

Abstract
The chronic myeloproliferative diseases (CMDs) are a group of conditions characterized by unregulated blood cell production, that due either to excessive numbers of erythrocytes, leukocytes or platelets, or their defective function cause symptoms and signs of fatigue, headache, ruddy cyanosis, hemorrhage, abdominal distension, and the complications of vascular thrombosis. In the late 19th century Vaquez provided the first description of polycythemia vera (PV) and Hueck defined idiopathic myelofibrosis (IMF). In 1920, di Guglielmo established criteria for patients with essential thrombocythemia (ET). In 1951 Dameshek argued that these disorders, along with chronic myelogenous leukemia (CML) display many similar clinical and laboratory features (1), and grouped them. In 2002 the World Health Organization expanded the definition of CMDs to also include chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia/hypereosinophilic syndrome (CEL/HES) and systemic mast cell disorder (SMCD; 2). While the molecular pathogenesis of CML is well known (3), and the causes of CEL/HES and SMCD have been identified in about half of all cases (4,5), until very recently the etiologies of the three classically defined CMDs, PV, IMF and ET, were poorly understood. Each of these disorders is characterized by excessive hematopoiesis, a process usually dependent on one or more hematopoietic growth factors (HGFs). This review will focus on how our knowledge of the molecular mechanisms by which HGFs are produced, bind cell surface receptors and transduce survival and proliferative signals have provided the platform on which the multiple origins of CMDs can be understood and novel therapeutic interventions designed.
The Regulation of Hematopoiesis
Each day a normal adult produces ∼2 × 1011 erythrocytes, ∼1 × 1011 leukocytes and ∼1 × 1011 platelets, rates of production that can increase more than ten-fold under conditions of increased need. Careful regulation of hematopoiesis is vital for normal health and well being. All the formed elements of the blood are derived from the hematopoietic stem cell (HSC), functionally defined as a cell that can repopulate all of hematopoiesis following transplantation into a lethally irradiated recipient (6). Active marrow stem cells undergo a series of developmental decisions, giving rise to cells that progressively lose multipotency while undergoing a large number of cell divisions; the magnitude of the cell expansion that characterizes basal hematopoiesis is best illustrated by considering that at any given time only a few thousand human HSCs (7) give rise to the 4 × 1011 blood cells produced daily. Three major mechanisms have been identified that influence both the developmental fate and level of proliferation of HSCs and their progeny: the stochastic rise and fall of lineage specific transcription factors (8), hematopoietic cell-marrow niche interactions (9,10), and carefully orchestrated changes in the levels of HGFs (11).
The HGFs are a group of acidic glycoproteins that bind to specific cell surface receptors and act in both endocrine and paracrine fashions to influence the growth and development of HSCs and their progeny. They are, for the most part, regulated by a physiological need for the blood cell type which they regulate. The HGFs bind to distinct members of the type I cytokine receptor family with high picomolar affinity, an event which triggers receptor conformational changes that transmit biochemical signals for cell survival, proliferation, differentiation and/or activation. A general model of the regulation of HGF production, their route of paracrine or endocrine action, and their target cell effects are depicted in Figure 1. Erythropoietin (EPO), granulocyte colony-stimulating factor (G-CSF), and thrombopoietin (TPO) are the primary regulators of erythrocyte, neutrophil and platelet production, respectively, and their plasma and marrow levels are inversely related to the total body mass of the respective mature blood cell types (12-14). HGF production is regulated at several molecular and cellular levels, including gene transcription, mRNA stability, protein translation and receptor mediated uptake and destruction. Many of the myeloproliferative disorders of man can now be understood as disorders of one of these steps in hematopoietic regulation.
Figure 1.
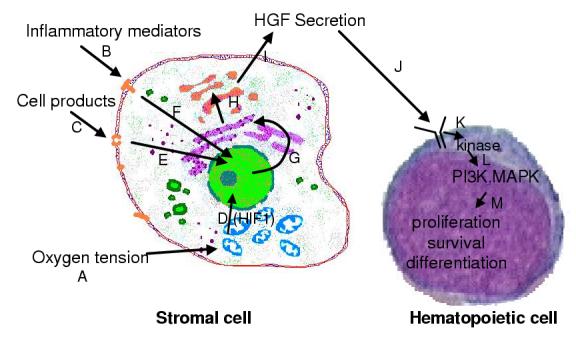
A general scheme of hematopoietic growth factor physiology. A number of modes of cytokine regulation have been identified, and include oxygen tension (acting via hypoxia inducible factor [HIF1; A] on EPO production), inflammatory mediators (acting on G-CSF and TPO production [B]) and blood cell constituents (enhancing TPO production [C]). Once these mediators bind to their corresponding cell surface receptors, or affect intracellular signaling (such as a prolyl hydroxylase [D]), signal transduction mechanisms ultimately result in enhanced growth factor gene transcription (D,E,F), mRNA translation (G,H) and protein secretion (I). Growth factors then act locally (TPO, G-CSF) or at a distance (EPO, G-CSF, TPO) by binding to their receptors on hematopoietic stem and progenitor cells (J), transducing signals that require immediate activation of a receptor-tethered kinase (K) and downstream mediators (L) including phosphoinositol-3-kianse (PI3K) and mitogen activated protein kinase (MAPK), which then alter transcription factors that affect cell survival, proliferation, differentiation or activation (M).
