

Summary
The triglycerides in chylomicrons are hydrolyzed by lipoprotein lipase (LpL) along the luminal surface of the capillaries. However, the endothelial cell molecule that facilitates chylomicron processing by LpL has not yet been defined. Here, we show that glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 (GPIHBP1) plays a critical role in the lipolytic processing of chylomicrons. Gpihbp1-deficient mice exhibit a striking accumulation of chylomicrons in the plasma, even on a low-fat diet, resulting in milky plasma and plasma triglyceride levels as high as 5,000 mg/dl. Normally, Gpihbp1 is expressed highly in heart and adipose tissue, the same tissues that express high levels of LpL. In these tissues, GPIHBP1 is located on the luminal face of the capillary endothelium. Expression of GPIHBP1 in cultured cells confers the ability to bind both LpL and chylomicrons. These studies strongly suggest that GPIHBP1 is an important platform for the LpL-mediated processing of chylomicrons in capillaries.
Introduction
Dietary fats in mammals are packaged by the intestine into chylomicrons, which are large, triglyceride-rich lipoproteins (Havel and Kane, 2001). After reaching the bloodstream, the triglycerides within chylomicrons are hydrolyzed by lipoprotein lipase (LpL) along the luminal surface of capillaries, mainly in heart, skeletal muscle, and adipose tissue (Havel and Kane, 2001; Nestel et al., 1962). In those tissues, LpL is synthesized and secreted by the parenchymal cells (i.e., myocytes and adipocytes) and then translocated to the lumen of capillaries, where lipolytic processing of chylomicrons occurs (Goldberg, 1996). The liver also secretes triglyceride-rich lipoproteins called very low density lipoproteins (VLDL). The initial phase of VLDL metabolism closely resembles that of chylomicrons, with LpL-mediated hydrolysis of triglycerides in the heart, skeletal muscle, and adipose tissue (Havel and Kane, 2001; Nestel et al., 1962).
LpL is thought to bind to the surface of capillaries through the interaction of its positively charged heparin-binding domains (Sendak and Bensadoun, 1998) with negatively charged heparan sulfate proteoglycans (HSPGs) expressed on the surface of endothelial cells (Goldberg, 1996; Parthasarathy et al., 1994). This concept stems from the observation that LpL can be released from the capillary endothelium with an injection of heparin (Korn, 1955), and also from the fact that normal endothelial cells bind more LpL than endothelial cells depleted of HSPGs (Shimada et al., 1981). However, the precise endothelial cell molecules required for the lipolytic processing of chylomicrons have remained obscure (Goldberg, 1996).
A deficiency in LpL causes familial chylomicronemia, which is characterized by extremely high levels of triglycerides in the plasma (>2,000 mg/dl) (Brunzell and Deeb, 2001). Familial chylomicronemia can also be caused by a deficiency in apolipoprotein CII, a cofactor for LpL (Bengtsson and Olivecrona, 1979; Breckenridge et al., 1978). No cases of chylomicronemia have yet been ascribed to defects in endothelial cells affecting the binding of either chylomicrons or LpL.
In the current study, we examined the functional importance of glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 (GPIHBP1) in plasma lipid metabolism. GPIHBP1 was initially identified, by expression cloning in Chinese hamster ovary (CHO) cells, as a glycosylphosphatidylinositol (GPI)-anchored cell-surface protein that conferred the ability to bind high density lipoproteins (Ioka et al., 2003). GPIHBP1 contains a highly negatively charged amino-terminal domain (with 17 of 25 consecutive residues in the mouse sequence being glutamate or aspartate) followed by a Ly-6 motif containing multiple cysteines (Ioka et al., 2003).
Because GPIHBP1 was identified as an HDL-binding protein, Ioka and coworkers (Ioka et al., 2003) speculated that it might be involved in reverse cholesterol transport. Here, we show that GPIHBP1 is a protein within the lumen of capillaries that plays a critical role in the lipolytic processing of triglyceride-rich lipoproteins.
Results
Generation of Gpihbp1-deficient mice
To investigate the biological role of GPIHBP1 in plasma lipid metabolism, we examined Gpihbp1-deficient mice (Gpihbp1−/− mice). Gpihbp1−/− mice were produced by replacing all of the exons and introns of Gpihbp1 with lacZ and neo cassettes (Figure 1A). Southern blots showing gene-targeting events are shown in Supplementary Online Figure 1. Northern blots confirmed a complete absence of Gpihbp1 transcripts in Gpihbp1−/− mice (Figure 1B). By western blot, GPIHBP1 could be detected in the adipose tissue of wild-type mice but not of Gpihbp1−/− mice (Figure 1C). Although the targeted Gpihbp1 allele contained a lacZ reporter, lacZ transcripts were undetectable in knockout mice (Supplementary Online Figure 2), and no β-galactosidase activity could be detected. The reason for the absence of lacZ expression in the Gpihbp1 knockout mice is not known with certainty, but presumably relates to the removal of important regulatory elements contained within the gene's introns.
Figure 1.
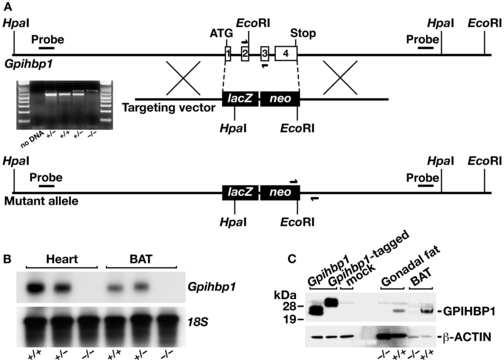
Generation of Gpihbp1 knockout mice. (A) Sequence-replacement gene-targeting strategy for inactivating Gpihbp1. Locations of the 5′ and 3′ probes (bars) for Southern blot analysis and PCR primers (arrows) for genotyping are shown. An ethidium-bromide stained agarose gel illustrates a PCR strategy for genotyping Gpihbp1+/+, Gpihbp1+/−, and Gpihbp1−/− mice. A description of the PCR genotyping strategy is contained in the Experimental Procedures. (B) A northern blot, probed with a Gpihbp1 cDNA probe spanning the complete open reading frame of Gpihbp1, shows the absence of Gpihbp1 expression in heart and brown adipose tissue (BAT) from Gpihbp1−/− mice. (C) Western blot with a GPIHBP1-specific rabbit antiserum showing GPIHBP1 in brown adipose tissue (BAT) and gonadal fat pad extracts from Gpihbp1+/+ mice but not Gpihbp1−/− mice. As a control, extracts from HeLa cells that had been transfected with an untagged or an S-protein–tagged GPIHBP1 construct were included.
Gpihbp1-deficient mice manifest severe chylomicronemia
Both male and female Gpihbp1−/− mice had milky plasma on a normal chow diet (Figure 2A). This phenotype appeared as early as four to five weeks of age, and was invariably present by 14 weeks of age. The plasma triglyceride levels were significantly increased at all ages, with nearly all mice having plasma triglyceride levels over 1,000 mg/dl by 7–10 weeks of age, and some having triglyceride levels as high as 5,000 mg/dl (Figure 2B). Prior to weaning, the plasma triglyceride levels in Gpihbp1−/− pups were significantly elevated compared with wild-type mice, but not markedly so (120 ± 12 vs. 23 ± 16 mg/dl; P = 0.0023). The plasma triglyceride levels in Gpihbp1+/− mice were indistinguishable from those in Gpihbp1+/+ mice (Figure 2B). Plasma cholesterol levels were also elevated in Gpihbp1−/− mice (Supplementary Online Figure 3A).
Figure 2.
