

Summary
The pathogenicity of many bacteria colonizing the gastrointestinal tract often depends on their ability to gain access to cells that are normally non-phagocytic. Helicobacter pylori colonizes the stomach of over half the world population and is the main cause of peptic ulcer disease and gastric cancer. It is generally considered to be a non-invasive pathogen present only in the lumen of the stomach and attached to gastric epithelial cells although a number of in vivo and in vitro studies have demonstrated that H. pylori is in fact invasive. In addition, H. pylori can repopulate the extracellular environment after complete elimination of extracellular bacteria with gentamicin, suggesting it may be considered a facultative intracellular bacterium. This review examines the validity of these observations and describes the evidence suggesting that the intracellular presence of H. pylori plays a role in the induction of diseases, in immune evasion, and in life-long persistence of the bacterium in the stomach of a majority of humans.
Introduction
The gastric lumen has long been considered a sterile environment, although it is now established that the presence of many bacteria can be detected in the stomach. Since the start of the 20th century, many authors observed the presence of spiral organisms in the stomach and suggested that these bacteria were causing gastric diseases. Their observations were mostly ignored until 1985, when Warren and Marshall demonstrated convincingly that Helicobacter pylori causes gastritis (Marshall et al., 1985a), a discovery for which they received the 2005 Nobel Prize in Physiology or Medicine. In addition, the presence of H. pylori is strongly associated with peptic ulcer disease (Marshall et al., 1985b; Anonymous, 1994a) and gastric cancer (Anonymous, 1994b). Today, we know that H. pylori colonizes the stomach of over half of the word population and that it is consistently associated with chronic active gastritis, although only 15–20% of H. pylori positive subjects experience gastric diseases and some patients with peptic ulcer or gastric cancer appear to be H. pylori negative.
This apparent paradox suggests that the H. pylori presence in the stomach is not sufficient to cause gastric disease and that one or several additional conditions need to be fulfilled. For example, H. pylori has to carry specific virulence genes (e.g. the cag pathogenicity island and the vacA and babA genes) (Peek and Blaser, 2002) and the host may have to be predisposed to develop disorders during infection [e.g. IL-1β or NOD gene polymorphism in certain populations (El-Omar et al., 2000; Rosenstiel et al., 2006)]. In addition, undefined environmental factors that precipitate the occurrence of disease may have to be present, and it is believed that protective factors such as vitamins are protective. Finally, other factors may play a role in H. pylori pathogenicity, life-long persistence, and resistance to antibiotic treatment.
Because the invasiveness of many pathogens is believed to play a role in their virulence and pathogenicity, it is possible that invasiveness plays a role in H. pylori pathogenicity, as reviewed in 2003 (Petersen and Krogfelt, 2003). The present review updates the information available at the time and summarizes the in vivo and in vitro data demonstrating that H. pylori can invade the gastric mucosa and be present within gastric epithelial cells and immunocytes. The review also discusses whether H. pylori invasiveness plays a role in the persistence and pathogenicity of the bacterium.
Observations in H. pylori-infected humans and animals
Helicobacter pylori infection is consistently associated with an intense cellular inflammatory response that is initiated by the innate and adaptive immune systems and is characterized by an influx of neutrophils, mononuclear cells, and T-helper 1 cells. Although this response is typically aimed at clearing intracellular infections (Meylan et al., 2006), the bacterium is not generally considered to be an invasive bacterium and some of the early publications did not report the intracellular presence of the bacterium (Hazell et al., 1986; Hessey et al., 1990; Morris et al., 1990; Thomsen et al., 1990). However, many early ultrastructural studies in patients with gastritis and ulcers demonstrated that, in addition to intimately adhering to epithelial cell surface, ‘Campylobacter-like organisms’ invaded the intercellular spaces and were present within some of the gastric and duodenal epithelial cells, parietal cells and immunocytes (Shousha et al., 1984; Buck et al., 1986; Chen et al., 1986; Tricottet et al., 1986; Bode et al., 1987; 1988); the presence of the bacteria was associated with the formation of pedestals on the surface of gastric epithelial cells, disruption of the mucus granules and of the tight-junction complexes and degeneration of intraepithelial polymorphonuclear neutrophils (PMNs). Subsequent studies confirmed the invasive nature of H. pylori in patients with gastritis, peptic ulcer, precancerous lesions and gastric cancer (Kazi et al., 1990; Wyle et al., 1990; Noach et al., 1994; el Shoura, 1995; Genta et al., 1996; Papadogiannakis et al., 2000; Semino-Mora et al., 2003; Necchi et al., 2007). Similar observations were made in rhesus monkeys naturally infected by H. pylori (Dubois et al., 1994) and in experimental infection of mice (Oh et al., 2005). Finally, the use of confocal microscopy combined with immunohistochemistry (IHC) or fluorescence in situ hybridization (FISH) (Semino-Mora et al., 2003) and transmission electron microscopy (TEM) studies combined with immunogold cytochemistry (Necchi et al., 2007) suggested that intracellular bacteria were viable because intracellular H. pylori observed in patient tissues can express H. pylori mRNA and antigens, and that their ultrastructural appearance is well preserved.
The lack of recognition that H. pylori can be intracellular may be due to the fact that electron microscopy is less adapted to screening large number of samples, is difficult to apply to archived material, and also requires excellent preservation of the samples. In contrast, the use of high-power histochemistry, IHC and FISH is efficient in screening large number of biopsies (including archived tissues) and in characterizing small organisms (Makristathis et al., 2004). When such methods are applied, H. pylori is detectable in the lamina propria and within gastric epithelial cells and immunocytes of patients with gastric diseases (Andersen and Holck, 1990; Genta et al., 1996; Engstrand et al., 1997; Semino-Mora et al., 2003; Necchi et al., 2007).
