

Abstract
We used gene knockout mice to explore the role of Angiopoietin-like-4 (Angptl4) in lipid metabolism as well as to generate anti-Angptl4 mAbs with pharmacological activity. Angptl4 −/− mice had lower triglyceride (TG) levels resulting both from increased very low-density lipoprotein (VLDL) clearance and decreased VLDL production and had modestly lower cholesterol levels. Also, both Angptl4 −/− suckling mice and adult mice fed a high-fat diet showed reduced viability associated with lipogranulomatous lesions of the intestines and their draining lymphatics and mesenteric lymph nodes. Treating C57BL/6J, ApoE −/−, LDLr −/−, and db/db mice with the anti-Angptl4 mAb 14D12 recapitulated the lipid and histopathologic phenotypes noted in Angptl4 −/− mice. This demonstrates that the knockout phenotype reflects not only the physiologic function of the Angptl4 gene but also predicts the pharmacologic consequences of Angptl4 protein inhibition with a neutralizing antibody in relevant models of human disease.
Keywords: monoclonal antibody, mouse, triglycerides, cholesterol
Angiopoietin-like-4 (Angptl4), a member of the angiopoietin family of secreted proteins, is a PPARα and -γ target gene (1–4). Studies of Angptl4 excess or deficiency in mice strongly suggest a role for Angptl4 in the metabolism of lipids, primarily triglycerides (TGs) (5–10). Injecting human Angptl4 (hAngptl4) into KK/San mice raised plasma TG and nonesterified fatty acids (NEFA), but not total cholesterol; the ability of Angptl4 to inhibit lipoprotein lipase (LPL) in vitro suggested this was the mechanism by which Angptl4 raised TG levels in vivo (5). Further studies using adenoviral- (6, 7) or transgene- (8) mediated overexpression of various mammalian Angptl4 forms in liver or transgene-mediated overexpression of rodent Angptl4 in heart (9) or fat and skeletal muscle (10) showed a marked increase in TG and a lesser increase in total cholesterol. The TG increase was due to a rise in circulating very low-density lipoprotein (VLDL) (6, 8, 10). This phenomenon likely resulted from decreased VLDL clearance secondary to the lower LPL activity measured in vivo (8, 9), because hepatic VLDL production was unaffected (6, 10). Consistent with these findings, Angptl4 knockout (−/−) mice exhibited 65–90% lower fasting TG levels and slightly lower total cholesterol levels, in addition to lower circulating VLDL and an increase in LPL activity (4, 8). Importantly, Angptl4 −/− mice were initially reported to be born at the expected Mendelian frequency and to have a normal lifespan without any phenotypic abnormalities, suggesting that exogenous Angptl4 inhibitors would cause a fairly specific fall in serum lipids, primarily TG, without significant on-target side-effects (8). Recent work suggests that Angptl4 may play a similar role in humans, because individuals carrying the Angptl4 E40K variant had significantly lower TG levels and higher high-density lipoprotein (HDL) cholesterol levels in multiple independent populations (11).
To more clearly define the physiologic role of Angptl4, we generated Angptl4 −/− mice, characterized the physiological consequences of Angptl4 inactivation and immunized Angptl4 knockout (−/−) mice to generate anti-Angptl4 mAbs. Angptl4 −/− mice were then studied alongside C57BL/6J, ApoE −/−, LDLr −/−, and db/db mice treated with the anti-Angptl4 mAb 14D12 to demonstrate that both genetic and antibody inhibition of Angptl4 resulted in lower TG levels, due to increased VLDL clearance and decreased VLDL production, and in modestly lower cholesterol levels. Also, both gene knockout and antibody neutralization of Angptl4 were associated with significant injury to the intestines and their draining lymphatics and lymph nodes when mice were maintained on high-fat diet (HFD).
Results
Generation of Angptl4 −/− Mice.
We used our sequence-tagged gene trap library to generate an initial line of Angptl4 −/− mice. Omnibank ES cell clone OST352973 was chosen because of sequence identity with published mouse Angptl4 EST sequence. Inverse genomic PCR of DNA from OST352973 ES cells confirmed that the gene trap vector inserted into intron 2 of mouse Angptl4 (accession NM_02081.1), as shown in supporting information (SI) Fig. 5A. Angptl4 heterozygous (+/−) mice were generated and then intercrossed, yielding viable Angptl4 −/− offspring. RT-PCR analysis revealed the presence of endogenous Angptl4 transcript in kidney and liver of Angptl4 wild-type (+/+) mice, but not in Angptl4 −/− littermates (SI Fig. 5B).
A second line of Angptl4 −/− mice was generated by homologous recombination. Exons 2 and 3 were targeted for deletion, as shown in SI Fig. 6. Targeted clones were confirmed by Southern blot analysis and used to generate Angptl4 +/− mice, which were then intercrossed, yielding viable Angptl4 −/− offspring. Because Angptl4 −/− mice from each line show an identical phenotype for serum lipids and for intestinal pathology, data presented are limited to studies using Angptl4 −/− mice generated by gene trapping.
