

Abstract
Neutrophil elastase (NE), a potent neutrophil inflammatory mediator, increases MUC5AC mucin gene expression through undefined pathways involving reactive oxygen species. To determine the source of NE-generated reactive oxygen species, we used pharmacologic inhibitors of oxidoreductases to test whether they blocked NE-regulated MUC5AC mRNA expression. We found that dicumarol, an inhibitor of the NADP(H) quinone oxidoreductase 1 (NQO1), inhibited MUC5AC mRNA expression in A549 lung adenocarcinoma cells and primary normal human bronchial epithelial (NHBE) cells. We further tested the role of NQO1 in mediating NE-induced MUC5AC expression by inhibiting NQO1 expression using siRNA. Transfection with short interfering RNA (siRNA) specific for NQO1 suppressed NQO1 expression and significantly abrogated MUC5AC mRNA expression. NE treatment caused lipid peroxidation in A549 cells; this effect was inhibited by pretreatment with dicumarol, suggesting that NQO1 also regulates oxidant stress in A549 cells following NE exposure. NE exposure increased NQO1 protein and activity levels; NQO1 expression and activity were limited to the cytosol and did not translocate to the plasma membrane. Our results indicate that NQO1 has an important role as a key mediator of NE-regulated oxidant stress and MUC5AC mucin gene expression.
Keywords: Neutrophil elastase, NADP(H) quinone oxidoreductase 1, MUC5AC, reactive oxygen species
Introduction
Cystic fibrosis (CF), one of the most common fatal hereditary disorders among Caucasians [1], and chronic bronchitis (CB) [2], are characterized by chronic mucus obstruction and severe neutrophilic inflammation in the airways. Expression of MUC5AC, a major secreted, gel-forming respiratory tract mucin, is closely linked to goblet cell metaplasia and mucus hypersecretion [3]. Therefore, regulation of MUC5AC is a critical component of the pathogenesis of mucus overproduction in CF and CB.
Neutrophils release neutrophil elastase (NE), a serine protease present in abnormally high concentrations in the lungs of patients with CF and CB [4, 5]. NE contributes to CF and CB pathology by inducing goblet cell metaplasia [6, 7], triggering mucin secretion [8-10], and increasing mucin mRNA and glycoprotein expression [11-13]. Results from this laboratory have shown that in airway epithelial cells, NE upregulates MUC5AC gene expression by increasing mRNA stability [11]. In addition, dimethylthiourea, a broad spectrum scavenger of free radicals, and desferrioxamine, an iron chelator, attenuate NE-induced MUC5AC expression. [14]. These data suggest that reactive oxygen species (ROS) mediate NE-induced MUC5AC expression; however, the source of ROS remains unknown.
There are several candidate pro-oxidant enzymes that may be induced by NE. NE is known to increase the conversion of xanthine dehydrogenase to xanthine oxidase in endothelial cells [15]. Aoshiba et. al. have also shown in epithelial cells that NE increases production of ROS via mitochondrial sources [16]. Duox1, a homologue of the gp91phox subunit of NADPH oxidase, has been shown to be highly expressed in epithelial cells of large airways. In cultured normal human bronchial epithelial cells (NHBE), Duox1 activity generates hydrogen peroxide [17], at the apical cell surface. Our report focuses on another oxidoreductase, NAD(P)H:quinone oxidoreductase 1 (NQO1, EC 1.6.99.2), which is highly expressed in the lung [18]. NQO1 uses either NADH or NADPH as a reducing cofactor to catalyze the obligate two-electron reduction of quinones to hydroquinones [19]. Depending on the type of hydroquinone formed, reduction by NQO1 can result in a more stable product or a redox-labile product that is able to form ROS [20]. Therefore, NQO1 is a potential source of NE-induced oxidant stress. Importantly, NQO1 has recently been linked epidemiologically to pulmonary susceptibility to ozone in healthy individuals [21], and to childhood asthma in Mexico city [22], suggesting that NQO1 may have important functions in the lung. In this report, we evaluate the role of NQO1 in mediating MUC5AC mRNA expression and oxidant stress in airway epithelial cells.