High-magnification observation of Genta-stained biopsies readily visualizes silver-stained spiral organisms not only in the lumen of the stomach, but also within normal and metaplastic gastric epithelial cells, in the lamina propria, and in post-venous capillaries where they appear to adhere to red blood cells (Fig. 1). In addition, the size and appearance of the silver-stained organisms in the lamina propria are strikingly similar to those of intraluminal H. pylori. However, silver may stain the host’s tissue membranes and is not specific for H. pylori and many pathologists discount such observations. In contrast, confocal microscopy observations of IHC and FISH preparations and ultrastructural studies with immunogold revealed the specific identity of the organisms and their intracellular localization (Semino-Mora et al., 2003; Necchi et al., 2007) and also confirmed H. pylori presence in gastric lamina propria capillaries (Aspholm et al., 2006; Necchi et al., 2007).
Fig. 1.

Gastric biopsy of a patient with intestinal metaplasia (Genta stain, original magnification ×1000). A blue-stained goblet cell is surrounded by more or less normal epithelial cells. Intraluminal H. pylori are either free-floating (1) or adherent to epithelial cells (2), but not to the goblet cell. Rod-, comma- and spiral-shaped H. pylori are visible in the goblet cell (3), in epithelial cells (4), in inflammatory cells (5), in the lamina propria (6), and in a post-capillary venule, where they are closely associated with erythrocytes (7). Interestingly, the silver straining of intraluminal H. pylori appears to be stronger than for intracellular and interstitial bacteria.
In vitro observations in cell cultures of epithelial cells and immunocytes
Helicobacter pylori were also observed inside epithelial cell lines infected with the bacterium (Evans et al., 1992; Segal et al., 1996; Lofman et al., 1997; Su et al., 1999; Amieva et al., 2002; Bjorkholm et al., 2000; Kwok et al., 2002). The fraction of cell-associated H. pylori entering cell lines was insignificant in HeLa cells (Rautelin et al., 1995) and was 0.1% in human embryonic kidney HEK293 (Viala et al., 2004) but was in the 1% range in epithelial cell lines such as gastric cancer AGS cells (Birkness et al., 1996; Amieva et al., 2002). Persistent intraepithelial H. pylori were located within large vacuoles (Amieva et al., 2002) and these bacteria were viable as demonstrated by the acridine orange method (Wilkinson et al., 1998), perhaps because intravacuolar H. pylori are protected against bactericidal components of the lysosomal pathway. In addition, approximately 1% of H. pylori infecting an AGS cell monolayer survived for up to 3 days of continuous gentamicin treatment (an antibiotic that is considered not to enter eukaryotic cells when in concentrations of 200 μg ml−1), and this tolerance was not due to selection for gentamicin resistance mutations. Furthermore, live internalized H. pylori exhibit stop and go directional movements within the confines of the vacuole for up to 3 h, and they can repopulate the extracellular environment upon removal of gentamicin (Amieva et al., 2002). Finally, H. pylori was also able to pass through a bilayer of AGS cells and an endothelial layer (Birkness et al., 1996). This latter observation provides further experimental support for the presence of the bacterium within capillaries of the lamina propria (Aspholm et al., 2006; Necchi et al., 2007), but the clinical significance of the observation is unknown, and H. pylori bacteraemia has been reported in only one case (Ndawula et al., 1994).
At this time, however, there is no direct proof that H. pylori can replicate intracellularly because no time-lapse study has shown H. pylori dividing within cells and colony-forming units (cfu) counts did not increase within AGS cells treated with gentamicin (Amieva et al., 2002). This latter observation indicates that these specific in vitro experimental conditions do not provide a self-sustaining environment but does not completely exclude intracellular replication of H. pylori. Indeed, intracellular cfu counts represent a complex balance between entry, intracellular growth, intracellular death, and exit from cells, and this delicate balance may be disrupted by extracellular gentamicin. In vivo, it is possible that H. pylori temporarily hides intracellularly and then egresses once the danger is passed, similar to what may happen with Campylobacter jejuni, another facultative intracellular organism.
The complex relationship between H. pylori and immunocytes that are tasked with their elimination may help explain why the infection usually persists for the life of the individual. In vitro, phagocytosed H. pylori remained ultrastructurally unimpaired when incubated with polymorphonuclear leucocytes and monocytes in the absence of complement or serum, whereas the addition of complement or serum to the incubation medium led to the destruction of the bacteria (Andersen et al., 1993; Kist et al., 1993). Similarly, addition of immune serum plus an excess of monocytes reduced the number of H. pylori, whereas an excess of PMNs resulted in complete killing of H. pylori (Andersen et al., 1993). However, some of the phagocytosed bacteria were unaffected after incubation with monocytes and serum, and often were surrounded by large aggregates of platelets (Andersen et al., 1993). This latter observation may be relevant to the normalization of platelet count observed in nearly half of patients with immune thrombocytopenic purpura after H. pylori eradication (Kuwana and Ikeda, 2006).
Helicobacter pylori-induced non-opsonic activation of human neutrophils occurs by lectinophagocytosis, i.e. adherence of the sialic acid binding adhesin SabA to neutrophils surface gangliosides (sialylated glycolipids) that activate phagocytosis (Unemo et al., 2005). SabA was recently identified as the long-sought sialic acid-dependent H. pylori haemagglutinin, which aggregates bacterial cells with erythrocytes in vitro. In addition, H. pylori is attached to erythrocytes within small vessels in the gastric mucosa, suggesting that haemagglutination may be of direct biological relevance and mechanistically active in vivo (Aspholm et al., 2006). In the gastric mucosa, H. pylori is often in excess compared with the phagocytes. If the in vitro results reflect the in vivo situation, the phagocytes may be ineffective in H. pylori killing, which may play a role in the persistence of H. pylori in the gastric epithelium.