Chronic Myeloproliferative Disorders due to Abnormal Regulation of Hematopoietic Cytokine Expression
Chuvash Polycythemia
Erythropoietin is produced primarily in the kidney in response to poor tissue oxygenation. The primary regulator of EPO transcription is hypoxia inducible factor 1 (HIF1; 15), a heterodimeric transcription factor composed of α and β subunits. Only HIF-1α is responsive to hypoxia, controlled by the level of its ubiquination and proteasomal degradation (16). HIF-1α is susceptible to ubiquination only when combined with the von Hippel Lindau (VHL) protein, which occurs only when the transcription factor is proline hydroxylated by an oxygen-sensitive, iron and reactive oxygen species-dependent prolyl hydroxylase (PHD; Figure 1). Thus, under normoxic conditions, HIF-1α is highly unstable due to chronic hydroxylation, VHL binding, ubiquination and destruction. In contrast, when the juxtamedullary renal tubular cells are hypoxic, the HIF-1α hydroxylase is inactive; VHL fails to target the transcription factor for degradation and HIF-1α levels rise, leading to transcription, translation and secretion of EPO. Thus, disorders of HIF-1α regulation would be expected to alter EPO production and erythropoiesis.
In the early 1970s a large number of patients with polycythemia were described in an isolated Chuvash population of Asian immigrants in the mid Volga region of Russia (17). Molecular studies revealed that virtually all affected individuals were homozygous for an Arg200Trp mutation in the VHL gene (18). The mutation was subsequently found to reduce VHL interaction with hydroxylated HIF-1α, resulting in increased stability of the transcription factor, increased EPO production and enhanced erythropoiesis. Since its discovery, additional pedigrees outside this ethnic group have been identified, although a founder effect has been established.
Familial Thrombocytosis
A second process altered in some patients with CMDs is the rate that HGF mRNA is translated into protein. The efficiency with which protein translation initiates is governed by the context in which the start (AUG) codon resides. In ∼90% of mRNA species the initiation codon is the first AUG present in the transcript, so that protein translation is efficiently initiated. However, if the initiation codon is not the first AUG within the mRNA, and resides within another, out of frame translational open reading frame (ORF),translation of the protein is very inefficient, as post-termination ribosomes cannot scan backwards to initiate at AUG codons positioned upstream of a stop codon (19). The gene for TPO encodes such a transcript; the mature protein is encoded by the 8th AUG present in the mRNA, which lies within the short, out of frame 7th ORF in the transcript. Thus, TPO protein production is inefficient and quite low under normal conditions.
Several patients with an autosomal dominant form of familial thrombocytosis have now been described who bear mutations in the TPO gene. In all such cases the mutation leads to altered translation efficiency of a constant amount of TPO mRNA. In two families, single nucleotide mutations result in abnormal splicing of the primary TPO transcript, resulting in mRNA devoid of the usual TPO initiation codon, but containing a highly efficient, upstream translation initiation codon now in frame with the TPO ORF (20,21). The result of the altered splicing is greatly enhanced TPO protein production. In another pedigree (22), a single nucleotide deletion between the 7th and 8th AUG codons shifts the reading frame of TPO to that of the 7th initiation codon in the mRNA, a highly efficient translation start site, resulting in overproduction of the protein. A third mechanism resulting in this same pathophysiology has been identified (23); a single nucleotide mutation of the 7th ORF creates a stop codon upstream of the 8th (TPO) ORF, allowing ribosomes that complete translation of the now shortened 7th ORF to re-initiate translation downstream, at the 8th AUG, greatly increasing the translation efficiency of the hormone. Overall, the abnormal regulation of TPO in some patients with Familial Thrombocytosis illustrates a second mechanism by which HGF production is increased, altered translational efficiency. Of interest, unlike patients with PV and ET, who for the most part display clonal hematopoiesis, hematopoiesis in individuals with Chuvash Polycythemia (J. Prchal, personal communication, April 2005) and Familial Thrombocytosis (21) is polyclonal, a finding that is expected given the cell extrinsic nature of these disorders.
Myeloproliferative Disorders due to Constitutive Hematopoietic Cytokine Receptor Activation
Familial Thrombocytosis
Hematopoietic growth factors act by binding to specific members of a highly related family of single pass transmembrane proteins. With the cloning of the EPO receptor (24), and numerous interleukin receptors shortly thereafter, the family of type I hematopoietic cytokine receptors was defined (25). These molecules are characterized by one or two 200 amino acid extracellular domains containing four conserved cysteine residues, 14 beta sheets and a five amino acid juxtamembrane domain, Trp-Ser-Xaa-Trp-Ser. These receptors also contain a single 20-25 amino acid transmembrane domain and an intracytoplasmic domain of 70 to over 500 residues. At present, at least 25 hematopoietic cytokines, including EPO, TPO and G-CSF, employ one or more members of this family of proteins to transduce signals for cell survival, proliferation, differentiation or activation.