Materials and Methods
Reagents
A549 cells (CCL-185) were obtained from ATCC (Rockville, MD). Ham's F-12 and F12K media, minimal essential medium (MEM), phenol red-free Dulbecco's modified eagle medium (DMEM), Opti-MEM, non-essential amino acids, sodium pyruvate, fetal bovine serum (FBS), penicillin, streptomycin, trypsin-EDTA, phosphate buffered saline (PBS), bovine serum albumin (BSA), and Trizol were from Gibco/Invitrogen (Carlsbad, CA). 6-well Transwell clear chambers (24 mm diameter) were from Corning Costar (Corning, NY). Neutrophil elastase (875 U/mg protein) was from Elastin Products, Co. (Owensville, MO). Nylon filter (Nytran plus) was from Schleicher and Schuell (Keene, NH). X-Omat AR film was purchased from Kodak (Rochester, NY). [α-32P]dCTP, [γ-32P]ATP, horseradish peroxidase-conjugated sheep anti-mouse IgG secondary antibody, and the enhanced chemiluminescence (ECL) developing reagent ECL Plus were from Amersham Biosciences/GE Health Care (Piscataway, NJ). Biospin columns, sodium dodecyl sulfate, DC Protein Assay Kit, 12% Tris-HCL ready gels, Mini-PROTEAN 3 system, Precision Plus Kaleidoscope ladder and nitrocellulose membrane were from Bio-Rad Laboratories (Hercules, CA) and cesium chloride from ICN/MP Biomedicals (Costa Mesa, CA). Monoclonal mouse anti-E-cadherin IgG was from BD Biosciences (San Jose, CA). Polyclonal goat anti-NQO1 antibody and horseradish peroxidase-conjugated bovine anti-goat secondary antibody used for A549 westerns were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal goat anti-NQO1 antibody and horseradish peroxidase-conjugated rabbit anti-goat secondary antibody used for primary normal human bronchial epithelial (NHBE) cell westerns were from Abcam (Cambridge, MA). Monoclonal anti-β-actin antibody was from Sigma Chemical Co. (St. Louis, MO). TaqMan gene expression assays for MUC5AC, NQO1 and 18s ribosomal RNA control, 2X universal PCR master mix, RNase inhibitor and MultiScribe reverse transcriptase were from Applied Biosystem (Foster City, CA). Lactate dehydrogenase assay was from Promega Corp. (Madison, WI). siPort Amine was from Ambion Inc. (Austin, TX). Short interfering RNA (siRNA) smartpool for NQO1 and negative control siRNA were from Dharmacon, Inc. (Lafayette, CO). Tris, and glycine were from EM Science (Kansas City, MO). NaCl and methanol were from Mallinckrodt (Hazelwood, MO). Prime-It Kit was from Stratagene (La Jolla, CA) and thymidine kinase end-labeling kit was from Gibco/Invitrogen (Carlsbad, CA). Statistical software package, Statistix 8.0 was from Analytical Software (Tallahassee FL). Dicumarol and all other chemicals were from Sigma Chemical Co. (St. Louis, MO).
Cell Culture
A549, a lung adenocarcinoma cell line, was cultured in Ham's F-12K medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were grown at 37°C in a humidified 5% CO2 atmosphere to 90-100% confluence, and then changed to serum-free medium for experiments. Primary normal human bronchial epithelial (NHBE) cells were harvested from human tracheobronchial tissues from 3 donors obtained from the National Disease Research Interchange or from the Dept. of Pathology, Duke University Medical Center. The protocol was approved by the Institutional Review Board for Clinical Investigations, Duke University Medical Center. After initial harvest and expansion, cells were plated on a 6-well Transwell Clear chambers (24 mm diameter) at 1–2 × 104 cells/cm2, in a Ham's F12:DMEM (1:1) supplemented with eight factors: insulin (5 μg/ml), transferrin (5 μg/ml), epidermal growth factor (10 ng/ml), dexamethasone (0.1 μM), cholera toxin (10 ng/ml), bovine hypothalamus extract (15 μg/ml), BSA (0.5 mg/ml), and all-trans-retinoic acid (RA) (30 nM). The RA was added fresh at each media change. After 4-7 days in immersed culture condition, cultured cells were shifted to an air–liquid interface culture condition (ALI). During ALI, 200 μl of media was on the apical surface to simulate normal mucociliary clearance. Experiments were performed at Day 10 after the change of the culture condition from immersed to ALI. NHBE cells were starved in media supplemented with only two factors, BSA and RA, for 24h before NE treatment [23]. Cells were preincubated with dicumarol (4-30 μM) or the equivalent volume of control vehicle (0.1 M sodium hydroxide) and then coincubated with NE or the equivalent volume of control vehicle (50:50, glycerol: 0.02 M sodium acetate, pH 5.0) for 1-6h. Lactate dehydrogenase assay was used to determine cytotoxicity under all treatment conditions [14].