Similar events have been observed in macrophages. Immunofluorescence and electron microscopy demonstrated that virulent H. pylori containing the cag pathogenicity island were rapidly internalized into actin-rich phagosomes (Allen et al., 2000). Homotypic phagosome fusion then led to formation of ‘megasomes’ containing multiple H. pylori that are viable for ≥ 24 h (Allen et al., 2000). In contrast, H. pylori strains that did not express CagA or VacA were readily phagocytosed and killed, and no megasomes were observed (Allen et al., 2000). Megasome formation and H. pylori survival were shown to depend on urease and urease-derived ammonia, and acidification of phagosomes (Schwartz and Allen, 2006). The intracellular presence of H. pylori within immunocytes was associated with production of apoptosis in T- and B-cell lines (Singh et al., 2006) and in vivo (Semino-Mora et al., 2005), which may play a role in the persistence of the infection (Singh et al., 2006).
Mechanism and time-course of H. pylori invasiveness
Before entry into a cell can occur, H. pylori needs to establish adherence with the epithelial cells, and the two best-described H. pylori adhesins BabA and SabA may be involved. Of particular relevance are the facts that the SabA-mediated binding to gangliosides stimulates G-proteins, activates phagocytosis and oxidative burst reactions of neutrophils and that the associated phosphorylation signalling triggers remodelling of the actin skeleton. The CagA-mediated reorganization of the host cell membrane and signalling probably forms the essential prerequisites for bacterial adhesins to accumulate in the bacterial–host cell interface. The multivalent interactions recruits additional glycolipids to the interface and local membrane density increases, which can lead to raft formation. Such reorganization of the cell membrane with local accumulation of rafts rich in cholesterol moieties may be used by H. pylori because, like some Mycoplasma species, it acquires cholesterol from the host cells. The high density of cholesterol and the resulting reorganization of local membrane melting point might develop to a critical combination that leads to intimate membrane association and phagocytosis (Wunder et al., 2006). Thus, lectinophagocytosis might allow ‘hitchhiking’ of H. pylori into the host cells during periods of activated and intense host responses, and may aid its immune evasion.
Time-lapsed scanning electron microscopy (SEM) and TEM documented the time-course of initial adhesion and subsequent entry of H. pylori into HEp2 cells (Fig. 2) (Lofman et al., 1997). Entry appears to involve a different mechanism in AGS cells, with intimate adhesion of H. pylori to pseudopods emerging from AGS cells, leading to the progressive engulfment of H. pylori in a zipper-like fashion (Fig. 3) (Kwok et al., 2002).
Fig. 2.

A. SEM picture of H. pylori entry in an HEp2 cell (with permission from M. Block, after Engstrand and Graham, 1997). B and C. Time-lapse observation of two H. pylori outlined in red showing the progressive entry into an HEp2 cell over a 49 s time. Adapted by permission from Macmillan Publishers Ltd. Nature Medicine 3: 930–931, copyright 1997.
Fig. 3.
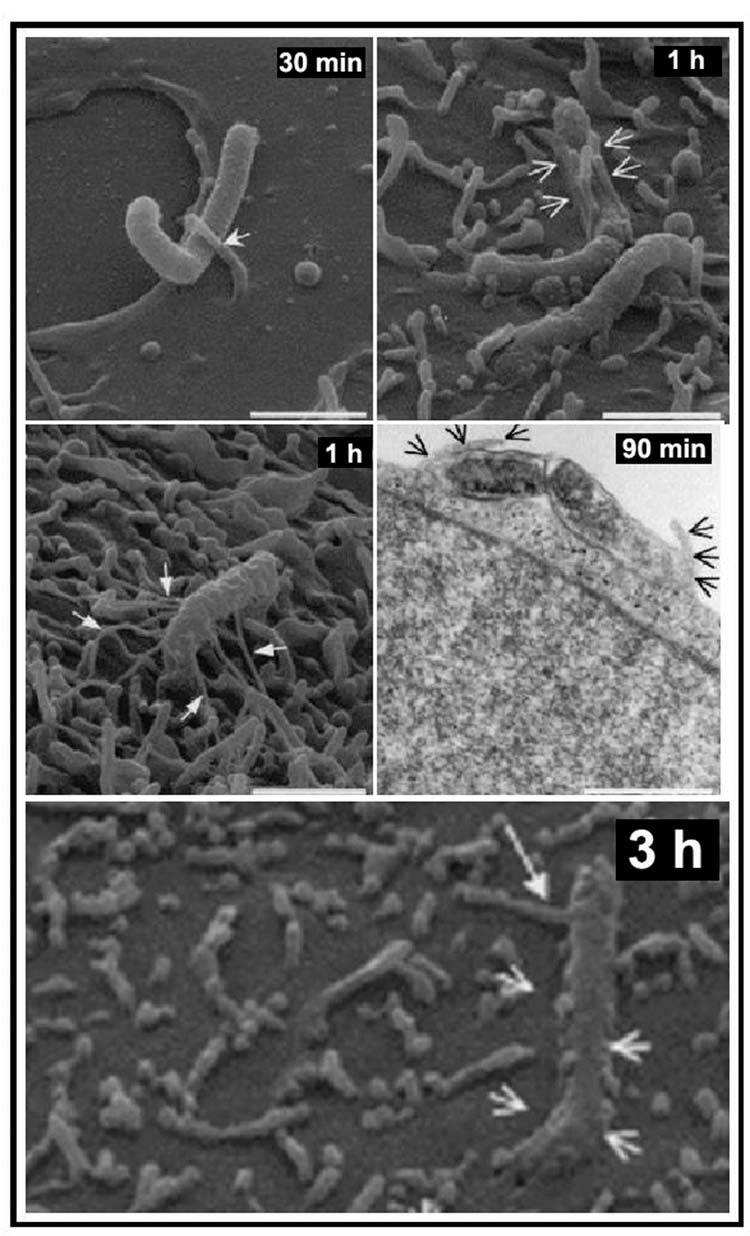
SEM and TEM analyses of H. pylori 26695 adherence to, and entry into, AGS cells. AGS cells infected with H. pylori 26695 at a multiplicity of infection of 100 for 12 h at 37°C. Samples obtained at various times were analysed by SEM and TEM. H. pylori adhered to AGS cells by intimate contact with the host cell microvilli (30 min, arrows). Features of zipper-like engulfment are observed at 1 h, 90 min and 3 h post infection. Bars, 1.5 μm. With permission from Kwok et al. (2002).