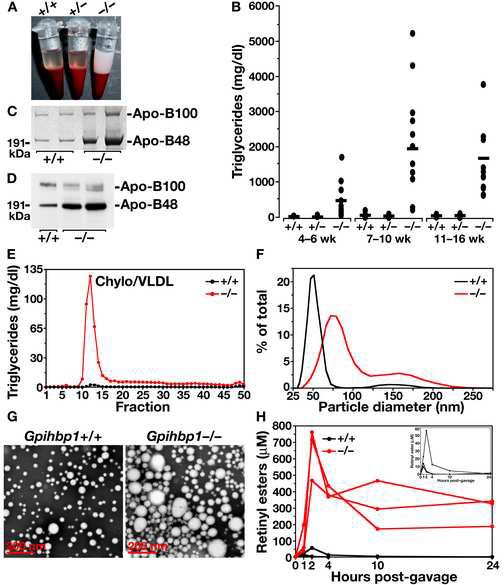
Analysis of Gpihbp1 knockout mice. (A) Photograph of blood samples from three two-month-old littermate chow-fed male mice (after low-speed centrifugation to sediment blood cells), revealing “milky” plasma in Gpihbp1−/− mice. (B) Plasma triglyceride levels in Gpihbp1+/+, Gpihbp1+/−, and Gpihbp1−/− mice at different ages, showing higher triglyceride levels in Gpihbp1−/− mice (P < 0.0001 in each age group). Triglyceride levels in Gpihbp1+/+ and Gpihbp1+/− mice were not different. (C) Coomassie blue–stained SDS-polyacrylamide gel of the d < 1.006 g/ml lipoproteins of wild-type and Gpihbp1−/− mice. (D) Western blot of mouse plasma (1.0 μl) with a mouse monoclonal antibody specific for apo-B48 and apo-B100 (Nguyen et al., 2006), showing increased amounts of apo-B48 in the plasma of Gpihbp1−/− mice. (E) Distribution of triglycerides in the plasma lipoproteins of Gpihbp1+/+ and Gpihbp1−/− mice. Plasma lipoproteins were separated by size on a Superose 6 FPLC column. Representative of two different experiments. (F) Distribution of lipoprotein sizes in the d < 1.022 g/ml lipoproteins from Gpihbp1−/− and Gpihbp1+/+ mice, as judged by dynamic laser light scattering methods. The median diameter of lipoproteins in Gpihbp1−/− mice (n = 3) was 157% larger than in Gpihbp1+/+ mice (n = 6). Shown here are representative data from a single Gpihbp1−/− and Gpihbp1+/+ mouse. Both Gpihbp1+/+ and Gpihbp1−/− mice had bimodal distributions of lipoproteins; the larger subpopulation of particles in Gpihbp1−/− mice, constituting 15.4% of the total particles, had diameters of 122–289 nm; in Gpihbp1+/+ mice, the larger population of particles, constituting 2.9% of particles, were 94–204 nm. The smaller subpopulation of particles in Gpihbp1−/− and Gpihbp1+/+ mice had diameters of 39–111 and 33–86 nm, respectively. (G) Electron micrographs of negatively stained d < 1.006 g/ml lipoproteins from the plasma of Gpihbp1−/− and Gpihbp1+/+ mice, showing the larger lipoproteins in Gpihbp1−/− mice. Representative images from two Gpihbp1−/− and two Gpihbp1+/+ samples. (H) Delayed clearance of retinyl esters in Gpihbp1−/− mice. Retinyl palmitate (5,000 IU in 50 μl of vegetable oil) was administered to mice by gavage, and retinyl esters in the plasma were measured over the next 24 h. The inset shows the retinyl ester levels in Gpihbp1+/+ mice plotted on a smaller scale, showing that the levels in Gpihbp1+/+ mice peaked between 1 and 3 h and had largely disappeared by 10 h. Retinyl palmitate clearance studies were also performed in Gpihbp1+/− mice, and they were indistinguishable from those of Gpihbp1+/+ mice. The retinyl palmitate clearance studies were performed in 7 Gpihbp1−/−, 6 Gpihbp1+/−, and 5 Gpihbp1+/+ mice. Shown here are representative curves for 3 Gpihbp1−/− and Gpihbp1+/+ mice. When the retinyl ester measurements for all of the mice were analyzed, the differences between the Gpihbp1−/− mice and the other two groups were statistically significant at each time point (P < 0.001). There were no differences between Gpihbp1+/− and Gpihbp1+/+ mice.
The plasma lipoproteins of adult Gpihbp1−/− mice contained increased amounts of apo-B48, as judged by a Coomassie blue–stained SDS-polyacrylamide gel (Figure 2C) and by a western blot with a mouse apo-B-specific monoclonal antibody (Nguyen et al., 2006) (Figure 2D). Fractionation of the lipoproteins by FPLC revealed that the vast majority of the triglycerides in the plasma of Gpihbp1−/− mice was in large lipoproteins (i.e., in the “chylomicron/VLDL” peak) (Figure 2E). Most of the cholesterol in the plasma was also located in the chylomicron/VLDL peak (Supplementary Online Figure 3B). The HDL cholesterol levels in Gpihbp1−/− mice were low (Supplementary Online Figure 3B), as is generally the case with hypertriglyceridemia (Brunzell and Deeb, 2001). The diameters of the triglyceride-rich lipoproteins in the plasma of Gpihbp1−/− mice were extremely large, much larger than in wild-type mice, as judged by laser light scattering (Figure 2F) and electron microscopy (Figure 2G).
The milky plasma in Gpihbp1−/− mice, the increased plasma levels of apoB48, and the large lipoproteins suggested a defect in the lipolytic processing of chylomicrons. To determine if the processing of chylomicrons were abnormal, a retinyl palmitate clearance study was performed. Following the oral administration of retinyl palmitate, retinyl esters are packaged into chylomicrons in the intestine and enter the circulation; the disappearance of these retinyl esters from the plasma indicates the rate of chylomicron clearance. In wild-type mice, plasma retinyl ester levels peaked after 1–3 h and had largely disappeared by 10 h (Figure 2H). In the Gpihbp1−/− mice, the peak retinyl ester levels were more than 10-fold higher than those in Gpihbp1+/+ mice, and very high levels persisted for 24 h (Figure 2H).
GPIHBP1 is expressed on the luminal surface of capillary endothelial cells in lipolytic tissues
The finding of chylomicronemia in mice lacking a GPI-anchored protein led us to hypothesize that GPIHBP1 might be involved in the processing of triglyceride-rich lipoproteins by LpL at the surface of capillary endothelial cells. In support of this idea, the tissue pattern of GPIHBP1 expression closely resembled that of LpL and CD36 [a putative fatty acid transporter (Greenwalt et al., 1995)]. By northern blot analysis, all three genes were expressed at particularly high levels in adipose tissue and heart (Supplementary Online Figure 4). In addition, immunofluorescence microscopy revealed that GPIHBP1 is expressed exclusively in endothelial cells, colocalizing with the endothelial cell marker CD31 (Figure 3A). Confocal immunofluorescence microscopy showed that GPIHBP1 was located on the luminal face of capillaries, both in brown adipose tissue and heart (Figures 3B and 3C). GPIHBP1 was also expressed on the capillaries of skeletal muscle, but in multiple experiments was undetectable in the capillaries of the brain (Supplementary Online Figure 5), a tissue that relies heavily on glucose uptake rather than on lipolysis of triglyceride-rich lipoproteins.
Figure 3.
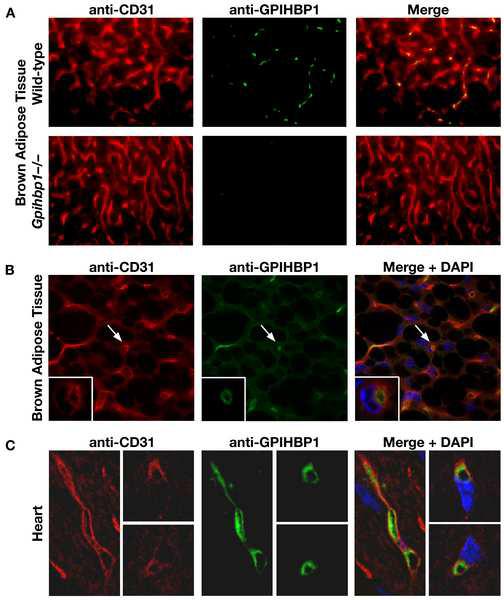
GPIHBP1 is located within the lumen of the capillary endothelium of brown adipose tissue and heart. (A) Epifluorescence microscopy showing the binding of a rat monoclonal antibody against mouse CD31 (red) and a rabbit antiserum against GPIHBP1 (green) to brown adipose tissue of Gpihbp1+/+ and Gpihbp1−/− mice. Images were taken with a 40× objective. (B) Confocal microscopy images showing the binding of antibodies against CD31 and GPIHBP1 to brown adipose tissue from a Gpihbp1+/+ mouse. Images were taken with a 100× objective. The arrow indicates the location of a capillary (shown at higher magnification in the insert; 100× objective with 4× digital zoom). The high-magnification micrograph shows a cross-section of a capillary, revealing that GPIHBP1 staining is particularly prominent on the luminal side of the capillary. (C) Confocal microscopy images showing the binding of antibodies against CD31 and GPIHBP1 to heart tissue from a Gpihbp1+/+ mouse. GPIHBP1 staining is particularly prominent on the luminal face of the capillary endothelium. Images were taken with a 100× objective.