Development of Monoclonal Antibodies to Angptl4.
Immunization of Angptl4 −/− mice with recombinant mAngptl4 protein resulted in multiple mice showing high serum titers to antigen by ELISA (data not shown). Hybridomas were generated by fusing isolated splenocytes from an immunoresponsive mouse with NS1 myeloma cells. From this fusion, 25 hybridomas were identified that expressed IgGs that specifically bound to mAngptl4 by ELISA.
To refine the anti-Angptl4 mAb set, we established an Angptl4 neutralization assay that was based on the ability of mAngptl4 to inhibit bovine LPL enzymatic activity. Sufficient mAngptl4 was added to the assay to inhibit LPL activity by 50%. We then used these conditions to test the ability of 15 anti-Angptl4 mAbs to rescue LPL activity. Three mAbs including 14D12, an IgG2a isotype, rescued LPL activity by at least 50% (SI Fig. 7).
Angptl4 +/− and −/− Mice and Mice Treated with 14D12 mAb are Hypolipidemic.
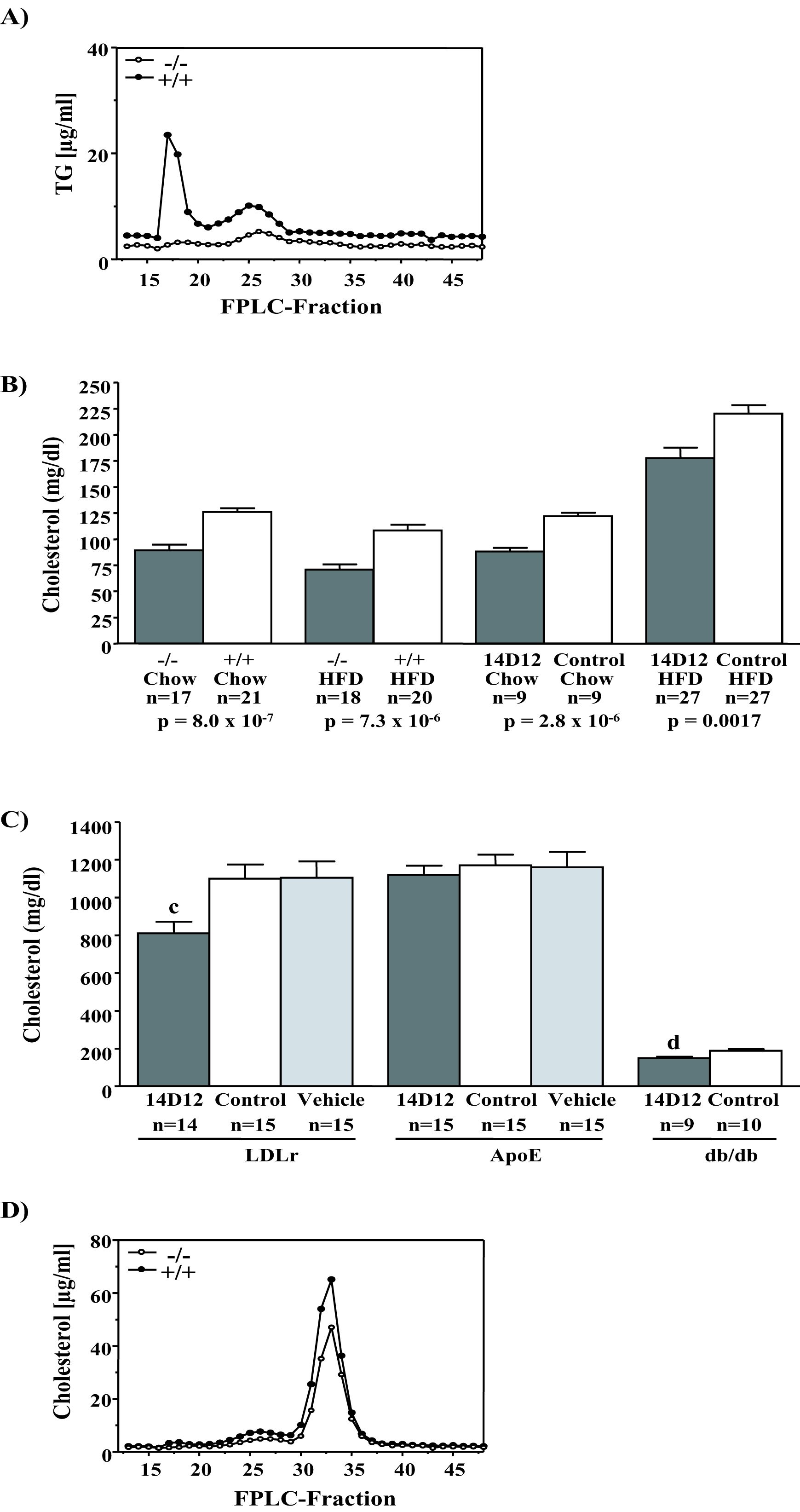
Fasting TG levels were lower in Angptl4 +/− and −/− mice relative to +/+ littermates (Fig. 1A and SI Fig. 8). Also, C57BL/6J mice treated with 14D12 mAb had lower fasting serum TG when compared with mice treated with control mAb (an isotype matched mouse mAb to the irrelevant antigen keyhole limpet hemocyanin) (Fig. 1A). Fractionation of plasma lipoproteins by FPLC showed that the lower TG level in Angptl4 −/− mice was due to less circulating VLDL (SI Fig. 9A). Time course and dose–response studies showed that a maximal sustained lowering of TG was achieved by weekly i.p. injection of 30 mg/kg 14D12 mAb (data not shown); unless otherwise stated, this is the mAb dose used in all studies. Under these conditions, 14D12 mAb lowered fasting TG by 50% and 59% in C57BL/6J mice maintained on chow and HFD, respectively, relative to levels in C57BL/6J mice maintained on the same diets but treated with control mAb (Fig. 1A). These values were similar to the 48% and 77% decreases in fasting TG measured in Angptl4 −/− mice maintained on chow and HFD, respectively. 14D12 mAb also lowered serum TG in LDLr −/−, ApoE −/−, and db/db mice (Fig. 1B).