Molecular studies showed that entry into epithelial cells was achieved by receptor-mediated endocytosis (Evans et al., 1992) and through an active process of bacterial motility and penetration of the cell membranes (Bjorkholm et al., 2000). The bacteria were then bound in tight vacuoles in close association with condensed filamentous actin and tyrosine phosphorylation signals. These events were inhibited dose-dependently by TNF-α and inhibitors of phosphatidylinositol 3-kinase and of protein kinase C, and enhanced by an inhibitor of tyrosine phosphatases and ATPases (Kwok et al., 2002).
Differential interference contrast video and immunofluorescence microscopy (Fig. 4) illustrated that H. pylori can enter large cytoplasmic vacuoles of multiple epithelial cell lines including gastric and colon adenocarcinoma and kidney cell line (Amieva et al., 2002). Cytochalasin D treatment did not affect vacuole formation but suppressed entry into the cells, suggesting that the actin cytoskeleton plays a role in cell entry (Amieva et al., 2002). Once inside the cells, H. pylori remained motile within vacuoles for several hours, as demonstrated by time-lapse microscopy. Pulsed treatment with gentamicin demonstrated that the half-life of intravacuolar bacteria was approximately 24 h, and that some intravacuolar bacteria remained viable, were released and did repopulate the extracellular environment after gentamicin was removed.
Fig. 4.

Illustration of differential immunostaining of AGS cells cocultured with H. pylori. As illustrated by the yellow arrow, most H. pylori are stained yellow and are located outside the cells. One of the H. pylori is intracellular (red arrow) and is stained red (after Amieva et al., 2002 with permission).
Vacuoles containing H. pylori have the same morphology as late endosomal multivesicular bodies induced by VacA (Amieva et al., 2002). They are formed by the fusion of late endocytic organelles in a VacA-dependent process involving retention of the small GTPase Rab7 and interactions with its downstream effector, the Rab-interacting lysosomal protein (Terebiznik et al., 2006). However, the role of H. pylori virulence factors in cell invasion in vitro is controversial at this time, probably due to differences in experimental design. On one hand, invasion was more prevalent with fresh clinical isolates (Segal et al., 1996) and with strains carrying the cag pathogenicity island (Allen et al., 2000), and a larger proportion of cagA- and VacA-positive strains attached to AGS cell membrane and also invaded the cells (Petersen et al., 2000). Furthermore, H. pylori resided in vacuoles formed through a VacA-dependent process that is responsible for the generation of the large vacuoles containing H. pylori, although VacA did not influence H. pylori capacity to invade AGS cells (Terebiznik et al., 2006). On the other hand, entry and intracellular survival were similar with isogenic vacA mutants and with the wild-type strain, suggesting that neither CagA nor VacA was required for these events to occur (Amieva et al., 2002). Therefore, it is likely that other, yet undefined, H. pylori virulence factors may be involved in cell invasion.
One of those could be an invasin (InvA) protein, similar to the one produced by enteropathogenic Yersinia and that binds to intestinal M cells integrin receptors and allows entry into these cells (Isberg et al., 2000). In the case of H. pylori, the J99 strain has a homologous invasin protein InvA and an invA gene, while similar sequences have been named NudA and nudA in the 26695 strain and also are present in the recently published HPAG1 complete genomes (access numbers CP000241 and NC_008086) although no name has been given (Oh et al., 2006).
The InvA protein was called NudA because it is a Nudix hydrolase. It belongs to the nucleoside polyphosphate hydrolase subgroup that hydrolyses diadenosine tetraphosphate with resulting asymmetrical cleavage of the molecule into ATP and AMP (Lundin et al., 2003). The biological importance of H. pylori NudA enzyme is illustrated by several observations. First, an insertion mutant has a 2.7-fold decrease in survival as compared with the wild type after hydrogen peroxide exposure but there was no difference in survival after heat shock or in spontaneous mutation frequency. In addition, NudA is constitutively expressed in H. pylori at different growth stages and during stress, thus indicating that this protein has a housekeeping function, and all the H. pylori strains tested to date harbour the nudA gene and show protein expression (Lundin et al., 2003). A gentamicin protection assay failed to demonstrate that a quantifiable difference in invasion frequency existed between the nudA mutant and the wild-type strain (Lundin et al., 2003). More recently, however, cell entry of an invA-cam mutant was found to be reduced by 50% relative to the wild-type H. pylori and TEM indicated a sevenfold reduction of intracellular H. pylori. Finally, the number of membrane-bound bacteria was 25-fold greater with the invA-cam mutants than with the wild-type strain (Liu et al., 2007). Thus, deletion of invA appears to limit H. pylori to the AGS cell surface, where, even if partially protected by pseudopodial embrace, it may remain more vulnerable to host defence or therapeutic intervention, or less prone to trigger normal immune or carcinogenic response pathways.