Post-heparin lipoprotein lipase activity is reduced in Gpihbp1-deficient mice
We considered the hypothesis that GPIHBP1 might be the sole binding site for LpL in capillaries and that the amount of LpL bound to capillaries might be dramatically reduced in Gpihbp1−/− mice. To examine this possibility, we measured the levels of LpL and hepatic lipase (HL) in the plasma after an injection of heparin (which releases LpL from capillaries into the circulation). To avoid methodological problems associated with measuring lipase activity in lipemic serum, we separated LpL and HL in post-heparin plasma by heparin-Sepharose chromatography and then quantified the HL and LpL activities (Figure 4). In four independent experiments with age and sex-matched plasma samples, LpL activity was reduced by 53.3, 27.2, 16.6 and 68.4% in Gpihbp1−/− mice when compared to wild-type mice, and the LpL to HL ratio in the Gpihbp1−/− mice was reduced by 71.3, 36.1, 56.6, and 90.4% (P = 0.03). The HL activities tended to be higher in the Gpihbp1−/− mice, but these changes did not achieve statistical significance (P > 0.05).
Figure 4.
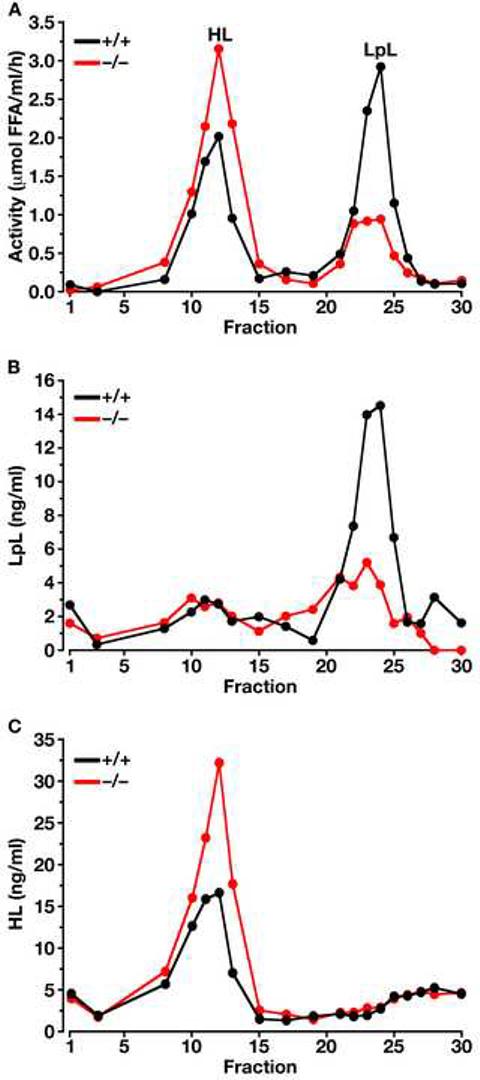
Separation of LpL and HL in the post-heparin plasma of Gpihbp1−/− and wild-type mice on a heparin-Sepharose column. (A) Lipase activities in the different fractions (representative data of four independent experiments). In these studies, the identities of the LpL and HL peaks were verified with immunoassays (B and C, respectively) (Cisar et al., 1989). The specific activity of the LpL in fractions 21–25 of the wild-type and Gpihbp1−/− mouse plasma was not different. Each data point represents the average of duplicate analyses.
We also measured LpL levels in whole plasma (i.e., not fractionated on a heparin-Sepharose column) following an injection of heparin. The post-heparin LpL protein mass levels were lower in Gpibhp1 knockout mice than in control mice (175 ± 111 ng/ml vs. 581 ± 98 ng/ml; n = 5 in each group; P = 0.00028). Levels of LpL mass in the pre-heparin plasma of wild-type and Gpihbp1−/− mice were very low and not different.
The fact that there was still some heparin-releasable LpL in Gpihbp1−/− mice suggested that other proteins, perhaps HSPGs, provide a reservoir for LpL binding. Nevertheless, the presence of GPIHBP1 within capillaries, along with the chylomicronemia in Gpihbp1−/− mice, strongly suggested that GPIHBP1 is likely an important “platform” for lipolysis. We hypothesized that GPIHBP1 might be capable of binding LpL or chylomicrons or both. This hypothesis seemed plausible given that GPIHBP1 contains a strongly negatively charged domain, and that both LpL and several apolipoproteins within chylomicrons contain positively charged domains that are known to mediate protein–protein interactions (Brown and Goldstein, 1986; Cardin et al., 1989).
GPIHBP1 binds both lipoprotein lipase and chylomicrons
To test the hypothesis that GPIHBP1 binds to LpL and/or chylomicrons, we constructed a full-length mouse Gpihbp1 cDNA expression vector. Transient transfection of CHO ldlA7 cells with a Gpihbp1 expression vector yielded high levels of GPIHBP1 expression at the surface of cells, as judged by confocal fluorescence microscopy (Figure 5A). This GPIHBP1 could be released from the surface of cells with a phosphatidylinositol-specific phospholipase C (PIPLC) (Low, 1992) (Figure 5B). To assess the ability of GPIHBP1 to bind LpL, we examined the binding of LpL to a mutant CHO cell line (pgsA-745) that lacks the ability to synthesize HSPGs (Lugemwa and Esko, 1991). CHO pgsA-745 cells stably transfected with Gpihbp1 bound 10–20-fold more LpL than cells transfected with empty vector, and the binding of LpL to cells was saturable, with nonspecific binding being negligible (Figure 5C). An analysis of LpL binding indicated that GPIHBP1 binds LpL tightly (calculated Kd = 3.6 × □10−8 M). The increased binding of LpL to Gpihbp1-transfected cells was eliminated by treating the cells with PIPLC (Figure 5D). Heparin was effective in releasing LpL from GPIHBP1 expressed on the surface of pgsA-745 cells (Figures 5E and 5F).
Figure 5.
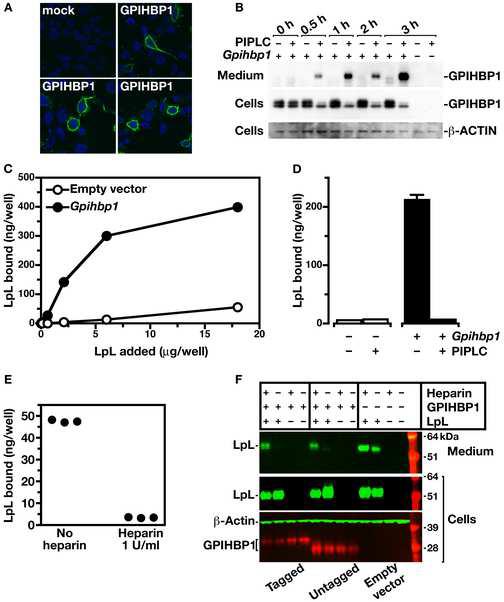
Generating cell lines expressing a Gpihbp1 cDNA and testing the ability of GPIHBP1 to bind LpL. In preliminary studies, we showed that the expression of Gpihbp1 was absent in wild-type CHO cells. (A) Confocal immunofluorescence microscopy showing the expression of an S-protein–tagged GPIHBP1 on the surface of CHO ldlA7 cells. GPIHBP1 was visualized with a FITC-conjugated goat antibody against the S-protein tag (green) and DNA was visualized with DAPI (blue). (B) Western blot analysis of cell extracts and cell culture medium from HeLa cells that had been transfected with a mouse Gpihbp1 cDNA, before and after treatment of the cells with PIPLC. (C) Binding of LpL to pgsA-745 CHO cells stably transfected with a cDNA encoding mouse Gpihbp1 (pcDNA3-Gpihbp1, see Experimental Procedures) or empty vector. Cells were incubated for 2 h with increasing amounts of avian LpL. Following the incubation, the amount of bound LpL was measured with an ELISA. Each data point represents the mean of triplicates (± S.D.). Standard deviations are too small to be seen. (D) Binding of avian LpL (2.5 μg/ml) to pgsA-745 CHO cells transfected with pcDNA3-Gpihbp1 or empty vector, before and after treatment with PIPLC. Each bar represents the mean (± S.D.) of triplicate determinations; in some cases, the standard deviations are too small to be seen. For several groups, the standard error bars are too small to be seen. In parallel experiments, pgsB-761 CHO cells transfected with the Gpihbp1 cDNA also bound 10-fold more LpL than cells transfected with the empty vector. In wild-type CHO cells, LpL binding to nontransfected cells was greater (as a result of the expected binding of LpL to HSPGs), but we consistently observed a 70% increase in LpL binding to Gpihbp1-transfected cells. (E) Binding of avian LpL (2.5 μg/ml) to pgsA-745 CHO cells transfected with pcDNA3-Gpihbp1 or empty vector, before and after treatment with heparin (1 U/ml). (F) Western blot analysis of cell extracts and cell culture medium from pgsA-745 CHO cells transfected with a mouse Gpihbp1 cDNA (tagged and untagged) or empty vector, alone or in combination with a human LPL cDNA (with a C-terminus V5 tag), before and after treatment of the cells with heparin (1 U/ml).