Fig. 1.

Lower TG levels in Angptl4 −/− mice and mice treated with 14D12 mAb. (A) Fasting total TG levels in Angptl4 −/− and +/+ mice maintained on HFD (TG measured by COBAS) or chow and 4 days after C57BL/6J mice maintained on HFD or chow received 14D12 or control mAb. (B) Fasting total TG levels 4 days after LDLr −/− mice received their 10th weekly dose of 14D12 mAb, control mAb, or vehicle; 4 days after ApoE −/− mice received their 9th weekly dose of 14D12 mAb, control mAb, or vehicle; and 6 days after db/db mice received a single dose of 14D12 or control mAb. The number (n) of mice in each group is shown. a, P < 0.0001; b, P = 0.0003.
Fasting total cholesterol levels trended lower in Angptl4 +/− mice and were significantly lower in Angptl4 −/− mice maintained on chow or HFD (SI Figs. 8 and 9B). 14D12 mAb lowered fasting total cholesterol significantly and to a similar extent in C57BL/6J mice maintained on these same diets (SI Fig. 9B). 14D12 mAb also modestly lowered total serum cholesterol in LDLr −/− and db/db mice, but not in ApoE −/− mice (SI Fig. 9C). Lipoprotein fractionation by FPLC showed that lower cholesterol levels in Angptl4 −/− mice were due mainly to lower HDL (SI Fig. 9D).
Angptl4 −/− Mice and Mice Treated with 14D12 mAb Rapidly Clear TG.
Intragastric loading of olive oil raised serum TG to a greater extent in Angptl4 +/+ mice than in −/− mice. Consistent with this finding, a comparable olive oil challenge increased serum TG to a greater extent in C57BL/6J mice treated with control mAb compared with those receiving 14D12 mAb (Fig. 2A).
Fig. 2.

Increased serum TG clearance and decreased VLDL production in Angptl4 −/− mice and mice treated with 14D12 mAb. (A) Intragastric lipid loading test. (Left) Serum TG measured at baseline (0) and 2, 4, and 6 h after a 15 ml/kg olive oil gavage in overnight-fasted Angptl4 −/− mice (n = 6) and +/+ mice (n = 7). (Right) Four days after receiving either 14D12 (n = 10) or control (n = 10) mAb, overnight-fasted C57BL/6J mice had serum TG measured at baseline (0) and 1, 3, and 6 h after a 15 ml/kg olive oil gavage. (B) i.v. lipid loading test. (Left) Serum TG measured at baseline (0) and 3, 15, and 30 min after Intralipid was given i.v. to overnight-fasted Angptl4 −/− mice (n = 8) and +/+ mice (n = 10). (Right) Four days after receiving either 14D12 (n = 7) or control (n = 9) mAb, overnight-fasted C57BL/6J mice had serum TG measured at baseline (0) and 3, 15, and 30 min after i.v. Intralipid delivery. (C) NEFA levels measured in the same samples presented in B. (D) VLDL production test. (Left) Serum TG measured at baseline (0) and 2, 4, and 6 h after Triton WR1339 was delivered i.v. to overnight-fasted Angptl4 −/− mice (n = 5) and +/+ mice (n = 6). (Right) Four days after receiving either 14D12 (n = 15) or control (n = 15) mAb, overnight-fasted C57BL/6J mice had serum TG measured at baseline (0) and 2, 4, and 6 h after i.v. delivery of Triton WR1339. ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001.
