 0.4%; 743 Brn3a+/10123 DAPI+) and Brn3b (p = 0.23; wild-type: 3.3 ± 0.7%; 269 Brn3b+/6813 DAPI+; Zac1 mutant: 5.0 ± 0.7%; 393 Brn3b+/7818 DAPI+).
Amacrine cell precursors do not undergo more apoptosis or divide ectopically in Zac1 mutant retinae. (a-d) E10.5 (a,b) and E15.5 (c,d) retinae immunostained for activated caspase-3 (ac-3) (red) in wild-type (a,c) and Zac1+m/-(b,d) embryos. Inserts in c,d are high magnification images of ac-3+ cells. (e-h) Ac-3 (red)/Pax6 (green) double+ cells label apoptotic amacrine cells in E18.5→4DIV explants. g and h are high magnification images of boxed areas in e and f, respectively. Ac-3+ amacrine cells are marked by arrowheads (g,h). (i) Percentage of Pax6+/ac-3+ apoptotic amacrine cells in wild-type (black bars; 45 ac-3/Pax6 double+/2925 Pax6+) and Zac1+m/- (white bars; 22 ac-3/Pax6 double+/2538 Pax6+) E18.5→4DIV explants. (j) Percentage of BrdU+/Pax6+ dividing amacrine cells in total Pax6+ population in E18.5 explants cultured 1DIV, 2DIV and 4DIV. 1DIV (p = 0.40; wild-type: 1.1 ± 0.3%; n = 3 explants; 12 BrdU/Pax6 double+/1071 Pax6+; Zac1 mutant: 0.7 ± 0.2%; n = 3 explants; 10 BrdU/Pax6 double+/1386 Pax6+), 2DIV (p = 0.76; wild-type: 0.9 ± 0.3%; n = 3 explants; 16 BrdU/Pax6 double+/1698 Pax6+; Zac1 mutant: 0.8 ± 0.3%; n = 3 explants; 16 BrdU/Pax6 double+/1983 Pax6+) and 4DIV (p = 0.44; wild-type: 0.2 ± 0.2%; n = 3 explants; 5 BrdU/Pax6 double+/712 Pax6+; Zac1 mutant: 0.4 ± 0.3%; n = 3 explants; 16 BrdU/Pax6 double+/2307 Pax6 single+). Blue is DAPI counterstain.
Misexpression of Zac1 in the retina does not affect amacrine cells genesis. (a-f) P0 retinae were electroporated with control pCIG2 (a,c,e) or pCIG2-Zac1 (b,d,f) and cultured 8DIV. Electroporated cells were detected by GFP epifluorescence (green; a,b) and amacrine cells were identified by anti- syntaxin (red; c,d). (e,e',f,f') Merged images show similar numbers of GFP-positive electroporated cells that expressed syntaxin (Syn) after control (e) and Zac1 (f) electroporations. Arrowheads indicate electroporated cells that differentiated into amacrine cells. e' and f' are high magnification images of boxed area in e and f. (g) Quantitation of the percentage of electroporated cells that differentiate into amacrine cells after control pCIG2 (black bar; n = 3) or pCIG2-Zac1 (white bar; n = 3) electroporations. pCIG2 at E15.5: 62.3 ± 6.5%; 519 syntaxin/GFP double+/787 GFP+; Zac1 at E15.5: 72.7 ± 5.4%; 379 syntaxin/GFP double+/680 GFP+; pCIG2 at E17.5: 39.3 ± 5.4%; 897 syntaxin/GFP double+/2520 GFP+; Zac1 at E17.5: 51.3 ± 2.0%; 456 syntaxin/GFP double+/928 GFP+; pCIG2 at P0: 11.7 ± 3.4%; 81 syntaxin/GFP double+/552 GFP+; Zac1 at P0: 10.2 ± 2.3%; 83 syntaxin/GFP double+/376 GFP+.
Molecular profile of Zac1-deficient retinal progenitors is unperturbed at E18.5. RNA in situ hybridization of E18.5 wild-type (non-prime) and Zac1-deficient (prime) retinae with Hes5 (a,a'), Hes1 (b,b'), Rx (c,c'), Chx10 (d,d'), Crx (e,e'), Barhl2 (f,f'), Mash1 (g,g'), Foxn4 (h,h'), NeuroD (i,i'), Math3 (j,j'), Math5 (k,k') and Pax6 (l,l') probes.
Amacrine cell marker expression domains are expanded in E18.5 Zac1 mutant retinal explants cultured 4 DIV. Marker expression in E18.5 retinal explants cultured 4 DIV from wild-type (a,c,e,g,i,k,m) and Zac1-deficient (b,d,f,h,j,l,n) embryos. Chx10 transcript (a,b) and Chx10 protein (c,d) distribution in retinal explants. Crx (e,f), Hes1 (g,h),Six3 (i,j), Barhl2 (k,l) and Math3(m,n) expression. Explants processed for Hes1 and Six3 RNA in situ hybridization were also immunolabeled with anti-BrdU (after 30 min exposure) to label dividing cells.