To determine if GPIHBP1 binds to triglyceride-rich lipoproteins, we isolated the d < 1.006 g/ml lipoproteins (“chylomicrons”) from the plasma of Gpihbp1−/− mice, labeled them with the fluorescent dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), and incubated them with CHO ldlA7 cells stably transfected with the mouse Gpihbp1 cDNA or empty vector. The Gpihbp1-transfected cells bound chylomicrons avidly, whereas the cells transfected with the empty vector did not (Figure 6A). The binding of chylomicrons was dramatically reduced by treating the transfected cells with PIPLC (Figure 6A). In transient transfection experiments, ∼10% of the cells expressed GPIHBP1, as judged by immunofluorescence microscopy, and only those cells bound chylomicrons (Figure 6B). In similar experiments, we found that Gpihbp1-transfected cells also bound apo-AV-phospholipid disks (Supplementary Online Figure 6).
Figure 6.
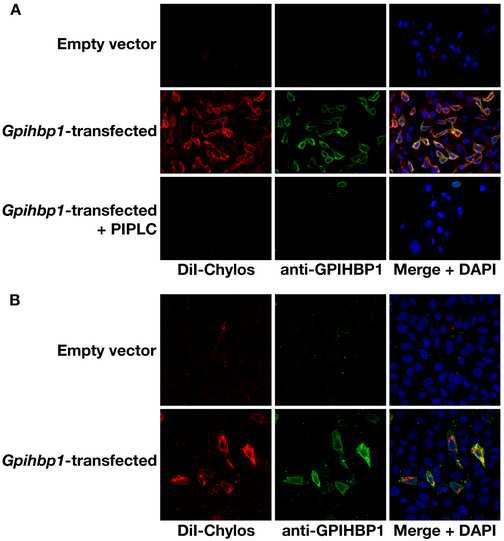
Binding of chylomicrons to CHO-ldlA7 cells expressing mouse GPIHBP1. (A) The binding of DiI-labeled chylomicrons (red) to nonpermeabilized CHO-ldlA7 cells that had been stably transfected with a mouse Gpihbp1 cDNA (or empty vector) was measured at 4°C, before and after PIPLC treatment. GPIHBP1 expression was detected with the rabbit anti-GPIHBP1 antiserum and a FITC-labeled anti-rabbit IgG antibody (green). DNA was visualized with a DAPI stain (blue). (B) Binding of DiI-labeled chylomicrons (red) to CHO-ldlA7 cells that had been transiently transfected with a mouse Gpihbp1 cDNA. GPIHBP1 expression was detected as described in Panel A.
Gpihbp1 expression levels in muscle change with feeding status
Lipolysis of triglyceride-rich lipoproteins is controlled at multiple steps, according to metabolic need. For example, LpL activity levels in muscle are controlled by feeding status (Lithell et al., 1978). We therefore tested whether Gpihbp1 expression in muscle changes with feeding status. Indeed, Gpihbp1 expression increased twofold in muscle during fasting (P < 0.001) (Figure 7).
Figure 7.
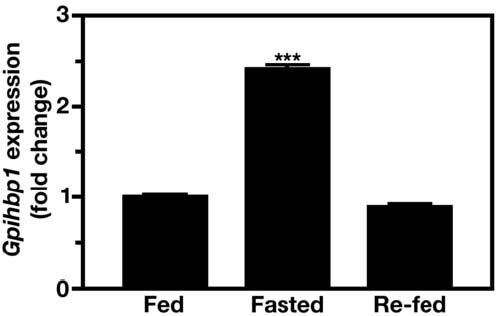
Regulation of Gpihbp1 expression in quadriceps according to feeding status. RNA was isolated from fed mice, fasted mice (16 h), and fasted/re-fed mice (6 h post-refeeding a high-carbohydrate diet) (10-week-old wild-type C57BL/6 male mice; n = 10 per group), and the expression of Gpihbp1 measured by quantitative RT-PCR (mean ± S.D.). β-2 microglobulin expression was used to normalize the data. Values show the amount of Gpihbp1 mRNA relative to that in fed mice. ***, P < 0.001.
Discussion
In this study, we show that adult Gpihbp1−/− mice have severe hypertriglyceridemia, associated with a marked delay in the clearance of retinyl palmitate from the plasma. Gpihbp1 is expressed at high levels in heart, adipose tissue, and skeletal muscle—the same tissues that express high levels of LpL. In these tissues, GPIHBP1 is located on the luminal face of the capillary endothelium. Expression of GPIHBP1 in cultured cells confers the ability to bind both LpL and chylomicrons. Together, these findings strongly suggest that GPIHBP1 is a key platform for the LpL-mediated processing of chylomicrons in capillaries.
We found a large difference in GPIHBP1 expression in different capillary beds. While GPIHBP1 was expressed highly in the capillaries of lipolytic tissues such as adipose tissue, heart, and skeletal muscle, it was virtually absent in the brain. These findings raise the possibility that differences in endothelial cell gene expression could play an active role in regulating the delivery of lipid nutrients to different tissues.
The ability of GPIHBP1 to bind both to LpL and chylomicrons favors the concept that GPIHBP1 serves as an important “platform” for lipolysis by drawing chylomicrons and LpL into close proximity. However, it is unlikely that Gpihbp1 deficiency completely eliminates LpL-mediated processing of lipoproteins. Lpl-deficient mice die shortly after birth with plasma triglyceride levels as high as 20,000 mg/dl (Weinstock et al., 1995), whereas Gpihbp1−/− mice survive and have far lower triglyceride levels. These differences between Gpihbp1- and Lpl-deficient mice strongly suggest that some LpL-mediated lipolysis occurs in the absence of GPIHBP1. Without question, LpL is present in Gpihbp1−/− mice; we clearly identified mouse LpL—with normal enzymatic activity—in the plasma of Gpihbp1−/− mice after an injection of heparin.
The fact that LpL was identified in the post-heparin plasma of Gpihbp1−/− mice indicates that, in vivo, there are additional binding sites for LpL—aside from GPIHBP1. Cell culture experiments have shown, quite convincingly, that LpL binds to HSPGs on the surface of endothelial cells (Cheng et al., 1981). However, at this point, we do not know with certainty whether the heparin-releasable LpL in Gpihbp1−/− mice is attached to HSPGs, and indeed whether the LpL in Gpihbp1−/− mice that is mobilized by heparin is actually located within the lumen of capillaries. At this time, is not known with certainty how much of the LpL found into the bloodstream after an injection of heparin is actually released from the luminal surface of capillaries versus other sites (e.g., parenchymal cells, basement membranes, and subendothelial spaces). Further, the percentage of post-heparin LpL that arises from endothelial cells versus those other sites could differ in Gpihbp1−/− and Gpihbp1+/+ mice. In any case, since the post-heparin plasma LpL levels are higher in wild-type mice than in Gpihbp1−/− mice, it seems possible that a significant fraction of the heparin-releasable LpL in wild-type mice is bound by GPIHBP1. In future studies, it will be of interest to determine the fraction of the total LpL pool on endothelial cells that is bound to GPIHBP1 and HSPGs, and whether LpL bound to GPIHBP1 is catalytically more efficient than LpL bound to HSPGs.
The degree of hypertriglyceridemia in Gpihbp1−/− pups before weaning was milder than that in 4—6 week-old Gpihbp1−/− mice, which in turn was milder than that in 7–16-week-old Gpihbp1−/− mice. The explanation for this finding is not known with certainty, but we suspect that it relates to the fact that Lpl mRNA levels are much higher in the liver during the suckling phase (Langner et al., 1989). We have confirmed higher Lpl expression levels by quantitative RT-PCR: suckling C57BL/6 mice have hepatic Lpl mRNA levels that are 50-times greater than those of 10-week-old mice, and hepatic Lpl mRNA levels in 4-week-old mice are approximately double those in 10-week-old mice (Supplementary Online Figure 7). Although we have not yet proven that the differences in hepatic expression of Lpl are responsible for the lower triglyceride levels in the younger mice, the time course of Lpl expression would be consistent with this possibility. In any case, we emphasize that the defect in LpL activity in adult Gpihbp1−/− mice is severe. When newborn Lpl knockout mice were injected with an LpL adenovirus, some survived the suckling period, owing to the adenoviral-mediated LpL expression in the liver (Strauss et al., 2001). However, this adenoviral-mediated LpL expression disappeared by 30 days of age. After the extinction of LpL expression, the plasma triglyceride levels in the rescued mice were 2,000–5,000 mg/dl—very similar to those in chow-fed Gpihbp1−/− mice in this study.