Lower serum TG levels after olive oil challenge in Angptl4 −/− mice and in 14D12 mAb-treated C57BL/6J mice could be due to increased TG clearance or decreased absorption. To test this, TGs were given parenterally by i.v. infusion of Intralipid. Serum TG peaked 3 min after Intralipid infusion and then fell more rapidly in Angptl4 −/− mice than in +/+ mice. A similar increase in TG clearance was seen in C57BL/6J mice treated with 14D12 mAb relative to those receiving control mAb (Fig. 2B). Consistent with the possibility that increased TG clearance is due to increased lipolysis with release of fatty acids, serum NEFA levels 3 min after Intralipid infusion were higher in serum of Angptl4 −/− mice than in +/+ mice and were higher in serum of mice treated with 14D12 mAb relative to those receiving control mAb (Fig. 2C). By 30 min, NEFA levels were lower in the Angptl4 −/− mice and the 14D12 mAb-treated mice relative to controls, consistent with their lower TG levels at this time. As shown in SI Fig. 10, stool analysis found no evidence of fat malabsorption, because there was no difference in fecal fat either between HFD-fed Angptl4 −/− mice vs. +/+ littermates (P = 0.69) or between C57BL/6J mice treated with 14D12 vs. control mAb and then challenged 4 days later with an intragastric olive oil load (P = 0.74).
Angptl4 −/− Mice and Mice Treated with 14D12 mAb Produce Less VLDL.
Low fasting serum TG levels could result from decreased hepatic VLDL production in addition to increased clearance. To test this, mice were injected with Triton WR1339 to inhibit TG clearance. As shown in Fig. 2D, TG levels rose more slowly in serum of Angptl4 −/− mice than in +/+ mice and more slowly in serum of mice treated with 14D12 mAb relative to those receiving control mAb, suggesting a significant decrease in hepatic VLDL production.
Intestinal Abnormalities in Angptl4 −/− Mice and Mice Treated with 14D12 mAb.
Although all Angptl4 +/− intercrosses produced apparently healthy Angptl4 −/− progeny, Mendelian ratios at weaning (549 +/+, 1,095 +/−, 301 −/−) indicated reduced viability of −/− mice. Multiple ill-appearing mice with bloated abdomens were identified preweaning and all were genotyped as −/−. Necropsy revealed that the mucosa of their entire small intestine was thickened, with mild to moderate infiltration of foamy macrophages in the lamina propria (SI Fig. 11 A and B). Foci of ruptured histiocytes and saponified lipids were associated with multinucleated giant cells. Lymphatics and lacteals were frequently dilated (SI Fig. 11C), and lipogranulomatous inflammation was present in the mesentery, draining lymphatics, and mesenteric lymph nodes (SI Fig. 11E).
Surviving Angptl4 −/− mice thrived when weaned onto chow diet and appeared to thrive during the first 20 weeks after weaning onto HFD. After 20 weeks on HFD, however, the −/− mice showed decreased survival relative to +/+ mice weaned onto HFD and also relative to −/− mice weaned onto chow (Fig. 3). Acutely ill mice often had abdominal distention due to accumulation of cloudy fluid with TG levels >2,500 mg/dl, consistent with chylous ascites. The major lesion, present in both healthy- and ill-appearing Angptl4 −/− mice after prolonged exposure to HFD was lipogranulomatous inflammation primarily affecting lymph vessels and mesenteric lymph nodes draining the intestine; the lymphatics were often fibrotic and dilated. Lipogranulomatous inflammation was also present in small intestinal lamina propria but was milder and more chronic than that seen in Angptl4 −/− pups.
Fig. 3.

Decreased survival of Angptl4 −/− mice maintained on HFD. Shown is the percentage of Angptl4 −/− mice fed HFD (n = 69) or chow (n = 76) from weaning and Angptl4 +/+ mice fed HFD (n = 140) or chow (n = 93) from weaning that remained alive at various time points up to 45 weeks of age. Because gender did not affect survival, male and female mice were combined.
ApoE −/− mice on a high-fat Western diet showed abdominal pathology after 15 weekly injections of 30 mg/kg 14D12 mAb. Intestinal mucosal lesions were rare and relatively mild (SI Fig. 11D), but 6 of 15 treated mice had chylous ascites (3 with distended abdomens), and 13 of 15 had pathologic findings in mesenteric lymphatics and lymph nodes identical to those seen in the HFD-challenged Angptl4 −/− mice (SI Fig. 11F). In contrast, none of the 29 ApoE −/− mice showed these abnormalities after 15 weekly injections of vehicle or control mAb. Similar pathology was noted in occasional HFD-challenged LDLr −/− mice and C57BL/6J mice after receiving multiple injections of 14D12 mAb, but never in mice receiving vehicle or control mAb. ApoE mice receiving four weekly injections of 14D12 mAb while on the high fat Western diet developed lipogranulomatous lesions first in the mesenteric lymph nodes draining the small intestine rather than in the intestine or lymphatic vessels.
14D12 mAb Does Not Lower Lipid Levels in Angptl4 −/− Mice.
As shown in Fig. 4A, Angptl4 +/+ mice receiving 14D12 mAb had lower TG and total cholesterol levels compared with those receiving control mAb. In contrast, Angptl4 −/− mice receiving 14D12 mAb showed no difference in TG or total cholesterol levels compared with those receiving control mAb.
Fig. 4.