Relevance of H. pylori invasiveness
Because pathogenicity of many invasive bacteria depends on their invasiveness, the in vitro observation that invasion frequency of viable H. pylori is similar to that of Yersinia and greater than that of Shigella (Wilkinson et al., 1998) supports the hypothesis that H. pylori invasiveness does play a role in its pathogenicity. Several clinical observations also are in favour of the hypothesis. First, the association of intramucosal H. pylori with histopathologic features was demonstrated in 104 H. pylori-positive human subjects and in the stomach of 17 mice persistently infected after inoculation with SS1 H. pylori strain (Min et al., 2003). By IHC, intraepithelial H. pylori were observed in 26% of the human biopsies and in 65% of the murine stomachs, and lamina propria H. pylori were present in 48% of the biopsies. Neutrophil-associated immunopositivity for H. pylori was observed in 24% of the biopsies with chronic active gastritis, and lamina propria and neutrophil-associated immunopositivity correlated significantly with the grade of acute inflammatory reaction gastritis. The lamina propria and/or neutrophil-associated H. pylori may play a more important role than intraepithelial H. pylori in the induction of acute inflammatory reactions (Min et al., 2003) and perhaps also in H. pylori persistence. In another study, gastric ulcers were associated with predominantly intracellular and intercellular colonization in 69% of the cases and with predominantly free-in-mucus H. pylori in only 39% of patients (P < 0.01) (Chan et al., 1992). Intracellular H. pylori were also found in the duodenal mucosa of some patients with duodenal ulcer (Bode et al., 1987). Finally, penetration of a large number of H. pylori into gastric epithelial cells was associated with cell damage and cell disintegration both in vivo (el Shoura, 1995) and in vitro (Wilkinson et al., 1998).
In addition, apoptosis of T lymphocytes can be induced through interactions between Yersinia pseudotuberculosis InvA protein and host transmembrane receptors such as β1-integrin (Arencibia et al., 2002). Similarly, interactions between H. pylori invA gene and host cells β1-integrin also may lead to apoptosis, as suggested by a study of gastric biopsies from patients with gastric precancerous and cancerous lesions (Semino-Mora et al., 2005). First, combined ISH (invA) and IHC (β1-integrin) showed that invA was expressed only by epithelial surface-bound and intracellular H. pylori, whereas 16S rRNA and cagA were expressed in all locations. β1-integrin was present in the cytoplasm (cytosol) and in the cellular membrane of mucus-secreting, immune, precancerous goblet, and pleomorphic neoplastic cells. Apoptosis and expression of invA and β1-integrin were all increased in intestinal metaplasia and in gastric cancer compared to gastritis biopsies, and these parameters were significantly correlated with each other (Semino-Mora et al., 2005). These observations suggest that the increased apoptosis in precancerous and cancerous lesions may be mediated by increased expression of H. pylori invA and of the host’s β1-integrin, and that this effect may play a role in persistence of the bacterium and perhaps in its carcinogenic effect.
The presence of H. pylori within epithelial cells may also play a role in the life-long persistence of the infection, as shown by recent experiments demonstrating the presence of intracellular H. pylori within progenitor neck cells in murine (Oh et al., 2005) and primate models (Semino-Mora et al., 2007). In a gnotobiotic transgenic mouse model of human chronic atrophic gastritis, a combination of TEM and multilabel immunohistochemi-cal methods demonstrated that intracellular H. pylori collections were present in dividing and non-dividing gastric epithelial progenitors cells (Oh et al., 2005). The number of intracellular H. pylori ranged from a few solitary bacteria to consolidated populations and to microorganisms traversing breaches in the plasma cell membranes. Importantly, 1–5% of epithelial cells surrounded by H. pylori contain an intracellular population of bacteria. It is possible that these cell-associated clusters of bacteria are formed by intracellular replication, but the authors did not comment on this possibility. In monkeys persistently infected with H. pylori, a FISH study recently demonstrated the presence of intracellular H. pylori within SOX-2 positive neck cells (Semino-Mora et al., 2007). Therefore, it is possible that H. pylori is sheltered from the host immune system within a cell population with a long life span. This stationary haven may be used for the slow-dividing H. pylori to multiply and be present in some of the epithelial cells that migrate towards the surface of the stomach and have a 2–4 days’ life span. These observations also suggest that other invasive pathogens may use a similar strategy to persist in the human gastrointestinal tract.
Finally, intracellular H. pylori are more resistant to antibiotic treatment and only high concentrations of antibiotics with intracellular activity can eliminate the bacterium from HEp2 cells. In vivo, IHC demonstrated that infection was not cured by antibiotics in patients with persistent intracellular H. pylori infection, even if elimination of extracellular bacteria was complete at 14 days after treatment (Engstrand et al., 1997). Thus, antibiotics with intracellular activity (e.g. macrolides) should be used preferentially for the treatment of H. pylori infection (Hulten et al., 1996; Pechere, 2001).
Conclusions
A quarter century after the discovery of H. pylori and the demonstration that the bacterium causes gastric adeno-carcinoma, MALT lymphoma, and peptic ulcers, convincing in vivo evidence demonstrates that the bacterium can enter and express mRNA and antigens within the gastric lamina propria. In addition, in vitro studies have shown that viable H. pylori survive within epithelial cells and immunocytes, exhibit directional movements within intra-cellular vacuoles, and repopulate the extracellular space after the extracellular bacterial population has been killed by gentamicin for up to 3 days. However, direct time-lapse observations have not provided direct proof of intracellular replication of H. pylori by visualization of bacteria dividing within epithelial cells, and no increase in intracellular counts have been observed. In addition, no H. pylori factor is known to be essential for invasion or intracellular survival. Finally, the fraction of intracellular versus intraluminal H. pylori was reported to be approximately 1% in gastrointestinal epithelial cells, and insignificant in HeLa cells, and it is tempting to consider that the invasiveness of the bacterium is not clinically important.