The plasma levels of apo-B48 were markedly elevated in Gpihbp1−/− mice. In the mouse, apo-B48 is synthesized by both the liver and the intestine (Hirano et al., 1996). Given the markedly delayed clearance of the retinyl palmitate in Gpihbp1−/− mice, we suspect that much of the apo-B48 accumulation is due to delayed clearance of intestinal lipoproteins. However, the lipolytic processing of chylomicrons and hepatic VLDL are similar, and there is no reason to believe that the accumulation of apo-B48 is exclusively due to an accumulation of intestinal lipoproteins.
The molecular basis for chylomicron binding to GPIHBP1 requires further study. Chylomicrons contain several apolipoproteins (e.g., apo-B48, apo-E, apo-AV) that have positively charged domains that bind to heparin sulfate and/or HSPGs (Cardin et al., 1984; Cardin et al., 1986; Lookene et al., 2005), so it is easy to imagine that one or more of these apolipoproteins could mediate the binding of chylomicrons to the strongly negatively charged domain of GPIHBP1. In particular, apo-AV would make a good candidate as a ligand for GPIHBP1, given that Apoav deficiency causes hypertriglyceridemia associated with decreased LpL-mediated lipolysis (Calandra et al., 2006; Grosskopf et al., 2005; Pennacchio et al., 2001). In support of this idea, our studies showed that apo-AV-phospholipid disks bind avidly to Gpihbp1-transfected proteoglycan-deficient cells (Supplementary Online Figure 6). Another interesting hypothesis would be that GPIHBP1 could act as a receptor for apo-B48, a key structural protein of large, triglyceride-rich lipoproteins. It would make sense that a receptor for apo-B48 would be located in the capillary endothelium of muscle and fat, where lipolysis occurs. Also, this hypothesis is in line with evolutionary considerations. Both apo-B48 and chylomicrons (defined as intestinal lipoproteins that are secreted into the lymph) are unique to mammals (Teng and Davidson, 1992). Similarly, GPIHBP1—defined as a GPI-anchored Ly-6-motif protein with a very strong acidic domain—is present in all mammals but is absent in fish, amphibians, and birds.
Transfection of a Gpihbp1 cDNA into cultured cells promotes the binding of both LpL and chylomicrons, but it is not yet clear whether a single GPIHBP1 molecule is capable of binding chylomicrons and LpL simultaneously. If a single GPIHBP1 molecule binds only a single ligand, chylomicrons and LpL could still be drawn together if the GPIHBP1 molecules were clustered together on the cell surface. This possibility seems plausible given that other GPI-anchored proteins are concentrated within lipid rafts on the plasma membrane (Varma and Mayor, 1998). Also, GPIHBP1 could exist as a homodimer on the cell surface and therefore bind to more than one ligand. Of note, urokinase-type plasminogen activator receptor, a related GPI-anchored Ly-6-motif protein, exists as a dimer on the cell surface (Cunningham et al., 2003).
In summary, GPIHBP1 is crucial for the lipolytic processing of triglyceride-rich lipoproteins. It is located on the luminal surface of the capillary endothelium and binds both LpL and chylomicrons. It likely forms a platform for lipolysis and plays an important role in the delivery of lipid nutrients to cells.
Experimental procedures
Gpihbp1-deficient mice
Gpihbp1−/− mice were created with a sequence-replacement gene-targeting vector designed to replace all of the exons of Gpihbp1 with a lacZ reporter and a PGK-neo cassette. Targeted cells were identified by Southern blotting of genomic DNA with 5′- and 3′-flanking probes. The 5′ probe was amplified from genomic DNA with oligonucleotides 5′–GGTCTGTAGCAAGCCTTCC–3′ and 5′–GTGTCCTTCCTGCCAAATCC–3′ (after cleaving genomic DNA with HpaI, the targeted band is 11.8 kb and the wild-type band is 17.1 kb). The 3′ probe was amplified with oligonucleotides 5′–CATCTGTACAGCCCTTGTCC–3′ and 5′–CCAGAAGCACAACAGCCAG–3′ (after cleaving genomic DNA with EcoRI, the targeted band is 7.1 kb and the wild-type band is 8.5 kb). Routine genotyping was performed by PCR; the mutant allele was detected by PCR with oligonucleotides 5′–GCAGCGCATCGCCTTCTATC–3′ and 5′–GGAGGCC TACACTAGAAGG–3′ (amplifying a 655-bp PCR product between the neo cassette and sequences 3′ to Gpihbp1). The wild-type allele was detected by PCR with oligonucleotides 5′–GGTGATGCGGACCCGGAG–3′ and 5′–GTGTCT GATTGCAGCTCTCC–3′ (amplifying a 519-bp PCR product spanning intron 2). All mice had a mixed genetic background (<10% 129/Sv and >90% C57BL/6); the mice were fed a chow diet and housed in a virus-free barrier facility with a 12-h light-dark cycle. All studies were approved by UCLA's Committee on Animal Research.
Northern blots
Total RNA was prepared from mouse tissues with Tri Reagent (Sigma, St. Louis, MO), size-fractionated on a 1% agarose/formaldehyde gel, and transferred to a sheet of Nytran SuPerCharge membrane (Schleicher and Schuell, Keene, NH). The blot was hybridized to 32P-labeled cDNAs for mouse Gpihbp1, human LPL, mouse Cd36, and mouse 18S ribosomal RNA.
Quantitative RT-PCR
Total RNA was prepared from mouse tissues with Tri Reagent (Sigma), treated with DNase I (Ambion, Austin, TX), and reversed transcribed into cDNA with a mixture of random primers and oligo dT and Superscript III (Invitrogen). Primers for each gene were designed with the Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi), and were assessed by DART-PCR (Peirson et al., 2003) to validate equivalent efficiencies between genes and the endogenous control gene (β2-microglobulin). Quantitative PCR analysis was performed on 50 ng cDNA, 200 nM of each primer, and 10 μl of SYBR green PCR master mix (QIAGEN, Valencia, CA). PCR reactions were performed in triplicate on a 7500 Fast Real-Time PCR system from Applied Biosystems (Foster City, CA). The relative expression of all gene-specific mRNA was calculated by the comparative CT method.
A rabbit antiserum against mouse GPIHBP1
A segment of the mouse Gpihbp1 cDNA (spanning nucleotides 88–636, corresponding to amino acids 23–205) (NM_026730) was cloned into the BamHI and SalI sites of pQE-30 (Qiagen, Valencia, CA) to create a bacterial expression vector for 6His-tagged GPIHBP1. This fragment was amplified from mouse adipose tissue cDNA with forward primer 5′–CCATCACGGATCCGCACAAGAAG ATGGTGATGCGGA–3′ and reverse primer 5′–GCTGCAGGTCGACGCCTGAAG GGTATCCTGCCCCAC–3′ (underlined sequences indicate restriction endonuclease sites). The plasmid was transformed into Escherichia coli Max 5αF′ cells; bacteria from 1.0-liter cultures were resuspended with 40 ml of lysis buffer (20 mM Na2HPO4, pH 7.0, 250 mM NaCl, 6M guanidinium chloride) and lysed by fifteen successive 10-min periods of sonication (at a power of 50 watts) alternating with 10-min incubations on ice. The lysate was stirred at room temperature for 30 min and then centrifuged at 4000 × g for 30 min in an SS34 rotor. The supernatant fluid was then incubated with 2 ml of TALON metal affinity resin (Clontech, Mountain View, CA) with continuous stirring for 60 min at 4° C. The resin was sedimented by low-speed centrifugation, loaded onto a column, and washed overnight with one liter of wash buffer containing 20 mM Na2HPO4 (pH 7.0), 250 mM NaCl, 8 M urea, and 5 mM imidazole. The recombinant GPIHBP1 fragment was eluted with a 30-ml linear gradient of imidazole (5–300 mM) in wash buffer. Three-ml fractions were collected, and aliquots were size-fractionated on SDS-polyacrylamide gels. Fractions containing the 6His–GPIHBP1 fragment (identified by Coomassie-blue staining) were pooled and dialyzed against PBS containing 0.02% sodium azide. Two rabbits were immunized with the GPIHBP1 fragment (75 μg/rabbit) in complete Freund's adjuvant and then boosted with the GPIHBP1 fragment (150 μg/rabbit) in incomplete Freund's adjuvant 3 and 6 weeks later.
Analysis of the plasma lipoproteins
Plasma cholesterol and triglyceride levels were measured on nonfasted plasma samples with the serum triglyceride determination kit (Sigma) and cholesterol E kit (Wako, Richmond, VA). Fractionation of mouse plasma lipoproteins was performed by fast phase liquid chromatography (FPLC) (Cole et al., 1988). The diameters of lipoprotein particles in the d < 1.022 g/ml fraction of plasma were measured with a Nanotrac 250 particle analyzer (Microtrac, Clearwater, FL) fitted with a flexible conduit–sheathed probe (Walzem et al., 1994). Lipoprotein particle diameters were recorded as frequency distributions of particles within a given diameter interval. Negatively stained lipoproteins (d < 1.006 g/ml fraction) from the plasma of Gpihbp1−/− and Gpihbp1+/+ mice were imaged by electron microscopy (Hamilton et al., 1980). The protein composition of lipoproteins was analyzed by SDS-PAGE followed by Coomassie-blue staining.