14D12 mAb lowers lipid levels in Angptl4 −/− mice expressing hAngptl4. (A) Fasting TG and cholesterol (chol) levels 4 days after Angptl4 +/+ and −/− mice received either 14D12 or control mAb. (B) Fasting TG levels measured in Angptl4 −/− mice at baseline on day 0 (d0) and then on day 4 (d4) and day 6 (d6). On d0, Angptl4 −/− mice were infected with Ad5-hAngptl4. After 4 days, serum TG levels were used to divide these mice into two groups with comparable TG levels. These groups then received 90 mg/kg of either 14D12 (n = 5) or control (n = 6) mAb on days 4 and 5.
14D12 mAb Lowers TG in Angptl4 −/− Mice Expressing hAngptl4.
Preliminary studies found that mice infected with Ad5-hAngptl4, a recombinant adenoviral vector expressing hAngptl4, had higher levels of serum TG and serum Angptl4 relative to empty vector and that higher doses of Ad5-hAngptl4 led to higher TG levels. Consistent with these findings, Angptl4 −/− mice infected with Ad5-hAngptl4 had increased serum TG levels (Fig. 4B). Mice that then received 14D12 mAb showed an 86% decrease in TG levels after 2 days, a value significantly greater than the 21% fall in TG noted for mice receiving control mAb (P = 0.0003).
Discussion
The data presented here demonstrate the power of combining knockout and monoclonal antibody technologies to determine the physiologic role of a secreted protein. Both genetic (knockout)- and antibody-mediated inhibition of Angptl4 led to lower serum TG and modestly lower serum cholesterol levels, confirming data reported in Angplt4 −/− mice (8). Also, both genetic and antibody-mediated Angptl4 inhibition led to lower serum TG levels after oral olive oil challenge. This result could represent increased systemic clearance, consistent with the decreased clearance found with Angptl4 overexpression (9, 10), but could also result from lipid malabsorption secondary to intestinal pathology. The fact that Angptl4 −/− mice and C57BL/6J mice treated with 14D12 mAb showed a more rapid drop in serum TG after i.v. lipid challenge indicates that Angptl4 inhibition results in increased TG clearance. The fact that these two groups of mice had increased serum NEFA levels when TG levels were comparable to those of control mice supports published data that LPL-mediated lipolysis underlies the increased clearance (4, 5, 8, 9).
Although Angptl4 inhibition results in lower serum TG levels by increasing clearance, it is possible that decreased hepatic VLDL production contributes to lower TG levels. This possibility is not predicted by published studies, which showed no increase in VLDL production during adenoviral-mediated overexpression of rat Angptl4 in liver (6) or transgene-mediated overexpression of mouse Angptl4 in adipose tissue and skeletal muscle (10). Nevertheless, studies presented here show that both genetic and antibody inhibition of Angptl4 resulted in decreased VLDL production. The reason for this discrepancy is not clear, but the effect of inhibition should be better predicted by direct gene and protein inactivation studies than by extrapolation from overexpression studies.
The mechanism behind the decrease in serum HDL cholesterol was not studied. However, systemic LPL activation after i.v. heparin injection (12) or transgenic LPL overexpression (13–15) is usually associated with decreased HDL cholesterol. Also, adenoviral-mediated overexpression of ApoAV, which may function as an LPL activator, also lowered HDL cholesterol (12). Thus, lower HDL cholesterol may result from the increased LPL activity associated with Angptl4 inhibition.
Lipogranulomatous lesions were often present in Angptl4 −/− mice. Lesions were more abundant in the intestines of suckling mice, whereas in older mice they were more abundant in mesenteric lymph nodes draining the small intestine. Increased mortality was seen in suckling mice and in adult mice fed HFD for at least 20 weeks, suggesting that dietary fat is a critical predisposing factor. The increased mortality in suckling mice confirms a recent study which reported 93% perinatal mortality for Angptl4 −/− mice on a 129/SvJ:C57BL/6J background (16). In both studies, Angptl4 −/− suckling mice displayed abdominal distention and abnormalities of intestinal lymphatics, underscoring the important role played by Angptl4 in intestinal physiology. Lipogranulomatous lesions and lymph node involvement were not described in the −/− mice from the previous study (16), whereas the striking abundance of red blood cells in lymphatic vessels of those −/− mice were not found in the lymphatics of the suckling Angptl4 −/− mice reported here. Further work is needed to reconcile the potential differences between these two lines.
In a number of independent HFD-fed mouse lines, long-term inhibition of Angptl4 with 14D12 mAb recapitulated the phenotype of lipogranulomatous lesions seen in HFD-fed Angptl4 −/− mice. This finding suggests that the lipogranulomatous lesions are an on-target result of Angptl4 inhibition, and that Angptl4 plays a role in the intestine that is not duplicated by other proteins. Although the mechanism behind this phenotype is unknown, the association of HFD intake with these lesions suggests that, without Angptl4, increased lipase activity may catalyze premature release of fatty acids from intestinal lipids before and/or during lymphatic transport. Chylous ascites and early mortality also occur in mice deficient for the related protein Angpt2 due to abnormal remodeling and maturation of lymphatic vessels (17). Short-term antibody studies presented here suggest that the lipogranulomatous lesions are first associated with the mesenteric lymph nodes draining the small intestine rather than with the lymphatic vessels themselves. This finding implies that lymph leakage and chylous ascites develops in Angptl4 −/− mice due to upstream obstruction of lymphatic flow rather than to a primary impairment in the integrity of lymphatic vessels as occurs in Angpt2 −/− mice.