Nonetheless, a small number of bacteria located within the cytoplasm of a few rapidly dividing and strategically positioned cells may have the potential to modify a number of cytoplasmic and nuclear processes and may cause CpG methylation and DNA mutations, especially because virulence genes are known to be expressed within epithelial cells (Semino-Mora et al., 2003; Necchi et al., 2007). Thus, as many cancers are believed to have evolved from one single cell, a few intracellular H. pylori may trigger immune, carcinogenic, or other developmental response pathways, especially when they are present in epithelial progenitor cells that divide rapidly (Oh et al., 2005; Semino-Mora et al., 2007). This process is intuitively attractive to explain neoplastic transformation of epithelial cells leading to adenocarcinoma and of immunocytes leading to MALT lymphoma. Furthermore, complex interactions between H. pylori and immunocytes may moderate the host’s immunity and play an important role in the persistence of the bacterium.
Despite the progress already accomplished, it is clear that additional in vitro and animal studies will have to be performed and the observations will have to be integrated with new clinical observations before we can ascertain that H. pylori invasiveness plays a role in the bacterium pathogenicity, persistence, and resistance to antibiotics.
Acknowledgments
We thank Dr V. Simko for providing gastric biopsies from human subjects and Dr Semino-Mora for providing Fig. 1. We also thank Drs Engstrand, Meyer and Amieva for providing and allowing us to use high-quality pictures used to construct Figs 2–4 respectively.
Supported in part by the National Institutes of Health (CA82312/AD).
References
- Allen LA, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000;191:115–128. doi: 10.1084/jem.191.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva MR, Salama N, Tompkins LS, Falkow S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol. 2002;4:677–690. doi: 10.1046/j.1462-5822.2002.00222.x. [DOI] [PubMed] [Google Scholar]
- Andersen LP, Holck S. Possible evidence of invasiveness of Helicobacter (Campylobacter) pylori. Eur J Clin Microbiol Infect Dis. 1990;9:135–138. doi: 10.1007/BF01963640. [DOI] [PubMed] [Google Scholar]
- Andersen LP, Blom J, Nielsen H. Survival and ultrastructural changes of Helicobacter pylori after phagocytosis by human polymorphonuclear leukocytes and monocytes. APMIS. 1993;101:61–72. [PubMed] [Google Scholar]
- Anonymous. Helicobacter pylori in peptic ulcer disease. NIH Consensus Conference. JAMA. 1994a;272:65–69. [PubMed] [Google Scholar]
- Anonymous. Schistosomes, Liver Flukes and Helicobacter Pylori. Lyon: International Agency for Research on Cancer; 1994b. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans. [PMC free article] [PubMed] [Google Scholar]
- Arencibia I, Frankel G, Sundqvist KG. Induction of cell death in T lymphocytes by invasin via beta1-integrin. Eur J Immunol. 2002;32:1129–1138. doi: 10.1002/1521-4141(200204)32:4<1129::AID-IMMU1129>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Aspholm M, Olfat FO, Norden J, Sonden B, Lundberg C, Sjostrom R, et al. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog. 2006;2:e110. doi: 10.1371/journal.ppat.0020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkness KA, Gold BD, White EH, Bartlett JH, Quinn FD. In vitro models to study attachment and invasion of Helicobacter pylori. Ann NY Acad Sci. 1996;797:293–295. doi: 10.1111/j.1749-6632.1996.tb52983.x. [DOI] [PubMed] [Google Scholar]
- Bjorkholm B, Zhukhovitsky V, Lofman C, Hulten K, Enroth H, Block M, et al. Helicobacter pylori entry into human gastric epithelial cells: a potential determinant of virulence, persistence, and treatment failures. Helicobacter. 2000;5:148–154. doi: 10.1046/j.1523-5378.2000.00023.x. [DOI] [PubMed] [Google Scholar]
- Bode G, Malfertheiner P, Ditschuneit H. Invasion of Campylobacter-like organisms in the duodenal mucosa in patients with active duodenal ulcer. Klin Wochenschr. 1987;65:144–146. doi: 10.1007/BF01728609. [DOI] [PubMed] [Google Scholar]
- Bode G, Malfertheiner P, Ditschuneit H. Pathogenetic implications of ultrastructural findings in Campylobacter pylori related gastroduodenal disease. Scand J Gastroenterol Suppl. 1988;142:25–39. [PubMed] [Google Scholar]
- Buck GE, Gourley WK, Lee WK, Subramanyam K, Latimer JM, DiNuzzo AR. Relation of Campylobacter pyloridis to gastritis and peptic ulcer. J Infect Dis. 1986;153:664–669. doi: 10.1093/infdis/153.4.664. [DOI] [PubMed] [Google Scholar]
- Chan WY, Hui PK, Leung KM, Thomas TM. Modes of Helicobacter colonization and gastric epithelial damage. Histopathology. 1992;21:521–528. doi: 10.1111/j.1365-2559.1992.tb00439.x. [DOI] [PubMed] [Google Scholar]
- Chen XG, Correa P, Offerhaus J, Rodriguez E, Janney F, Hoffmann E, et al. Ultrastructure of the gastric mucosa harboring Campylobacter-like organisms. Am J Clin Pathol. 1986;86:575–582. doi: 10.1093/ajcp/86.5.575. [DOI] [PubMed] [Google Scholar]
- Dubois A, Fiala N, Heman-Ackah LM, Drazek ES, Tarnawski A, Fishbein WN, et al. Natural gastric infection with Helicobacter pylori in monkeys. A model for human infection with spiral bacteria. Gastroenterology. 1994;106:1405–1417. doi: 10.1016/0016-5085(94)90392-1. [DOI] [PubMed] [Google Scholar]