A large body of evidence indicates that cytokines transduce growth and differentiation signals primarily by phosphorylating, and thereby activating intracellular second messenger proteins. The cytoplasmic domains of the cytokine receptor family contain binding motifs for cytoplasmic protein tyrosine kinases of the Janus (JAK) family, and for both positively- and negatively-acting secondary signaling molecules bearing src homology (SH)2 or phosphotyrosine binding (PTB) domains (26). Much is known of the molecular changes that ensue upon HGF binding to their cognate receptors, based in large measure on studies of EPO and the EPO receptor. X-ray crystallography studies have established that the EPO receptor is a homodimer, and upon ligand binding undergoes a major conformational shift such that the cytoplasmic domains of the receptors shift from 73 Å to 39 Å apart (27), bringing the tethered JAK kinases into closer juxtaposition and allowing their mutual cross-phosphorylation and activation. Once active, JAK phosphorylates one or more Tyr residues within the cytoplasmic domain of the receptor, creating docking sites for both positive and negatively acting secondary signaling molecules that are responsible for the cellular effects of cytokine binding. The conformational change that ensues upon ligand binding to receptor could theoretically be mimicked by alterations in the receptors themselves. In fact, at least in some families, inherited excessive myeloproliferation is due to genetic changes in a hematopoietic cytokine receptor, in either the extracellular or transmembrane domains.
The helical packing that affects the conformation of transmembrane domains of several proteins that transmit signals from outside to inside cells has been extensively studied. Based on these studies the positions in the transmembrane helixes that can lead to either gain- or loss-of-function have been mapped (28,29). In a recent study, 8 members of a family with thrombocytosis were described in which the transmembrane domain of the TPO receptor carried a Ser505Asn mutation (30). Our modeling of the wild-type and mutant transmembrane domains of the receptor predicts that the amino acid change stabilizes the close juxtaposition of the mutant form of the homodimeric receptor, which should enhance its signaling. Other mutations of the TPO receptor closely adjacent to the transmembrane domain are also associated with its constitutive activation, including acquired mutations of W515 that are cause chronic activation of the JAK/STAT signaling pathway and are found in 5-10% of patients with idiopathic myelofibrosis, a chronic myeloproliferative disease (31,32).
The TPO receptor contains two cytokine receptor motifs in its extracellular domain. Structure-function studies have shown that the membrane distal motif is necessary for binding of TPO, and that its elimination leads to constitutive activation of the receptor (33). Based on these findings the membrane distal motif appears to constrain the receptor in a non-signaling conformation; binding of TPO is thought to relieve this conformational constraint. Recently, a single nucleotide polymorphism has been described in Americans of African descent with thrombocytosis; the mutation causes a Lys39Asn mutation in the membrane distal motif of the extracellular domain of the TPO receptor (34). Population studies indicate that the heterozygous state for the Lys39Asn allele is found in 7% of American Blacks, and that its presence is associated with a statistically significantly higher platelet count (mean 424 × 109/L) than individuals lacking the allele (mean 242 × 109/L). It is very tempting to speculate that this mutation inactivates the conformational constraint levied by the membrane distal cytokine receptor motif of the TPO receptor, a hypothesis that requires experimental verification. Although not accounting for a substantial number of patients with thrombocytosis, these examples of mutational receptor activation illustrate how genetic changes can activate a receptor based on mimicry of a ligand-induced conformational change.
Myeloproliferative Disorders of Hematopoietic Cytokine Receptor Down-modulation
Familial Erythrocytosis
Once growth factor receptors have been activated, an efficient mechanism must be in place to limit cell signaling, lest growth control be lost. At least three mechanisms have been identified that blunt or eliminate signaling from HGF receptors minutes to hours following ligand binding (Figure 2): 1) receptor down-modulation (35), 2) activation of protein tyrosine phosphatases (36,37) that remove the activating phosphorylation of receptor and secondary signaling mediators, and 3) induction of suppressors of cytokine signaling (SOCS) molecules (38) that directly inhibit JAK kinase activity or the binding of SH2-containing signaling molecules to phosphotyrosine containing receptor motifs, and that lead to destruction of the signaling complex. Several patients with CMDs have now been described in which these down-modulatory influences on cytokine signaling are lost.
Figure 2.

Mechanisms of hematopoietic cytokine signaling and down-modulation.The three predominant mechanisms that down modulate hematopoietic growth factor (HGF) signaling are illustrated. A. Upon ligand binding the ligand-receptor complex undergoes endocytosis in a clathrin mediated process leading to receptor down-modulation from the cell surface; ligand is ultimately destroyed and the receptors recycled to the cell surface. B. Once the cytoplasmic domain of the HGF receptor is phosphorylated it serves as a docking site for both positive and negatively acting signaling molecules. Amongst the latter are phosphatases, including SHP1 and SHIP1, which remove the activating phosphate groups on JAK2, several signaling intermediates and the receptor itself. C. One of the pro-survival and proliferation mediators, signal transducers and activators of transcription (STAT), also leads to the expression of one or more members of the suppressors of cytokine signaling (SOCS) family. Once transcribed and translated, SOCS proteins inhibit further signaling either by blockade of JAK kinase action or by interfering with the interaction of signaling molecules and phosphorylated docking sites.