The lipid phenotype of Angptl4 −/− mice was highly predictive for the target-based effects of the anti-Angptl4 mAb. This observation supports the hypothesis that knockouts are useful for modeling the pharmacologic activity of agents that inhibit the action of specific proteins in vivo (18–20). Also, the Angptl4 −/− mice provided additional advantages in the study of Angptl4 physiology. First, their lack of Angptl4 made them ideal for mAb generation because breaking immune tolerance was not an issue, resulting in a variety of anti-Angptl4 mAbs, including those like 14D12 mAb, which neutralized Angptl4 activity in vitro. Second, Angptl4 −/− mice allowed direct testing of whether the lipid-lowering effects of 14D12 mAb were on-target. Indeed, the ability of 14D12 mAb to lower TG and cholesterol in Angptl4 +/+ mice, but not −/− mice, suggested that the entire lipid-lowering effect of 14D12 was due to specific inhibition of Angptl4. This is important because knockout of the closely related family member Angptl3 also results in lower TG levels and because mice lacking both Angptl3 and Angptl4 have nearly undetectable TG levels (8). The inability of 14D12 mAb to lower TG in Angptl4 −/− mice suggests that the TG-lowering effect of 14D12 mAb is not, even in part, due to Angptl3 inhibition. Third, adenoviral-mediated overexpression of hAngptl4 in Angptl4 −/− mice showed that the human form of this protein, which is secreted by the liver, raises TG levels in mice in vivo, confirming previous findings (5, 8). Fourth, Angptl4 −/− mice that overexpressed hAngptl4 responded to 14D12 mAb with lower TG levels, suggesting that knockout mice may provide a useful model for testing whether mAbs regulate the in vivo function of the human form of their target protein.
Humans heterozygous for the Angptl4 variant E40K were recently found to have significantly lower fasting plasma TG levels in three independent studies (11). E40K homozygotes identified in the two largest studies had TG levels that were even lower than those measured in heterozygotes, reminiscent of the pattern of TG levels reported here for Angptl4 +/− and −/− mice. Also in the two largest studies, HDL cholesterol levels were significantly higher in heterozygotes. The association of high TG and low HDL cholesterol with increased risk of coronary artery disease is well established (21, 22), suggesting that Angptl4 inhibition may have utility in lowering the risk of adverse cardiovascular events for patients with this lipid profile. Although Angptl4-null alleles exist in humans (11), the intestinal pathology of Angptl4 −/− mice has not been reported. If humans with complete Angptl4 deficiency are not at risk for the intestinal complications found in Angptl4 −/− mice, then the appropriate Angpltl4 neutralizing antibody could have therapeutic applications in dyslipidemia and cardiovascular disease.
Materials and Methods
Generation of Angptl4 Knockout Mice.
The Angptl4 knockout mice were produced in a collaboration between Genentech (South San Francisco, CA) and Lexicon Pharmaceuticals to analyze the function of ≈500 secreted and transmembrane proteins. The first line of Angptl4-deficient mice was generated by gene trapping. Methods for gene trapping in ES cells, identification of trapped genes by using OmniBank sequence tags (OSTs), and characterization of retroviral gene-trap vector insertion points are published in refs. 23–26. OmniBank ES cell clone OST352973 was used to generate Angptl4 −/− mice as described in ref. 24. All mice were of mixed genetic background (129/SvEvBrd and C57BL/6J). To confirm mutation of the Angptl4 locus, RNA was extracted from kidney and liver of Angptl4 −/− and littermate +/+ mice, and RT-PCR was performed as described in ref. 24. Genotyping was performed on tail DNA by quantitative dot blots (Bio-Rad, Hercules, CA) as described in refs. 25 and 26. A second line of Angptl4 −/− mice was generated by homologous recombination. The conditional targeting vector, derived using the Lambda KOS system (27), targeted exons 2 and 3 for deletion as described in SI Methods.
Mouse Care and Study.
All procedures involving animals were conducted in conformity with Institutional Animal Care and Use Committee guidelines that are in compliance with state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals (28). Unless stated otherwise, adult male mice were used. Mice were housed at 24°C on a fixed 12-h light/12-h dark cycle and had free access to water and rodent chow. Angptl4 +/+, Angptl4 −/−, and C57BL/6J mice were fed either chow (product no. 5021; Purina, St. Louis, MO) or 45% HFD (D12451i; Research Diets, New Brunswick, NJ). LDLr −/− mice (catalog no. 002207; The Jackson Laboratory, Bar Harbor, ME) were fed Clinton diet (D12107; Research Diets), ApoE −/− mice (catalog no. APOE-M; Taconic, Hudson, NY) were fed Western diet (D12079B; Research Diets), and db/db mice (BKS.Cg-m +/+ Leprdb/ J, catalog no. 000642; The Jackson Laboratory) were fed chow diet.