- El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- Engstrand L, Graham DY. Ulcers in a new light. Stockholm: Sparre Medical AB; 1997. Photos by C. Lofman and M. Block. [Google Scholar]
- Engstrand L, Graham D, Scheynius A, Genta RM, El Zaatari F. Is the sanctuary where Helicobacter pylori avoids antibacterial treatment intracellular? Am J Clin Pathol. 1997;108:504–509. doi: 10.1093/ajcp/108.5.504. [DOI] [PubMed] [Google Scholar]
- Evans DG, Evans DJ, Graham DY. Adherence and internalization of Helicobacter pylori by HEp-2 cells. Gastroenterology. 1992;102:1557–1567. doi: 10.1016/0016-5085(92)91714-f. [DOI] [PubMed] [Google Scholar]
- Genta RM, Gurer IE, Graham DY, Krishnan B, Segura AM, Gutierrez O, et al. Adherence of Helicobacter pylori to areas of incomplete intestinal metaplasia in the gastric mucosa. Gastroenterology. 1996;111:1206–1211. doi: 10.1053/gast.1996.v111.pm8898634. [DOI] [PubMed] [Google Scholar]
- Hazell SL, Lee A, Brady L, Hennessy W. Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J Infect Dis. 1986;153:658–663. doi: 10.1093/infdis/153.4.658. [DOI] [PubMed] [Google Scholar]
- Hessey SJ, Spencer J, Wyatt JI, Sobala G, Rathbone BJ, Axon AT, et al. Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut. 1990;31:134–138. doi: 10.1136/gut.31.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulten K, Cars O, Hjelm E, Engstrand L. In-vitro activity of azithromycin in against intracellular Helicobacter pylori. J Antimicrob Chemother. 1996;37:483–489. doi: 10.1093/jac/37.3.483. [DOI] [PubMed] [Google Scholar]
- Isberg RR, Hamburger Z, Dersch P. Signaling and invasin-promoted uptake via integrin receptors. Microbes Infect. 2000;2:793–801. doi: 10.1016/s1286-4579(00)90364-2. [DOI] [PubMed] [Google Scholar]
- Kazi JL, Sinniah R, Zaman V, Ng ML, Jafarey NA, Alam SM, et al. Ultrastructural study of Helicobacter pylori-associated gastritis. J Pathol. 1990;161:65–70. doi: 10.1002/path.1711610111. [DOI] [PubMed] [Google Scholar]
- Kist M, Spiegelhalder C, Moriki T, Schaefer HE. Interaction of Helicobacter pylori (strain 151) and Campylobacter coli with human peripheral polymorpho-nuclear granulocytes. Zentralbl Bakteriol. 1993;280:58–72. doi: 10.1016/s0934-8840(11)80941-2. [DOI] [PubMed] [Google Scholar]
- Kuwana M, Ikeda Y. Helicobacter pylori and immune thrombocytopenic purpura: unsolved questions controversies. Int J Hematol. 2006;84:309–315. doi: 10.1532/IJH97.06188. [DOI] [PubMed] [Google Scholar]
- Kwok T, Backert S, Schwarz H, Berger J, Meyer TF. Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect Immun. 2002;70:2108–2120. doi: 10.1128/IAI.70.4.2108-2120.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Semino-Mora C, Berg DE, Dubois A. Role of the invA invasin gene in H. pylori entry into AGS cells. Gastroenterology. 2007;132 (Suppl 2):S1511. [Google Scholar]
- Lofman C, Rigo R, Block M, Hulten K, Enroth H, Engstrand L. Bacterium–host interactions monitored by time-lapse photography. Nat Med. 1997;3:930–931. doi: 10.1038/nm0897-930. [DOI] [PubMed] [Google Scholar]
- Lundin A, Nilsson C, Gerhard M, Andersson DI, Krabbe M, Engstrand L. The NudA protein in the gastric pathogen Helicobacter pylori is an ubiquitous and constitutively expressed dinucleoside polyphosphate hydrolase. J Biol Chem. 2003;278:12574–12578. doi: 10.1074/jbc.M212542200. [DOI] [PubMed] [Google Scholar]
- Makristathis A, Hirschl AM, Lehours P, Megraud F. Diagnosis of Helicobacter pylori infection. Helicobacter. 2004;9 (Suppl 1):7–14. doi: 10.1111/j.1083-4389.2004.00254.x. [DOI] [PubMed] [Google Scholar]
- Marshall BJ, Armstrong JA, McGechie DB, Glancy RJ. Attempt to fulfill Koch’s postulates for pyloric Campylobacter. Med J Aust. 1985a;142:436–439. doi: 10.5694/j.1326-5377.1985.tb113443.x. [DOI] [PubMed] [Google Scholar]
- Marshall BJ, McGechie DB, Rogers PA, Glancy RJ. Pyloric Campylobacter infection and gastroduodenal disease. Med J Aust. 1985b;142:439–444. doi: 10.5694/j.1326-5377.1985.tb113444.x. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- Min K, Hong SM, Kim KR, Ro JY, Park MJ, Kim JS, et al. Intramucosal Helicobacter pylori in the human and murine stomach: its relationship to the inflammatory reaction in human Helicobacter pylori gastritis. Pathol Res Pract. 2003;199:1–8. doi: 10.1078/0344-0338-00345. [DOI] [PubMed] [Google Scholar]
- Morris A, Ali MR, Thomsen L, Hollis B. Tightly spiral shaped bacteria in the human stomach: another cause of active chronic gastritis? Gut. 1990;31:139–143. doi: 10.1136/gut.31.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndawula EM, Owen RJ, Mihr G, Borman P, Hurtado A. Helicobacter pylori bacteraemia. Eur J Clin Microbiol Infect Dis. 1994;13:621. doi: 10.1007/BF01971319. [DOI] [PubMed] [Google Scholar]