“Erythropoietin receptor mutations and Olympic glory” read the title of an editorial (39) commenting upon identification of the cause of erythrocytosis in a large kindred of ruddy Finns (40). Eero Mäntyranta won gold medals in the 1960 Squaw Valley and 1964 Innsbruck Olympics in cross country skiing, and is a member of a >200 person pedigree characterized by autosomal dominant erythrocytosis. Sequencing of the EPO receptor in affected individuals in the family revealed a non-sense codon, prematurely terminating the cytoplasmic domain of the receptor. Several additional individuals or families with polycythemia have been described with frameshift mutations in the same region of the receptor (41-43). Structure-function studies of artificially truncated EPO receptors revealed that the distal 40-70 amino acids of the receptor bind one or more negatively regulating signaling molecules; while the nature of the negative regulator of signaling responsible for these causes of familial erythrocytosis have not yet been definitely established, experiments in mice have shown that genetic elimination of two signal terminating phosphatases, SHP1 (44) and SHIP1 (45), which bind to this region of EPO receptor, also lead to chronic myeloproliferation.
Severe Congenital Neutropenia associated Acute Myelogenous Leukemia
A second example of pathological myeloproliferation accompanying the loss of the negative regulatory domain of a cytokine receptor is found in patients with Severe Congenital Neutropenia (SCN), or Kostmann syndrome. Now known to be caused by missense mutations of the neutrophil elastase gene (ELA2; 46), Kostmann described neutropenia in several families in Northwestern Sweden. Since the initial description of the disorder, SCN has been described in numerous populations, and while considered a rate disorder, nearly 1000 patients have been followed in a single registry (47). SCN is genetically heterogeneous (48), with most cases arising sporadically, consistent with its transmission as an often lethal, autosomal dominant disorder. While previously fatal during childhood or early adolescence due to the development of antibiotic resistant bacterial infections, patients with SCN treated with G-CSF do remarkably well, living many decades, if not a completely normal life. While no abnormalities are seen in germ line DNA other than in the elastase gene, as a consequence of the increased longevity afforded by treatment with the cytokine, or because of cytokine induced selection, ∼20% of patients with SCN are found to harbor a variety of non-sense mutations resulting in a prematurely truncated G-CSF receptor (49). Of considerable interest, most of these patients develop myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML; 50), and when this occurs, virtually all the leukemic cells display the truncated receptor. Like that for EPO receptor, the G-CSF receptor encodes a carboxyl-terminal negative regulatory domain, lost in the MDS/AML associated receptor truncations, and shown in vitro to mediate G-CSF down modulation by binding both the signal terminating phosphatase SHIP1 and CIS, a SOCS protein (51). However, the G-CSF receptor truncation is not sufficient for the development of MDS/AML, as mice engineered to express the mutant allele remain free of disease (52). It has been postulated that rather than being leukemogenic alone, the altered receptor cooperates with additional transforming events during the development of MDS or AML (53). Additional work is required to more fully understand the precise mechanisms of leukemogenesis in the presence of a truncated G-CSF receptor, but it is clear that the MDS and AML that occurs in ∼20% of patients with SCN is very likely related, at least in part, to the truncated cytokine receptor.
Myeloproliferative Disorders of the Cytokine Signal Transduction Apparatus
Acquired Polycythemia Vera, Idiopathic Myelofibrosis and Essential Thrombocythemia
As noted earlier, hematopoietic cytokine receptor signal transduction is initiated by the juxtaposition and cross activation of JAK kinases. While four members of this family of kinases have been described, JAK2 is particularly important for myeloproliferation, as the EPO, TPO, G-CSF, stem cell factor, interleukin (IL)-3, and IL-5 receptors all employ this kinase for signal transduction (26). In addition to our understanding of the mechanism of JAK activation provided by the crystallographic studies of the EPO receptor (27), additional insights into the molecular basis of kinase activation have come from studies in which truncated forms of JAK2 were characterized, and from molecular modeling studies of the protein. Three major structural domains have been identified in all members of the JAK kinase family, 1) the JH1 or kinase domain, 2) the JH2 or pseudokinase domain, and 3) the FERM (Four-point-one, Ezrin, Radixin, Moesin) or receptor binding domain (54). While the function of JH1 and FERM are well known, the function of the pseudokinase domain, so named because of its imperfect homology to protein tyrosine kinases, is devoid of kinase activity due to alteration of critical active site residues. Recent studies have shown that while expression of JH1 alone leads to substantial kinase activity, when coupled to JH2 the kinase domain activity is substantially blunted, implying that JH2 represents a physiologically relevant regulatory domain (55). Molecular modeling studies have provided additional insights into the natural mechanism of JAK kinase activation; a model of JH1-JH2 based on the x-ray crystallography based tertiary structure of the fibroblast growth factor receptor kinase dimer suggests that JH2 stabilizes the kinase activation loop of JH1 in the inactive conformation (56).