Recombinant Adenovirus Construction and Virus Dosing of Mice.
Full-length hAngptl4 cDNA was inserted into the Ad E1-deleted region of pAd5(dE1) under the control of the CMV promoter. To generate control vector, a similar construct without the expression cassette was used. Recombinant adenoviruses Ad5-hAngptl4 and Ad5-empty were generated, propagated, and purified as described in ref. 29. Angptl4 −/− mice received 5 × 108 infectious units by tail vein injection.
mAb Generation, Purification, and Effect on LPL Activity.
Methods for immunization, hybridoma generation and scale-up, and mAb purification are provided in SI Methods. The ability of individual mAbs to rescue Angptl4-inhibited LPL activity was tested using a modification of an assay (30) that measured 3H-labeled oleic acid released by LPL-mediated hydrolysis of glycerol tri[9,10(n)-3H]oleate, as described in SI Methods.
Angptl4 Protein Production and Purification.
Methods for Angptl4 expression in CHO cells and purification from conditioned medium are provided as SI Methods.
Analyses of Serum and Plasma Lipids.
Blood samples for lipid analysis were obtained from the retro-orbital plexus. Total cholesterol levels were measured using a COBAS Integra 400 serum chemistry analyzer (Roche, Indianapolis, IN). TG levels were occasionally measured by COBAS but usually by kit (Serum TG determination kit, catalog no. TR0100; Sigma–Aldrich, St. Louis, Missouri). This kit estimates either true TG by measuring only glycerol released from TG by lipoprotein lipase or total TG by measuring this glycerol fraction plus free glycerol. Unless stated otherwise, TG values represent true TG. NEFA levels were measured enzymatically by kit (NEFA-C kit, catalog no. 994-75409; Wako Chemicals, Richmond, VA). Size distribution of plasma lipoproteins was determined on samples from fasted mice by using FPLC as described in SI Methods.
Intragastric Lipid Loading Test.
After an overnight fast, mice received 15 ml/kg olive oil by oral gavage. Blood samples for TG levels were drawn at baseline and various time points up to 6 h after olive oil gavage.
IV Lipid Loading Test.
After an overnight fast, mice received 150 μl of Intralipid (phospholipid-stabilized soybean oil as 20% fat emulsion, catalog no. I-141; Sigma–Aldrich) i.v. via tail vein. Blood samples for TG and NEFA were obtained at baseline and at 3, 15, and 30 min after Intralipid infusion.
In Vivo VLDL Production Test.
After an overnight fast, mice received 100 μl of Triton WR1339 [20% (wt/vol) solution in 0.9% NaCl, catalog no. T-8761; Sigma–Aldrich] i.v. via tail vein. Blood samples for TG were obtained at baseline and at 2, 4, and 6 h after Triton WR1339 infusion.
Pathology.
Tissues collected from −/− mice and age-matched +/+ mice were immersion fixed in 10% neutral buffered formalin. Bones and skulls were decalcified in 10% EDTA (pH 6.95) for 7 days, and all tissues were embedded in paraffin, sectioned at 4 μm, mounted on positively charged glass slides (Superfrost Plus; Fisher Scientific, Pittsburgh, PA), and stained with H&E for histopathologic examination.
Statistics.
Data are presented as the mean ± SEM unless otherwise stated. Comparisons between two groups were analyzed by unpaired Student's t test. Comparisons among multiple groups were analyzed by one-way ANOVA followed by Scheffé posthoc test.
Supplementary Material
Abbreviations
- Angptl
angiopoietin-like
- hAngptl4
human Angptl4
- HDL
high-density lipoprotein
- HFD
high-fat diet
- LPL
lipoprotein lipase
- mAb
monoclonal antibody
- NEFA
nonesterified fatty acids
- TG
triglycerides
- VLDL
very low-density lipoprotein.
Footnotes
Conflict of interest statement: All authors are or have been employed by Lexicon Pharmaceuticals, Inc., and may own publicly traded stock in Lexicon. The research was supported by Lexicon.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705041104/DC1.