MUC5AC mRNA expression by Northern Analysis
Following pretreatment with dicumarol (30 μM, 1h) or control vehicle and then coincubation with NE (100 nM, 6h) or control vehicle, RNA was isolated from A549 cells as previously described [11] by the guanidinium thiocynanate/cesium chloride method. Total RNA (10μg) was separated by electrophoresis on a 1.2% agarose-formaldehyde gel and transferred by capillary blot to a nylon filter in 1M ammonium acetate. After UV crosslinking, the filters were hybridized at 62°C with 32P- labeled probes for MUC5AC and 28s rRNA. Radiolabeled cDNA probes for MUC5AC were prepared using Prime-It Kit according to manufacturer's instructions using a 340bp template for MUC5AC [11]. Radiolabeled probes for 28s rRNA [24] were prepared by end-labeling using thymidine kinase according to manufacturer's instructions. Filters were washed twice with 2× saline sodium citrate (SSC; 0.3M NaCl, 0.03M sodium citrate) and 0.1% sodium dodecyl sulfate at room temperature for 30 min, and then with 0.1× SSC and 0.1% SDS at 62°C for 15 min. Filters were exposed for autoradiography at −80°C. Band density on autoradiographs was determined by digitalization with Fotolook and Photoshop softwares and quantitation using NIH image software and ImageQuant TL (Amersham Biosciences/GE Health care, Piscataway, NJ).
siRNA Transfections
A549 cells (6 well plate) were transfected with Opti-MEM (1 ml) containing siPort Amine (9μl) and NQO1 siRNA (20 nM) or negative control siRNA (20 nM) at 37 °C, 4 h, and then OptiMEM (1 ml) was added to each well for an additional 18h incubation. After a total of 22h, transfected A549 cells were treated with NE (100 nM) or control vehicle for 6h, and total RNA was isolated using Trizol reagent. Suppression of NQO1 expression was evaluated at the mRNA and protein level at 24 h by real-time RT-PCR and western analysis. The effect of NQO1 knockdown on NE-induced MUC5AC expression was evaluated by real-time RT-PCR. Each condition was performed in triplicate wells and transfection reagent master mix was made for each condition to minimize well-to-well variation.
Real-Time Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR) for NQO1, MUC5AC, and 18s rRNA
NHBE cells were preincubated with dicumarol (4 or 10μM, 1hr) or dicumarol solvent (0.1M NaOH) at both the apical (total volume 350 μl) and basolateral compartments. NE (500 nM) [14] or control vehicle were then added to both the apical and basolateral compartments, and incubated for 1hr. A549 were preincubated with dicumarol (4-30 μM, 1 h) or control vehicle and then coincubated with NE (100 nM, 6 h) or control vehicle. Following cell treatments with NE, dicumarol, or control vehicles, RNA was isolated using Trizol reagent per the manufacturer's instructions. After RNA quantitation, one-step real-time RT-PCR was performed on an SDS 7300 machine (Applied Biosystems) in a 25μl reaction which contained 25-150 ng total RNA, 1X universal PCR master mix, RNase inhibitor (0.4U/μl), 1× TaqMan gene expression assay (mixture of PCR primer set and a FAM dye-labeled TaqMan MGB probe) and Multiscribe reverse transcriptase (5U/μl), using universal amplification conditions: 50°C for 30 min, followed by 95°C for 2 min, and then 40 cycles of 95°C, 15 sec followed by 60°C, 1 min. Amplification reaction of 18s rRNA control contained 25ng total RNA, 1X universal PCR master mix, RNase inhibitor (0.4U/μl), 0.2μM forward primer, 0.2μM reverse primer, 0.8μM VIC dye-labeled probe and Multiscribe reverse transcriptase (5U/μl). Each sample was amplified in duplicate reactions for both the gene of interest and 18s rRNA control. The relative gene expression level was calculated by the ∆∆Ct method which represents the fold difference in gene expression corrected for 18s rRNA control expression and normalized to the control treated sample [7].