The three classic chronic myeloproliferative disorders, PV, IMF and ET share many common clinical features, first recognized over 50 years ago by Dameshek (1). More recently, this concept has been strengthened by the observation that all three disorders represent clonal myeloproliferation of an HSC (57-59), marrow and blood-derived hematopoietic progenitor cells display enhanced sensitivity to multiple hematopoietic cytokines (EPO, TPO, IL-3; 60-62), the three disorders can transform into acute myelogenous leukemia or to an end-stage fibrotic marrow, albeit with different frequencies, and the three are clinically characterized by a hypercoagulable state (63). Even more recently, multiple lines of investigation have indicated that the chronic myeloproliferative diseases represent disorders of the hematopoietic cytokine signaling apparatus. For example, numerous animal models of chronic myeloproliferation can be developed by altered expression of hematopoietic cytokine signaling elements, including transgenic expression of murine TPO, that results in thrombocytosis or aggressive myelofibrosis, depending on the site of expression (64,65). Mutation of either the EPO or TPO receptors results in autonomous cell growth in cytokine dependent murine cell lines and their transformation to leukemia when transplanted into mice (66,67). Loss of a Ras regulatory protein, RasGAP, or overexpression of an activated form of Ras, a key component of hematopoietic cytokine signaling, results in a murine myeloproliferative syndrome (68,69). And loss of regulatory hematopoietic phosphatases results in murine models of chronic myeloproliferative syndromes (44,45). Consistent with these animal studies, several secondary hematopoietic cytokine signaling molecules are constitutively active in marrow cells derived from patients with chronic myeloproliferative syndromes, including STAT3, BclXL and Akt (70-72).
Based on these findings, on finding loss of heterozygosity on the short arm of chromosome 9 in some patients with CMDs, the effects of JAK kinase inhibitors on hematopoietic growth in PV, and a mutational survey of multiple kinases in patients with myeloproliferative disorders, several groups began to explore whether JAK2 might be responsible for the chronic myeloproliferative disorders. Recently, multiple groups reported that from 65 to 97% of patients with PV, from 35 to 57% of patients with IMF, and from 23 to 57% of patients with ET express the same, acquired, mutant form of JAK2 in hematopoietic, but not non-hematopoietic cells (73-78). Additional studies have established that the altered kinase, bearing a Val617Phe missense mutation in the JH2, pseudokinase domain, is constitutively active (74-76), leads to chronic activation of multiple secondary mediators of hematopoietic cytokine receptor signaling (75,76), mutant kinase is found in all growth factor hypersensitive marrow cells derived from patients with these disorders (73), and when expressed in murine marrow cells, the mutant kinase but not a wild type control JAK2 leads to polycythemia (75). Of clinical interest, in one of the clinical studies patients that carry the mutant JAK2 gene have a more aggressive disease than those patients devoid of the abnormal gene product (76), as measured by a near doubling of the rates for developing myelofibrosis, hemorrhage and/or thrombosis. While these remarkable studies establish that most patients with chronic myeloproliferative disorders carry a mutation in the primary signal transducing kinase of hematopoiesis, and provide an unparalleled opportunity to develop enhanced diagnostic tools and prognostication for patients with CMDs, they raise additional questions. For example, how can one genetic lesion lead to three rather distinct clinical entities? Do the ∼35% of patients with two Val617Phe alleles (due to mitotic recombination) do more poorly than those with a single mutant gene? Can a specific therapy designed to inhibit the constitutively active mutant of JAK2 be designed, one that avoids inhibiting the wild type kinase? Is the constitutive activation of JAK2 responsible for the hypercoagulable state that characterizes CMDs, and is it due to activation of platelets, neutrophils or endothelial cells? With all remarkable discoveries come great opportunities to further our understanding of human physiology and pathology.
Knowledge of how cytokines lead to hematopoietic cell survival and proliferation has lead to multiple examples of therapeutic intervention in disorders of blood cell production; the use of EPO, G-CSF and pending use of TPO mimetics to bolster erythrocyte, neutrophil and platelet production serve as landmarks of pharmacology. By applying our understanding of the molecular basis for hematopoiesis, from cytokine production, to receptor binding and activation, and through intracellular signaling provides an outstanding platform to understand chronic disorders of overproduction of one or more hematopoietic cell lineages. Moreover, these pathophysiological insights are almost certain to lead to additional rationally designed therapeutic opportunities, such as the generation of a Val617Phe JAK2 kinase inhibitor for the CMDs associated with the mutant kinase, inhibitors of EPO or its production for the erythrocytosis seen in Chuvash-type polycythemia, or strategies to enhance the negative regulators of hematopoietic cytokine signaling seen in children with SCN and secondary MDS/AML, or for the ruddy Finns.
Biography
Biographical sketch of the author

Dr. Kenneth Kaushansky received his medical training at the University of California, Los Angeles, School of Medicine, performed his internship, residency and chief residency in Internal Medicine at the University of Washington, and received fellowship training in Hematology at the University of Washington before joining the faculty there in 1985. In Seattle, Dr. Kaushansky developed a research interest in hematopoietic growth factors, cloning human GM-CSF, interleukin-3, and most recently thrombopoietin, studying their structure-function relationships, regulation of production and physiological effects. For this work he has received several awards, including the Dameshek Award from the American Society of Hematology and the Outstanding Investigator Award from the American Society for Medical Research, and was elected to membership in the American Society for Clinical Investigation, the Association of American Physicians, the Institute of Medicine and the American Academy of Arts and Sciences. In 2002 Dr. Kaushansky moved to the University of California, San Diego to become the Helen M. Ranney Professor and Chair of the Department of Medicine, but continues his research interests in hematopoietic growth factors.