Western blots
Proteins from tissue or cell culture extracts (20 μg) were size-fractionated on 4–12% Bis-Tris gradient SDS-polyacrylamide gels and then transferred to nitrocellulose membranes for western blots. The antibody dilutions were 1:500 for a mouse monoclonal antibody against mouse apo-B48 and apo-B100 (Nguyen et al., 2006); 1:500 for a rabbit antiserum against GPIHBP1; 1:500 for a mouse antibody against V5 tag (Invitrogen, Carlsbad, CA); 1:500 for a mouse monoclonal antibody against β-actin (Abcam, Cambridge, MA); 1:5,000 for an HRP-labeled anti-mouse IgG (NA931V, GE Healthcare, Piscataway, NJ); 1:5,000 for an HRP-labeled anti-rabbit IgG (NA934V, GE Healthcare); 1:2,000 for an IRdye680-conjugated goat anti-rabbit IgG (Li-Cor, Lincoln, NE); 1:2,000 for an IRdye800-conjugated goat anti-mouse IgG (Li-Cor). Antibody binding was detected with the ECL Plus chemiluminescence system (GE Healthcare) and exposure to X-ray film. When the IRdye-conjugated secondary antibodies were used, binding was detected with the Odyssey infrared scanner (Li-Cor).
Measurement of retinyl esters
Mice were given 5,000 IU of retinyl palmitate (all trans) or vehicle alone (soybean oil) in 50 μl by oral gavage. A blood sample was taken immediately after gavage and 1, 2, 4, 10, and 24 hours later. Plasma levels of retinyl esters were measured by HPLC on a 250 × 4.6 mm Beckman Ultrasphere C18 column (Palo Alto, CA) (Mills et al., 1992; Redlich et al., 1996). Retinyl esters were separated in a mobile phase consisting of acetonitrile:methanol:dichloromethane (70:15:15 v/v) at a flow rate of 1.8 ml/min and detected by UV absorbance at 325 nm with a Waters 996 photodiode array detector (Milford, MA). Retinyl acetate was used as an internal standard. Retinyl ester standards were synthesized from authentic all-trans retinol and the corresponding fatty acyl chloride (Blaner et al., 1994). The reported plasma retinyl ester concentrations represent the sum of individual retinyl ester concentrations (retinyl linoleate, retinyl oleate, retinyl palmitate, and retinyl stearate). All extraction and HPLC procedures were carried out under N2 and reduced light to prevent oxidation of the compounds. Plasma samples (25–200 μl) were denatured with an equal volume of absolute ethanol containing known amounts of retinyl acetate and then extracted into hexane. Following phase separation, the hexane extract was evaporated under a stream of N2 and the residue resuspended in benzene for injection onto the HPLC column. The lower limit of detection for retinyl esters was 2 ng/ml (Burger et al., 1997).
Expression of mouse Gpihbp1 in CHO cells
Two independent strategies were used to express a mouse Gpihbp1 cDNA in cultured cells. Strategy 1: the complete coding sequence of Gpihbp1 (nucleotides 1–711) was amplified from a mouse adipose cDNA library with primer 5′–CTCGAGCGGCCGCGACAGAACAGCCTGAGCCAGGAT–3′ (underlined segment is a NotI site) and 5′–TTAAGCTTGGTACCTCTTCAGGCC CCTGACGCCCAC–3′ (underlined segment is a KpnI site). The fragment was ligated into the NotI and KpnI sites of pcDNA3.1(−) (Invitrogen, Carlsbad, CA) to produce pcDNA3-Gpihbp1. CHO-K1 cells, Ldlr-deficient CHO cells (ldlA7 cells, a generous gift from Dr. Monty Krieger) (Kingsley and Krieger, 1984), and CHO cells deficient in the ability to synthesize heparan-sulfate proteoglycans (pgsA-745 and pgsB-761 cells, a generous gift from Dr. Jeff Esko) (Esko, 1991) were transfected with pcDNA3-Gpihbp1 or empty vector using FuGENE 6 Transfection Reagent (Roche, Indianapolis, IN) according to the manufacturer's instructions. Stably transfected cells were obtained following selection with G418 (0.5 mg/ml). The cells were then subjected to a second round of transfection with pcDNA3-Gpihbp1 and pCMV/Bsd (Invitrogen) and selected with blasticidin (5 μg/ml). Strategy 2: an IMAGE clone (ID #30298145) containing the complete open reading frame of mouse Gpihbp1 in pDNR-LIB was purchased from Open Biosystems (Huntsville, AL). An NcoI restriction site immediately upstream to the translational start site was created by site-directed mutagenesis (QuickChange, Stratagene, La Jolla, CA). An NcoI–HindIII fragment containing the complete open reading frame was subcloned into the mammalian expression vector pTriEx-4 (Novagen, San Diego, CA). To facilitate subsequent cloning steps, the ApaI site in pTriEx-4 was replaced with an SfoI restriction site by site-directed mutagenesis. An ApaI site was subsequently created by silent mutation at nucleotide 66 of the Gpihbp1 cDNA (between the signal peptide and the acidic domain). A Gpihbp1 construct with an amino-terminal S-protein tag was created by inserting the S-protein coding sequence (AAAGAAACCGCTGCTGCGAAATT TGAACGCCAGCACATGGACTCG) into the newly created ApaI site. The integrity of all constructs was verified by DNA sequencing.
Immunofluorescence microscopy
For the detection of GPIHBP1 in cultured cells, cells were plated on coverslips at ∼25,000 cells per well in 24-well plates, fixed in 3% paraformaldehyde, blocked with 10% sheep serum, and incubated with the rabbit anti-GPIHBP1 antiserum diluted in blocking buffer (1:800). Detergents were omitted to facilitate the detection of only cell surface GPIHBP1 protein. Bound rabbit immunoglobulin (IgG) was detected with a FITC-conjugated donkey anti-rabbit IgG (1:800; Jackson ImmunoResearch). After washing, cells were stained with DAPI to visualize DNA. For cells transfected with the S-protein–tagged GPIHBP1 construct, the fusion protein was detected with a FITC-conjugated goat antibody against the S-protein tag (Abcam, Cambridge, MA; 1:400). Images were obtained with an Axiovert 200 MOT microscope (Zeiss, Germany) with a 63×/1.25 oil-immersion objective and processed with AxoVision 4.2 software (Zeiss). Confocal fluorescence microscopy was performed with a Leica TCS-SP MP confocal inverted microscope (Heidelberg, Germany) equipped with an argon laser (488 nm blue excitation), diode laser (561 nm green excitation), and a two-photon laser tuned at 768 nm for UV excitation. Individual images were captured sequentially with a 63×/1.32 planapo objective and merged images were generated with Leica confocal software (version 2.5).
For the detection of GPIHBP1 in mouse tissues, 10-μm thick frozen sections were fixed with 4% formaldehyde and blocked with 10% donkey serum. The samples were incubated overnight with the rabbit antiserum against mouse GPIHBP1 (1:200) and a rat monoclonal antibody against mouse CD31 (1:50, BD Pharmigen, San Diego, CA). Samples were subsequently washed and incubated for one hour with a FITC-conjugated antibody against rabbit IgG (1:200, Jackson ImmunoResearch, West Grove, PA) and an Alexa Flour 568-conjugated antibody against rat IgG (1:800, Molecular Probes, Eugene, OR). After washing, the sections were stained with DAPI to visualize DNA. Microscopy was performed as described above.
Binding of lipoprotein lipase to Gpihbp1-transfected CHO cells
CHO pgsA-745 cells transfected with pcDNA3-Gpihbp1 or empty vector were grown to confluence in Ham's F12 medium containing 5% fetal bovine serum. The cells were placed on ice, washed twice with ice-cold PBS, and incubated for 2 h at 4°C with 1.0 ml of Ham's F12 medium supplemented with 0.5% BSA and highly purified chicken LpL (Bensadoun et al., 1999). After this incubation, the cells were washed once with PBS supplemented with 0.5% BSA and lysed with 400 μl of CHAPS lysis buffer (4 mM CHAPS, 50 mM NH4Cl, and 3 U heparin/ml). The amount of LpL in the medium and cell lysates was measured by ELISA (Cisar et al., 1989).