Western Analysis
A549 or NHBE lysates (10 or 30 μg) were separated by electrophoresis on a 12% SDS-polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane, and stained with Ponceau S to evaluate total protein levels. Ponceau S staining was imaged and bands were quantitated using ImageQuant TL. The nitrocellulose membrane was then blocked with 5% non-fat milk in 15 mM Tris, 150 mM NaCl, 0.1% Tween 20 (4°C, overnight or 1h, room temperature). Membranes were incubated with polyclonal goat anti-NQO1 antibodies (1:200 dilution for Santa Cruz antibody, 1:4000 for Abcam antibody, room temperature, 1h), followed by horseradish peroxidase (HRP)-conjugated anti-goat IgG (1:5000 for Santa Cruz antibody, 1:20,000 for Abcam antibody, room temperature, 1h). A monoclonal mouse anti-E-cadherin antibody (1:250, room temperature, 1h), followed by HRP-conjugated sheep anti-mouse IgG (1:5000, room temperature, 1h) was used as a positive control for the membrane fraction. Antigen-antibody complexes were visualized by enhanced chemiluminescence (ECL). Non-immune goat IgG was used as a negative control in place of primary NQO1 antibody.
Assessment of Oxidative Injury
In serum-free medium, A549 cells were pre-incubated with dicumarol (10μM) or 0.1M NaOH (control vehicle for dicumarol) for 1h and then co-incubated with NE (100 nM) or an equivalent volume of control vehicle (50% glycerol: 50% 0.02 M sodium acetate, pH 5.0) for 2h. At the end of the treatment protocol, cell lysates were collected for lipid carbonyl analysis by HPLC as a measure of lipid peroxidation [25]. In detail, the cells were washed twice with Dulbecco's Phosphate Buffered Saline (DPBS) and scraped and collected in DPBS: 2,4-dinitrophenylhydrazine in acetonitrile (5:1). Cells were lysed and then analyzed for carbonyl content as an end product of lipid peroxidation after separation from protein and nucleic acids using a 2690 Separation Module HPLC as previously described. Briefly, separations were performed on a Waters' Xterra C18 column (2.1 × 15 mm; 3.5 μM). Carbonyl analysis was performed using a 2487 dual wavelength absorbance detector (at 365 nm) and a ZMD mass spectrometer (Waters' Associates, Milford, MA). The mass spectrometry was run in electrospray ionization negative mode with a capillary voltage of 2 kV, cone voltage of −18V, extractor voltage of −3V, source block temperature of 150°C, desolvation temperature of 350°C, and radio frequency lens of 0.3. The DNPH derivative of cis-11-hexadecenal (m/z 417; Aldrich Chemical) was added as an internal standard. Data was analyzed using Waters' MassLynx software (version 3.2).
Whole Cell Lysate Preparation
Following treatment with NE or control vehicle, A549 or NHBE cells were lysed in buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1% TX-100, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1× protease inhibitor and 1× phosphatase inhibitor. Following sonication and incubation in lysis buffer (4°C, 1h), the lysate was clarified by centrifugation (16,000×g, 4°C, 15 min). The resulting supernatant corresponded to the whole cell lysate, and was quantitated using the DC Protein Assay following the manufacturer's instructions.
Cell fractionation
Following treatment with NE (100 nM, 1h) or control vehicle, A549 cells were lysed in buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1× protease inhibitor and 1× phosphatase inhibitor. Following cell lysis by sonication and a low-speed centrifugation to remove nuclei (512×g, 5 min, 4° C), the supernatant was clarified by centrifugation (16,000×g, 30 min, 4° C). The resulting supernatant constituted the cytosolic fraction. The pellet remaining was resuspended in the above lysis buffer plus 1% TX-100, sonicated, incubated in lysis buffer (4°C, 1 h), and then centrifuged at 16,000×g, 30 min, 4°C. The resulting supernatant constituted the soluble plasma membrane fraction. The DC Protein Assay was used to quantitate protein concentrations for the cytosolic and soluble plasma membrane fractions.