References
- 1.Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372–375. [PubMed] [Google Scholar]
- 2.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 3.Melo JV, Deininger MW. Biology of chronic myelogenous leukemia--signaling pathways of initiation and transformation. Hematol Oncol Clin North Am. 2004;18:545–568. doi: 10.1016/j.hoc.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Gotlib J, Cools J, Malone JM, 3rd, Schrier SL, Gilliland DG, Coutre SE. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: implications for diagnosis, classification, and management. Blood. 2004;103:2879–2891. doi: 10.1182/blood-2003-06-1824. [DOI] [PubMed] [Google Scholar]
- 5.Valent P, Akin C, Sperr WR, Horny HP, Metcalfe DD. Mast cell proliferative disorders: current view on variants recognized by the World Health Organization. Hematol Oncol Clin North Am. 2003;17:1227–1241. doi: 10.1016/s0889-8588(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 6.Shizuru JA, Negrin RS, Weissman IL. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu Rev Med. 2005;56:509–538. doi: 10.1146/annurev.med.54.101601.152334. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt M, Zickler P, Hoffmann G, Haas S, Wissler M, Muessig A, Tisdale JF, Kuramoto K, Andrews RG, Wu T, Kiem HP, Dunbar CE, von Kalle C. Polyclonal long-term repopulating stem cell clones in a primate model. Blood. 2002;100:2737–2743. doi: 10.1182/blood-2002-02-0407. [DOI] [PubMed] [Google Scholar]
- 8.Cantor AB, Orkin SH. Hematopoietic development: a balancing act. Curr Opin Genet Dev. 2001;11:513–519. doi: 10.1016/s0959-437x(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 9.Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105:2631–2639. doi: 10.1182/blood-2004-06-2480. [DOI] [PubMed] [Google Scholar]
- 10.Arai F, Hirao A, Suda T. Regulation of hematopoietic stem cells by the niche. Trends Cardiovasc Med. 2005;15:75–79. doi: 10.1016/j.tcm.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Kaushansky K. Hematopoietic stem cells, progenitor cells, and cytokines, chapter 15 in Williams Hematology. 7th ed. Accepted for publication; [Google Scholar]
- 12.Krantz SB. Erythropoietin. Blood. 1991;77:419–434. [PubMed] [Google Scholar]
- 13.Anderlini P, Przepiorka D, Champlin R, Korbling M. Biologic and clinical effects of granulocyte colony-stimulating factor in normal individuals. Blood. 1996;88:2819–2825. [PubMed] [Google Scholar]
- 14.Kaushansky K. Thrombopoietin. (Drug Therapy Series).New Engl J Med. 1998;339:746–754. doi: 10.1056/NEJM199809103391107. [DOI] [PubMed] [Google Scholar]
- 15.Ratcliffe PJ, O’Rourke JF, Maxwell PH, Pugh CW. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol. 1998;201:1153–1162. doi: 10.1242/jeb.201.8.1153. [DOI] [PubMed] [Google Scholar]
- 16.Maxwell PH, Ratcliffe PJ. Oxygen sensors and angiogenesis. Semin Cell Dev Biol. 2002;13:29–37. doi: 10.1006/scdb.2001.0287. [DOI] [PubMed] [Google Scholar]
- 17.Poliakova LA. Familial erythrocytosis among the residents of the Chuvash ASSR. Probl Gematol Pereliv Krovi. 1974;19:30–33. [PubMed] [Google Scholar]
- 18.Pastore YD, Jelinek J, Ang S, Guan Y, Liu E, Jedlickova K, Krishnamurti L, Prchal JT. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood. 2003;101:1591–1595. doi: 10.1182/blood-2002-06-1843. [DOI] [PubMed] [Google Scholar]
- 19.Kozak M. Constraints on reinitiation of translation in mammals. Nucleic Acids Res. 2001;29:5226–5232. doi: 10.1093/nar/29.24.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet. 1998;18:49–52. doi: 10.1038/ng0198-49. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen MJ, Raskind WH, Wolff JF, Bachrach HR, Kaushansky K. Familial thrombocytosis associated with overproduction of thrombopoietin due to a novel splice donor site mutation. Blood. 1998;92(Suppl 1):205a. [Google Scholar]
- 22.Ghilardi N, Wiestner A, Kikuchi M, Ohsaka A, Skoda RC. Hereditary thrombocythaemia in a Japanese family is caused by a novel point mutation in the thrombopoietin gene. Br J Haematol. 1999;107:310–316. doi: 10.1046/j.1365-2141.1999.01710.x. [DOI] [PubMed] [Google Scholar]
- 23.Kondo T, Okabe M, Sanada M, Kurosawa M, Suzuki S, Kobayashi M, Hosokawa M, Asaka M. Familial essential thrombocythemia associated with one-base deletion in the 5′-untranslated region of the thrombopoietin gene. Blood. 1998;92:1091–1096. [PubMed] [Google Scholar]
- 24.D’Andrea AD, Lodish HF, Wong GG. Expression cloning of the murine erythropoietin receptor. Cell. 1989;57:277–285. doi: 10.1016/0092-8674(89)90965-3. [DOI] [PubMed] [Google Scholar]
- 25.Cosman D. The hematopoietin receptor superfamily. Cytokine. 1993;5:95–106. doi: 10.1016/1043-4666(93)90047-9. [DOI] [PubMed] [Google Scholar]
- 26.Ihle JN. Cytokine receptor signaling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 27.Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–993. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- 28.Weiner DB, Liu J, Cohen JA, Williams WV, Greene MI. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]