Experiments were also performed to determine if the binding of LpL to cells could be prevented by treating cells with a phosphatidylinositol-specific phospholipase C (PIPLC). CHO-pgsA-745 cells transfected with either pcDNA3-Gpihbp1 or empty vector were grown to confluence in 35-mm dishes. The cells were incubated in 2 ml of Ham's F12 in the absence or presence of 10 U of PIPLC at 37°C for 1 h. Cells were then placed on ice and incubated in Ham's F12 supplemented with 0.5% BSA and 1.5 μg of avian LpL for 2 h. Cells were then washed once with PBS/0.5% BSA and lysed with CHAPS buffer. Bound LpL was determined by ELISA (Cisar et al., 1989). Recombinant PIPLC was purified from the media of Bacillus subtilis transfected with a PIPLC gene from B. thuringiensis (a generous gift from Dr. Martin Low at Columbia University) (Low, 1992). The enzyme was purified from media by ion-exchange chromatography on Amberlite CG-50, followed by ammonium sulfate precipitation, gel filtration on Sephadex G-75, and a final anion-exchange chromatography step on MonoQ. PIPLC activity was documented with l-3-phosphatidyl[2-3H]inositol (GE Healthcare) (Low, 1992). The final PIPLC preparation had an activity of 199 units/ml.
Measurements of LpL and hepatic lipase activities
To estimate LpL and HL activities in post-heparin plasma, mice were injected intraperitoneally with 0.5 U heparin sodium/g body weight, and blood was collected under anesthesia from the retro-orbital sinus 15 min later. To avoid the possibility of the elevated plasma triglyceride levels interfering with the lipase assays, the lipases were partially purified from the plasma samples by heparin-Sepharose chromatography. Post-heparin plasma (200 μl) was loaded onto 1.0-ml heparin-Sepharose HiTrap columns (GE Healthcare) equilibrated with 0.25 M NaCl, 20% glycerol, 1% BSA, 10 mM sodium phosphate, pH 6.5. The column was washed with 10 ml of the equilibration buffer and eluted with a 30 ml NaCl gradient (0.25–1.5 M in 20% glycerol, 1% BSA, 10 mM sodium phosphate, pH 6.5). Fractions were assayed for lipolytic activity with [3H]triolein as described previously (Hocquette et al., 1998). The activities in fractions corresponding to the HL and LpL peaks were added together to calculate the lipase activities per ml of plasma.
Binding of DiI-labeled chylomicrons to cells
DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) was purchased from Invitrogen. DiI was dissolved in DMSO (3 mg/ml) and 10 μl of this solution was added to 70 μl of serum from a Gpihbp1−/− mouse and 250 μl of human lipoprotein-deficient serum. This mixture was then incubated at 37°C overnight. The d < 1.006 g/ml lipoproteins (“chylomicrons”) were isolated by centrifugation at 100,000 rpm in a Beckman TLA-100.3 rotor for 6 h at 4°C. The DiI-labeled chylomicrons were harvested and dialyzed against PBS containing 5.0 μM EDTA.
To evaluate the binding of the DiI-labeled chylomicrons to GPIHBP1, transfected CHO ldlA7 cells were incubated with 1 μg/ml of DiI-labeled chylomicrons in PBS containing 1.0 mM CaCl2, 1.0 mM MgCl2, and 0.5% BSA for 2 h at 4°C. The cells were then washed five times in the same buffer, fixed in 3% paraformaldehyde, and chylomicron binding detected by fluorescence microscopy. Cells expressing GPIHBP1 on the cell surface were detected by immunofluorescence microscopy as described above.
Binding of apolipoprotein AV-phospholipid disks to cells
An apo-AV-FLAG expression vector was constructed by replacing the apo-AV coding sequence in the apo-AV/pET vector (Beckstead et al., 2003) with sequences encoding apo-AV-FLAG. Recombinant apo-AV with a carboxyl-terminal, 8-residue FLAG extension was expressed and purified as described (Beckstead et al., 2003). To generate apo-AV-FLAG-phospholipid complexes, dimyristoylphosphatidylcholine (DMPC) was dissolved in chloroform:methanol (3:1 v/v) and the solvent evaporated under a stream of nitrogen. The sample was then incubated in vacuo for 16 h to remove residual solvent. DMPC was resuspended in a solution containing 50 mM sodium citrate and 150 mM NaCl (pH 3.0), followed by the addition of apo-AV-FLAG (6:1 DMPC:apoA-V; w/w). The mixture was sonicated until clear, dialyzed against 50 mM phosphate, pH 7.4, 150 mM NaCl and filter-sterilized (0.22 μM). Protein concentration was determined with the BCA Protein Assay Kit (Pierce), and the phospholipid content was determined with an enzyme-based colorimetric assay (Wako). To evaluate the binding of the apo-AV-DMPC disks to GPIHBP1, pgsA-745 CHO cells were transiently transfected with the S-protein-tagged Gpihbp1 cDNA and incubated with 5 μg/ml of apo-AV-DMPC disks in PBS containing 1.0 mM CaCl2, 1.0 mM MgCl2, and 0.5% BSA for 2 h at 4°C. Apoprotein binding was detected with a mouse anti-FLAG antibody (and a Cy5-conjugated goat anti-mouse antibody) and the S-protein–tagged GPIHBP1 was assessed as described above.
Supplementary Material
Acknowledgments
This work was supported by a Beginning Grant-in-Aid from the American Heart Association, Western States Affiliate (to A.P.B.), by BayGenomics (HL66621 and HL66600 from the National Heart, Lung, and Blood Institute) (to S.G.Y.), and by HL073061 (to R.O.R.). We thank Jennifer A. Beckstead for discussions and advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beckstead JA, Oda MN, Martin DDO, Forte TM, Bielicki JK, Berger T, Luty R, Kay CM, Ryan RO. Structure-Function Studies of Human Apolipoprotein A-V: A Regulator of Plasma Lipid Homeostasis. Biochemistry. 2003;42:9416–9423. doi: 10.1021/bi034509t. [DOI] [PubMed] [Google Scholar]
- Bengtsson G, Olivecrona T. Apolipoprotein CII enhances hydrolysis of monoglycerides by lipoprotein lipase, but the effect is abolished by fatty acids. FEBS Lett. 1979;106:345–348. doi: 10.1016/0014-5793(79)80529-3. [DOI] [PubMed] [Google Scholar]
- Bensadoun A, Hsu J, Hughes B. Large-scale lipoprotein lipase purification from adipose tissue. Methods Mol. Biol. 1999;109:145–150. doi: 10.1385/1-59259-581-2:145. [DOI] [PubMed] [Google Scholar]
- Blaner WS, Obunike JC, Kurlandsky SB, al-Haideri M, Piantedosi R, Deckelbaum RJ, Goldberg IJ. Lipoprotein lipase hydrolysis of retinyl ester. Possible implications for retinoid uptake by cells. J. Biol. Chem. 1994;269:16559–16565. [PubMed] [Google Scholar]
- Breckenridge WC, Little JA, Steiner G, Chow A, Poapst M. Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N. Engl. J. Med. 1978;298:1265–1273. doi: 10.1056/NEJM197806082982301. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. [Google Scholar]
- Burger H, Kovacs A, Weiser B, Grimson R, Nachman S, Tropper P, van Bennekum AM, Elie MC, Blaner WS. Maternal serum vitamin A levels are not associated with mother-to-child transmission of HIV-1 in the United States. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997;14:321–326. doi: 10.1097/00042560-199704010-00003. [DOI] [PubMed] [Google Scholar]
- Calandra S, Oliva CP, Tarugi P, Bertolini S. APOA5 and triglyceride metabolism, lesson from human APOA5 deficiency. Curr. Opin. Lipidol. 2006;17:122–127. doi: 10.1097/01.mol.0000217892.00618.54. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Barnhart RL, Witt KR, Jackson RL. Reactivity of heparin with the human plasma heparin-binding proteins thrombin, antithrombin III, and apolipoproteins E and B-100. Thromb. Res. 1984;34:541–550. doi: 10.1016/0049-3848(84)90258-5. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Hirose N, Blankenship DT, Jackson RL, Harmony JAK. Binding of a high reactive heparin to human apolipoprotein E: Identification of two heparin-binding domains. Biochem. Biophys. Res. Commun. 1986;134:783–789. doi: 10.1016/s0006-291x(86)80489-2. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Jackson RL, Sparrow DA, Sparrow JT. Interaction of glycosaminoglycans with lipoproteins. Ann. N.Y. Acad. Sci. 1989;556:186–193. doi: 10.1111/j.1749-6632.1989.tb22503.x. [DOI] [PubMed] [Google Scholar]