References
- 1.Yoon JC, Chickering TW, Rosen ED, Dussault B, Qin Y, Soukas A, Friedman JM, Holmes WE, Spiegelman BM. Mol Cell Biol. 2000;20:5343–5349. doi: 10.1128/mcb.20.14.5343-5349.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, Gonzalez FJ, Desvergne B, Wahli W. J Biol Chem. 2000;275:28488–28493. doi: 10.1074/jbc.M004029200. [DOI] [PubMed] [Google Scholar]
- 3.Ge H, Cha JY, Gopal H, Harp C, Yu X, Repa JJ, Li C. J Lipid Res. 2005;46:1484–1489. doi: 10.1194/jlr.M500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida K, Shimizugawa T, Ono M, Furukawa H. J Lipid Res. 2002;43:1770–1772. doi: 10.1194/jlr.c200010-jlr200. [DOI] [PubMed] [Google Scholar]
- 6.Ge H, Yang G, Yu X, Pourbahrami T, Li C. J Lipid Res. 2004;45:2071–2079. doi: 10.1194/jlr.M400138-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Xu A, Lam MC, Chan KW, Wang Y, Zhang J, Hoo RL, Xu JY, Chen B, Chow WS, Tso AW, et al. Proc Natl Acad Sci USA. 2005;102:6086–6091. doi: 10.1073/pnas.0408452102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koster A, Chao YB, Mosior M, Ford A, Gonzalez-DeWhitt PA, Hale JE, Li D, Qiu Y, Fraser CC, Yang DD, et al. Endocrinology. 2005;146:4943–4950. doi: 10.1210/en.2005-0476. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Burgess SC, Ge H, Wong KK, Nassem RH, Garry DJ, Sherry AD, Malloy CR, Berger JP, Li C. Proc Natl Acad Sci USA. 2005;102:1767–1772. doi: 10.1073/pnas.0409564102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Muller M, Kersten S. J Biol Chem. 2006;281:934–944. doi: 10.1074/jbc.M506519200. [DOI] [PubMed] [Google Scholar]
- 11.Romeo S, Pennacchio LA, Fu Y, Boerwinkle E, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Nat Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaap FG, Rensen PC, Voshol PJ, Vrins C, van der Vliet HN, Chamuleau RA, Havekes LM, Groen AK, van Dijk KW. J Biol Chem. 2004;279:27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- 13.Levak-Frank S, Weinstock PH, Hayek T, Verdery R, Hofmann W, Ramakrishnan R, Sattler W, Breslow JL, Zechner R. J Biol Chem. 1997;272:17182–17190. doi: 10.1074/jbc.272.27.17182. [DOI] [PubMed] [Google Scholar]
- 14.Zsigmond E, Scheffler E, Forte TM, Potenz R, Wu W, Chan L. J Biol Chem. 1994;269:18757–18766. [PubMed] [Google Scholar]
- 15.Liu MS, Jirik FR, LeBoeuf RC, Henderson H, Castellani LW, Lusis AJ, Ma Y, Forsythe IJ, Zhang H, Kirk E, et al. J Biol Chem. 1994;269:11417–11424. [PubMed] [Google Scholar]
- 16.Backhed F, Crawford PA, O'Donnell D, Gordon JI. Proc Natl Acad Sci USA. 2007;104:606–611. doi: 10.1073/pnas.0605957104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, et al. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 18.Zambrowicz BP, Sands AT. Nat Rev Drug Discov. 2003;2:38–51. doi: 10.1038/nrd987. [DOI] [PubMed] [Google Scholar]
- 19.Zambrowicz BP, Turner CA, Sands AT. Curr Opin Pharmacol. 2003;3:563–570. doi: 10.1016/j.coph.2003.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Powell DR. Obesity Rev. 2006;7:89–108. doi: 10.1111/j.1467-789X.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- 21.Manninen V, Tenkanen L, Koskinen P, Hurttunen JK, Manttari M, Heinonen OP, Frick MH. Circulation. 1992;85:37–45. doi: 10.1161/01.cir.85.1.37. [DOI] [PubMed] [Google Scholar]
- 22.Hopkins PN, Wu LL, Hunt SC, Brinton EA. J Am Coll Cardiol. 2005;45:1003–1012. doi: 10.1016/j.jacc.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 23.Zambrowicz BP, Friedrich GA, Buxton EC, Lilleberg SL, Person C, Sands AT. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- 24.Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, BeltrandelRio H, Buxton EC, Edwards J, Finch RA, et al. Proc Natl Acad Sci USA. 2003;100:14109–14114. doi: 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, Mathur BN, Turner CA, Geske R, Montgomery CA, et al. Mol Cell Biol. 2001;21:4829–4836. doi: 10.1128/MCB.21.14.4829-4836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Powell DR, Desai U, Sparks MJ, Hansen G, Gay J, Schrick J, Shi ZZ, Hicks J, Vogel P. Pediatr Nephrol. 2005;20:432–440. doi: 10.1007/s00467-004-1696-5. [DOI] [PubMed] [Google Scholar]
- 27.Wattler S, Kelly M, Nehls M. Biotechniques. 1999;26:1150–1160. doi: 10.2144/99266rr02. [DOI] [PubMed] [Google Scholar]
- 28.Council NR. Guide for the Care and Use of Laboratory Animals. Washington, DC: Natl Acad Press; 1996. [Google Scholar]
- 29.Hitt M, Bett AJ, Prevec L, Graham FL. In: Cell Biology: A Laboratory Handbook. Celis JE, editor. Vol 1. San Diego: Academic; 1998. pp. 500–512. [Google Scholar]
- 30.Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, Ueda K, Inaba T, Minekura H, Kohama T, et al. J Biol Chem. 2002;277:33742–33748. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}