- 29.Govaerts C, Lefort A, Costagliola S, Wodak SJ, Ballesteros JA, VanSande J, Pardo L, Vassart G. A conserved Asn in transmembrane helix 7 is an on/off switch in the activation of the thyrotropin receptor. J Biol Chem. 2001;276:22991–22999. doi: 10.1074/jbc.M102244200. [DOI] [PubMed] [Google Scholar]
- 30.Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, Tsuboi K, Nitta M, Miyazaki H, Iida S, Ueda R. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103:4198–4200. doi: 10.1182/blood-2003-10-3471. [DOI] [PubMed] [Google Scholar]
- 31.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, Deangelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is a novel somatic activating mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006 Jul 18;3(7):e270. doi: 10.1371/journal.pmed.0030270. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006 Jul 25; doi: 10.1182/blood-2006-04-018879. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Sabath DF, Kaushansky K, Broudy VC. Deletion of the membrane-distal cytokine receptor homology domain of MPL results in constitutive cell growth and loss of thrombopoietin binding. Blood. 1999;94:365–367. [PubMed] [Google Scholar]
- 34.Moliterno AR, Williams DM, Gutierrez-Alamillo LI, Salvatori R, Ingersoll RG, Spivak JL. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci U S A. 2004;101:11444–11447. doi: 10.1073/pnas.0404241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verdier F, Walrafen P, Hubert N, Chretien S, Gisselbrecht S, Lacombe C, Mayeux P. Proteasomes regulate the duration of erythropoietin receptor activation by controlling down-regulation of cell surface receptors. J Biol Chem. 2000;275:18375–18381. doi: 10.1074/jbc.275.24.18375. [DOI] [PubMed] [Google Scholar]
- 36.Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- 37.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997;15:302–307. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 38.Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem. 2004;279:821–824. doi: 10.1074/jbc.R300030200. [DOI] [PubMed] [Google Scholar]
- 39.Longmore G. Erythropoietin receptor mutations and Olympic glory. Nat Genet. 1993;4:108–110. doi: 10.1038/ng0693-108. [DOI] [PubMed] [Google Scholar]
- 40.de la Chapelle A, Traskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci U S A. 1993;90:4495–4499. doi: 10.1073/pnas.90.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokol L, Luhovy M, Guan Y, Prchal JF, Semenza GL, Prchal JT. Primary familial polycythemia: A frameshift mutation in the erythropoietin receptor gene and increased sensitivity of erythroid progenitors to erythropoietin. Blood. 1995;86:15–22. [PubMed] [Google Scholar]
- 42.Watowich SS, Xie X, Klingmuller U, Kere J, Lindlof M, Berglund S, de la Chapelle A. Erythropoietin receptor mutations associated with familial erythrocytosis cause hypersensitivity to erythropoietin in the heterozygous state. Blood. 1999;94:2530–2532. [PubMed] [Google Scholar]
- 43.Arcasoy MO, Karayal AF, Segal HM, Sinning JG, Forget BG. A novel mutation in the erythropoietin receptor gene is associated with familial erythrocytosis. Blood. 2002;99:3066–3069. doi: 10.1182/blood.v99.8.3066. [DOI] [PubMed] [Google Scholar]
- 44.Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 45.Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, Boxer LA, Kannourakis G, Zeidler C, Welte K, Benson KF, Horwitz M. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–2322. [PubMed] [Google Scholar]
- 47.Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Freedman MH, Kannourakis G, Kinsey SE, Davis R, Scarlata D, Schwinzer B, Zeidler C, Welte K. Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol. 2003;72:82–93. doi: 10.1002/ajh.10255. [DOI] [PubMed] [Google Scholar]
- 48.Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, Vaury C, Barkaoui M, Fenneteau O, Maier-Redelsperger M, Chomienne C, Donadieu J. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004;103:4119–4125. doi: 10.1182/blood-2003-10-3518. [DOI] [PubMed] [Google Scholar]
- 49.Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333:487–493. doi: 10.1056/NEJM199508243330804. [DOI] [PubMed] [Google Scholar]
- 50.Freedman MH, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol. 2002;39:128–133. doi: 10.1053/shem.2002.31912. [DOI] [PubMed] [Google Scholar]
- 51.Hunter MG, Jacob A, O’donnell LC, Agler A, Druhan LJ, Coggeshall KM, Avalos BR. Loss of SHIP and CIS recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J Immunol. 2004;173:5036–5045. doi: 10.4049/jimmunol.173.8.5036. [DOI] [PubMed] [Google Scholar]
- 52.Hermans MH, Ward AC, Antonissen C, Karis A, Lowenberg B, Touw IP. Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood. 1998;92:32–39. [PubMed] [Google Scholar]
- 53.van de Geijn GJ, Aarts LH, Erkeland SJ, Prasher JM, Touw IP. Granulocyte colony-stimulating factor and its receptor in normal hematopoietic cell development and myeloid disease. Rev Physiol Biochem Pharmacol. 2003;149:53–71. doi: 10.1007/s10254-003-0014-0. [DOI] [PubMed] [Google Scholar]
- 54.Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19:5662–5679. doi: 10.1038/sj.onc.1203925. [DOI] [PubMed] [Google Scholar]