- Cheng C-F, Oosta GM, Bensadoun A, Rosenberg RD. Binding of lipoprotein lipase to endothelial cells in culture. J. Biol. Chem. 1981;256:12893–12898. [PubMed] [Google Scholar]
- Cisar LA, Hoogewerf AJ, Cupp M, Rapport CA, Bensadoun A. Secretion and degradation of lipoprotein lipase in cultured adipocytes. Binding of lipoprotein lipase to membrane heparan sulfate proteoglycans is necessary for degradation. J. Biol. Chem. 1989;264:1767–1774. [PubMed] [Google Scholar]
- Cole T, Kitchens R, Daugherty A, Schonfeld G. An improved method for separation of triglyceride–rich lipoproteins by FPLC. FPLC BioCommuniqué. 1988;4:4–6. [Google Scholar]
- Cunningham O, Andolfo A, Santovito ML, Iuzzolino L, Blasi F, Sidenius N. Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. Embo J. 2003;22:5994–6003. doi: 10.1093/emboj/cdg588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD. Genetic analysis of proteoglycan structure, function and metabolism. Curr. Opin. Cell Biol. 1991;3:805–816. doi: 10.1016/0955-0674(91)90054-3. [DOI] [PubMed] [Google Scholar]
- Goldberg IJ. Lipoprotein lipase and lipolysis: Central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 1996;37:693–707. [PubMed] [Google Scholar]
- Greenwalt DE, Scheck SH, Rhinehart-Jones T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J. Clin. Invest. 1995;96:1382–1388. doi: 10.1172/JCI118173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskopf I, Baroukh N, Lee SJ, Kamari Y, Harats D, Rubin EM, Pennacchio LA, Cooper AD. Apolipoprotein A-V deficiency results in marked hypertriglyceridemia attributable to decreased lipolysis of triglyceride-rich lipoproteins and removal of their remnants. Arterioscler. Thromb. Vasc. Biol. 2005;25:2573–2579. doi: 10.1161/01.ATV.0000186189.26141.12. [DOI] [PubMed] [Google Scholar]
- Hamilton RL, Jr., Goerke J, Guo LSS, Williams MC, Havel RJ. Unilamellar liposomes made with the French pressure cell: A simple preparative and semiquantitative technique. J. Lipid Res. 1980;21:981–992. [PubMed] [Google Scholar]
- Havel RJ, Kane JP. Introduction: Structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2705–2716. [Google Scholar]
- Hirano K-I, Young SG, Farese RV, Jr., Ng J, Sande E, Warburton C, Powell-Braxton LM, Davidson NO. Targeted disruption of the mouse apobec-1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J. Biol. Chem. 1996;271:9887–9890. doi: 10.1074/jbc.271.17.9887. [DOI] [PubMed] [Google Scholar]
- Hocquette JF, Graulet B, Olivecrona T. Lipoprotein lipase activity and mRNA levels in bovine tissues. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 1998;121:201–212. doi: 10.1016/s0305-0491(98)10090-1. [DOI] [PubMed] [Google Scholar]
- Ioka RX, Kang M-J, Kamiyama S, Kim D-H, Magoori K, Kamataki A, Ito Y, Takei YA, Sasaki M, Suzuki T, Sasano H, Takahashi S, Sakai J, Fujino T, Yamamoto TT. Expression Cloning and Characterization of a Novel Glycosylphosphatidylinositol-anchored High Density Lipoprotein-binding Protein, GPI-HBP1. J. Biol. Chem. 2003;278:7344–7349. doi: 10.1074/jbc.M211932200. [DOI] [PubMed] [Google Scholar]
- Kingsley DM, Krieger M. Receptor-mediated endocytosis of low density lipoprotein: Somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc. Natl. Acad. Sci. USA. 1984;81:5454–5458. doi: 10.1073/pnas.81.17.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn ED. Clearing factor, a heparin-activated lipoprotein lipase. I. Isolation and characterization of the enzyme from normal rat heart. J. Biol. Chem. 1955;215:1–14. [PubMed] [Google Scholar]
- Langner CA, Birkenmeier EH, Ben-Zeev O, Schotz MC, Sweet HO, Davisson MT, Gordon JI. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J. Biol. Chem. 1989;264:7994–8003. [PubMed] [Google Scholar]
- Lithell H, Boberg J, Hellsing K, Lundqvist G, Vessby B. Lipoprotein-lipase activity in human skeletal muscle and adipose tissue in the fasting and the fed states. Atherosclerosis. 1978;30:89–94. doi: 10.1016/0021-9150(78)90155-7. [DOI] [PubMed] [Google Scholar]
- Lookene A, Beckstead JA, Nilsson S, Olivecrona G, Ryan RO. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J. Biol. Chem. 2005;280:25383–25387. doi: 10.1074/jbc.M501589200. [DOI] [PubMed] [Google Scholar]
- Low M. Phospholipases that degrade the glycosylphosphatidylinositol anchor of membrane proteins. In: Hooper N, Turner A, editors. Lipid Modifications of Proteins: a practical approach. Oxford University Press; Oxford: 1992. pp. 117–154. [Google Scholar]
- Lugemwa FN, Esko JD. Estradiol beta-D-xyloside, an efficient primer for heparan sulfate biosynthesis. J. Biol. Chem. 1991;266:6674–6677. [PubMed] [Google Scholar]
- Mills JL, Tuomilehto J, Yu KF, Colman N, Blaner WS, Koskela P, Rundle WE, Forman M, Toivanen L, Rhoads GG. Maternal vitamin levels during pregnancies producing infants with neural tube defects. J. Pediatr. 1992;120:863–871. doi: 10.1016/s0022-3476(05)81951-1. [DOI] [PubMed] [Google Scholar]
- Nestel PJ, Havel RJ, Bezman A. Sites of initial removal of chylomicron triglyceride fatty acids from the blood. J. Clin. Invest. 1962;41:1915–1921. doi: 10.1172/JCI104648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Braschi S, Geoffrion M, Fong LG, Crooke RM, Graham MJ, Young SG, Milne R. A mouse monoclonal antibody specific for mouse apoB48 and apoB100 produced by immunizing “apoB39-only” mice with mouse apoB48. Biochim. Biophys. Acta. 2006;1761:182–185. doi: 10.1016/j.bbalip.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Parthasarathy N, Goldberg IJ, Sivaram P, Mulloy B, Flory DM, Wagner WD. Oligosaccharide sequences of endothelial cell surface heparan sulfate proteoglycan with affinity for lipoprotein lipase. J. Biol. Chem. 1994;269:22391–22396. [PubMed] [Google Scholar]
- Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucl. Acids Res. 2003;31:e73. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart J-C, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- Redlich CA, Grauer JN, Van Bennekum AM, Clever SL, Ponn RB, Blaner WS. Characterization of carotenoid, vitamin A, and alpha-tocopheral levels in human lung tissue and pulmonary macrophages. Am. J. Respir. Crit. Care Med. 1996;154:1436–1443. doi: 10.1164/ajrccm.154.5.8912761. [DOI] [PubMed] [Google Scholar]
- Sendak RA, Bensadoun A. Identification of a heparin-binding domain in the distal carboxyl-terminal region of lipoprotein lipase by site-directed mutagenesis. J. Lipid Res. 1998;39:1310–1315. [PubMed] [Google Scholar]
- Shimada K, Gill P, Silbert J, Douglas W, Fanburg B. Involvement of cell surface heparin sulfate in the binding of lipoprotein lipase to cultured bovine endothelial cells. J. Clin. Invest. 1981;68:995–1002. doi: 10.1172/JCI110354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JG, Frank S, Kratky D, Hammerle G, Hrzenjak A, Knipping G, von Eckardstein A, Kostner GM, Zechner R. Adenovirus-mediated rescue of lipoprotein lipase-deficient mice. Lipolysis of triglyceride-rich lipoproteins is essential for high density lipoprotein maturation in mice. J. Biol. Chem. 2001;276:36083–36090. doi: 10.1074/jbc.M104430200. [DOI] [PubMed] [Google Scholar]
- Teng B, Davidson NO. Evolution of intestinal apolipoprotein B mRNA editing. Chicken apolipoprotein B mRNA is not edited, but chicken enterocytes contain in vitro editing enhancement factor(s) J. Biol. Chem. 1992;267:21265–21272. [PubMed] [Google Scholar]
- Varma R, Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- Walzem RL, Davis PA, Hansen RJ. Overfeeding increases very low density lipoprotein diameter and causes the appearance of a unique lipoprotein particle in association with failed yolk deposition. J. Lipid Res. 1994;35:1354–1366. [PubMed] [Google Scholar]
- Weinstock PH, Bisgaier CL, Aalto-Setälä K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, Zechner R, Breslow JL. Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired low density lipoprotein clearance in heterozygotes. J. Clin. Invest. 1995;96:2555–2568. doi: 10.1172/JCI118319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.