- Necchi V, Candusso ME, Tava F, Lunetti O, Ventura U, Fiocca R, et al. Intracellular, intercellular and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by H. pylori. Gastroenterology. 2007;132:1009–1023. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Noach LA, Rolf TM, Tytgat GN. Electron microscopic study of association between Helicobacter pylori and gastric and duodenal mucosa. J Clin Pathol. 1994;47:699–704. doi: 10.1136/jcp.47.8.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci USA. 2005;102:5186–5191. doi: 10.1073/pnas.0407657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JD, Kling-Backhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc Natl Acad Sci USA. 2006;103:9999–10004. doi: 10.1073/pnas.0603784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadogiannakis N, Willen R, Carlen B, Sjostedt S, Wadstrom T, Gad A. Modes of adherence of Helicobacter pylori to gastric surface epithelium in gastroduodenal disease: a possible sequence of events leading to internalisation. APMIS. 2000;108:439–447. doi: 10.1034/j.1600-0463.2000.d01-80.x. [DOI] [PubMed] [Google Scholar]
- Pechere JC. New perspectives on macrolide antibiotics. Int J Antimicrob Agents. 2001;18 (Suppl 1):S93–S97. doi: 10.1016/s0924-8579(01)00393-4. [DOI] [PubMed] [Google Scholar]
- Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- Petersen AM, Blom J, Andersen LP, Krogfelt KA. Role of strain type, AGS cells and fetal calf serum in Helicobacter pylori adhesion and invasion assays. FEMS Immunol Med Microbiol. 2000;29:59–67. doi: 10.1111/j.1574-695X.2000.tb01506.x. [DOI] [PubMed] [Google Scholar]
- Petersen AM, Krogfelt KA. Helicobacter pylori: an invading microorganism? A review. FEMS Immunol Med Microbiol. 2003;36:117–126. doi: 10.1016/S0928-8244(03)00020-8. [DOI] [PubMed] [Google Scholar]
- Rautelin H, Kihlstrom E, Jurstrand M, Danielsson D. Adhesion to and invasion of HeLa cells by Helicobacter pylori. Zentralbl Bakteriol. 1995;282:50–53. doi: 10.1016/s0934-8840(11)80796-6. [DOI] [PubMed] [Google Scholar]
- Rosenstiel P, Hellmig S, Hampe J, Ott S, Till A, Fischbach W, et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol. 2006;8:1188–1198. doi: 10.1111/j.1462-5822.2006.00701.x. [DOI] [PubMed] [Google Scholar]
- Schwartz JT, Allen LA. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J Leukoc Biol. 2006;79:1214–1225. doi: 10.1189/jlb.0106030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal ED, Falkow S, Tompkins LS. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc Natl Acad Sci USA. 1996;93:1259–1264. doi: 10.1073/pnas.93.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of H. pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis. 2003;187:1165–1177. doi: 10.1086/368133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semino-Mora C, Liu H, Mog SR, Dubois A. Gastric carcinogenesis in rhesus monkeys. Role of a carcinogen and intracellular H. pylori in stem cells. Gastroenterology. 2007;132 (Suppl 2):S1536. [Google Scholar]
- Semino-Mora MC, Liu H, Olsen C, Simko V, Dubois A. H. pylori invA gene, host’s 1-integrin and apoptosis in gastric cancer. Gastroenterology. 2005;128:112. [Google Scholar]
- el Shoura SM. Helicobacter pylori. I. Ultrastructural sequences of adherence, attachment, and penetration into the gastric mucosa. Ultrastruct Pathol. 1995;19:323–333. doi: 10.3109/01913129509064237. [DOI] [PubMed] [Google Scholar]
- Shousha S, Bull TB, Parkins RA. Gastric spiral bacteria. Lancet. 1984;2:101. [Google Scholar]
- Singh M, Prasad KN, Saxena A, Yachha SK. Helicobacter pylori induces apoptosis of T- and B-cell lines and translocates mitochondrial apoptosis-inducing factor to nucleus. Curr Microbiol. 2006;52:254–260. doi: 10.1007/s00284-005-0103-1. [DOI] [PubMed] [Google Scholar]
- Su B, Johansson S, Fallman M, Patarroyo M, Granstrom M, Normark S. Signal transduction-mediated adherence and entry of Helicobacter pylori into cultured cells. Gastroenterology. 1999;117:595–604. doi: 10.1016/s0016-5085(99)70452-x. [DOI] [PubMed] [Google Scholar]
- Terebiznik MR, Vazquez CL, Torbicki K, Banks D, Wang T, Hong W, et al. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect Immun. 2006;74:6599–6614. doi: 10.1128/IAI.01085-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen LL, Gavin JB, Tasman-Jones C. Relation of Helicobacter pylori to the human gastric mucosa in chronic gastritis of the antrum. Gut. 1990;31:1230–1236. doi: 10.1136/gut.31.11.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricottet V, Bruneval P, Vire O, Camilleri JP, Bloch F, Bonte N, et al. Campylobacter-like organisms and surface epithelium abnormalities in active, chronic gastritis in humans: an ultrastructural study. Ultrastruct Pathol. 1986;10:113–122. doi: 10.3109/01913128609014587. [DOI] [PubMed] [Google Scholar]
- Unemo M, spholm-Hurtig M, Ilver D, Bergstrom J, Boren T, Danielsson D, et al. The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J Biol Chem. 2005;280:15390–15397. doi: 10.1074/jbc.M412725200. [DOI] [PubMed] [Google Scholar]
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- Wilkinson SM, Uhl JR, Kline BC, Cockerill FR. Assessment of invasion frequencies of cultured HEp-2 cells by clinical isolates of Helicobacter pylori using an acridine orange assay. J Clin Pathol. 1998;51:127–133. doi: 10.1136/jcp.51.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, et al. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med. 2006;12:1030–1038. doi: 10.1038/nm1480. [DOI] [PubMed] [Google Scholar]
- Wyle FA, Tarnawski A, Schulman D, Dabros W. Evidence for gastric mucosal cell invasion by C. pylori: an ultrastructural study. J Clin Gastroenterol. 1990;12:S92–S98. doi: 10.1097/00004836-199001001-00016. [DOI] [PubMed] [Google Scholar]