- 55.Saharinen P, Vihinen M, Silvennoinen O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol Biol Cell. 2003;14:1448–1459. doi: 10.1091/mbc.E02-06-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14:27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 57.Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 1976;295:913–916. doi: 10.1056/NEJM197610212951702. [DOI] [PubMed] [Google Scholar]
- 58.Jacobson RJ, Salo A, Fialkow PJ. Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood. 1978;51:189–194. [PubMed] [Google Scholar]
- 59.Fialkow PJ, Faguet GB, Jacobson RJ, Vaidya K, Murphy S. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood. 1981;58:916–919. [PubMed] [Google Scholar]
- 60.Zanjani ED, Lutton JD, Hoffman R, Wasserman LR. Erythroid colony formation by polycythemia vera bone marrow in vitro. Dependence on erythropoietin. J Clin Invest. 1977;59:841–848. doi: 10.1172/JCI108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai CH, Krantz SB, Dessypris EN, Means RT, Jr, Horn ST, Gilbert HS. Polycythemia vera. II. Hypersensitivity of bone marrow erythroid, granulocyte-macrophage, and megakaryocyte progenitor cells to interleukin-3 and granulocyte-macrophage colony-stimulating factor. Blood. 1992;80:891–899. [PubMed] [Google Scholar]
- 62.Axelrad AA, Eskinazi D, Correa PN, Amato D. Hypersensitivity of circulating progenitor cells to megakaryocyte growth and development factor (PEG-rHu MGDF) in essential thrombocythemia. Blood. 2000;96:3310–3321. [PubMed] [Google Scholar]
- 63.Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, Marilus R, Villegas A, Tognoni G, Barbui T. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23:2224–2232. doi: 10.1200/JCO.2005.07.062. [DOI] [PubMed] [Google Scholar]
- 64.Zhou W, Toombs CF, Zou T, Guo J, Robinson MO. Transgenic mice overexpressing human c-mpl ligand exhibit chronic thrombocytosis and display enhanced recovery from 5-fluorouracil or antiplatelet serum treatment. Blood. 1997;89:1551–1559. [PubMed] [Google Scholar]
- 65.Yan X-Q, Lacey D, Hill D, Chen Y, Fletcher F, Hawley RG, McNiece IK. A model of myelofibrosis and osteosclerosis in mice induced by overexpressing thrombopoietin (mpl ligand): Reversal of disease by bone marrow transplant. Blood . 1996;88:402–409. [PubMed] [Google Scholar]
- 66.Pharr PN, Hankins D, Hofbauer A, Lodish HF, Longmore GD. Expression of a constitutively active erythropoietin receptor in primary hematopoietic progenitors abrogates erythropoietin dependence and enhances erythroid colony-forming unit, erythroid burst-forming unit, and granulocyte/macrophage progenitor growth. Proc Natl Acad Sci U S A. 1993;90:938–942. doi: 10.1073/pnas.90.3.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Onishi M, Mui ALF, Morikawa Y, Cho L, Kinoshita S, Nolan GP, Gorman DM, Miyajima A, Kitamura T. Identification of an oncogenic form of the thrombopoietin receptor using retrovirus-mediated gene transfer. Blood. 1996;88:1399–1406. [PubMed] [Google Scholar]
- 68.Gitler AD, Kong Y, Choi JK, Zhu Y, Pear WS, Epstein JA. Tie2-Cre-induced inactivation of a conditional mutant Nf1 allele in mouse results in a myeloproliferative disorder that models juvenile myelomonocytic leukemia. Pediatr Res. 2004;55:581–584. doi: 10.1203/01.PDR.0000113462.98851.2E. [DOI] [PubMed] [Google Scholar]
- 69.Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, Johnson L, Akashi K, Tuveson DA, Jacks T, Gilliland DG. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roder S, Steimle C, Meinhardt G, Pahl HL. STAT3 is constitutively active in some patients with Polycythemia rubra vera. Exp Hematol. 2001;29:694–702. doi: 10.1016/s0301-472x(01)00637-3. [DOI] [PubMed] [Google Scholar]
- 71.Silva M, Richard C, Benito A, Sanz C, Olalla I, Fernandez-Luna JL. Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. N Engl J Med. 1998;338:564–571. doi: 10.1056/NEJM199802263380902. [DOI] [PubMed] [Google Scholar]
- 72.Dai C, Chung IJ, Krantz SB. Increased erythropoiesis in polycythemia vera is associated with increased erythroid progenitor proliferation and increased phosphorylation of Akt/PKB. Exp Hematol. 2005;33:152–158. doi: 10.1016/j.exphem.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 73.Baxter EJ, Scott LM, Campbell PJ, East C, Fouroudas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative diseases. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 74.Levine RL, Wadleigh M, Cools J, Ebert BL, e Wernig G, Huntly BJP, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 75.James C, Ugo V, Le Couedic JP, Staerk J, Delmonneau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 76.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 77.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, Zhao ZJ. Identification of an acquired JAK2 mutation in Polycythemia vera. J Biol Chem. 2005 Apr 29; doi: 10.1074/jbc.C500138200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, White H, Zoi C, Loukopoulos D, Terpos E, Vervessou EC, Schultheis B, Emig M, Ernst T, Lengfelder E, Hehlmann R, Hochhaus A, Oscier D, Silver RT, Reiter A, Cross NC. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005 May 26; doi: 10.1182/blood-2005-03-1320. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]