NQO1 Activity Assay [19]
Cell lysate protein (15-30 μg) was added to Tris-HCl buffer, pH 7.76 containing 0.2% Tween-20, 2,6-Dichlorophenolindophenol (DCPIP, 80 μM), 0.075% BSA, NADPH (200 μM), in the presence or absence of dicumarol (10 μM), with a total reaction volume of 1 mL. An additional 0.05% TX-100 was added to the cytosolic fractions so that the final concentration of TX-100 would be equivalent to that of the soluble plasma membrane fractions and the whole cell lysate samples. Enzyme activity was determined by spectrophotometric assay of the dicumarolinhibitable reduction of DCPIP at 600 nm over time (20 sec), and was corrected for protein concentration. Activity in the presence of dicumarol was approximately 10% of total activity, demonstrating that the assay system predominantly reflected NQO1 activity.
Statistical Analysis
NQO1 protein concentrations and activity in NE- versus control-treated samples were compared with the Wilcoxon rank sum test. All other data was analyzed using the Kruskal-Wallis one-way nonparametric analysis of variance and post hoc comparisons by the Wilcoxon rank sum test [26] using Statistix Software. Differences were considered significant at P<0.05.
Results
NQO1 is required for NE-induced MUC5AC expression
To determine whether NQO1 is involved in NE-regulated MUC5AC expression, we used dicumarol, a pharmacologic inhibitor of NQO1. Dicumarol is a coumarin derivative that acts as a competitive inhibitor of NQO1 by binding reversibly to the NADH or NADPH binding site [27]. Dicumarol significantly inhibited NE-induced MUC5AC mRNA expression in a concentration-dependent manner in A549 cells (Figure 1), and in primary NHBE cells (Figure 2). Neither the inhibitor alone nor its solvent affected levels of MUC5AC expression. Treatment conditions did not cause cytotoxicity by LDH assay (data not shown).
Figure 1.

Dicumarol blocks the NE-induced increase in MUC5AC mRNA expression in A549 cells. A549 cells were treated with dicumarol (4-30 μM) 1h prior to and then during NE (100 nM, 6h) or control vehicle treatment. RNA was isolated using Trizol reagent as per the manufacturer's instructions, or by the GTC/CsCl method, and used for quantitative real-time RT-PCR (A) or for Northern analyses (10 μg) (B). MUC5AC mRNA expression determined by real-time RT-PCR was normalized to 18s ribosomal RNA and expressed relative to control treated cells using the ΔΔCt method (n=4-8, 3 separate experiments, *, significantly different from control, p=0.0002: #, significantly different from NE alone, p=0.014,A). A representative northern analysis for MUC5AC mRNA expression is shown [C, Control; D, 30μM dicumarol ; NE, 100nM neutrophil elastase; ND, neutrophil elastase and dicumarol] (B). The filter was stripped and reprobed for 28s rRNA. Molecular markers (9.5 & 4.4 kb) are shown with arrows. The graph summarizes the northern densitometry data of MUC5AC mRNA levels normalized to 28s rRNA levels and expressed as a percentage of control (C). Data are presented as mean ±SEM (n=6, 3 separate experiments). The dashed line at 100% represents control levels. *, significantly different from control (P < 0.05). #, significantly different from NE alone (P < 0.05).
Figure 2.

Dicumarol blocks the NE-induced increase in MUC5AC mRNA expression in NHBE cells. NHBE cells grown on Transwell clear chambers (10 days at ALI) were starved in media supplemented with only two factors, BSA and RA, for 24h and treated with dicumarol (4 or 10 μM) or vehicle 1h prior to and then during NE (500 nM, 1h) or control vehicle treatment. Total RNA was collected by Trizol. MUC5AC mRNA expression was evaluated by quantitative real-time RT-PCR. The relative MUC5AC mRNA level was normalized to 18S rRNA and expressed as percent of control treated cells. Data are presented as mean ± SEM (3 separate experiments with total n of 4-6). The dashed line at 100% represents control levels in the absence of dicumarol and NE.*, significantly different from control treated cells (P=0.002). #, significantly different from NE alone (P = 0.041).
Knock-down of NQO1Expression inhibits NE-increased MUC5AC expression
To confirm that NQO1 activity is required for NE regulation of MUC5AC expression, siRNA transfection was used to specifically decrease NQO1 mRNA and protein expression in A549 cells. NQO1-specific siRNA decreased mRNA expression by 80% (Figure 3A), and protein levels by 50% of control siRNA transfected levels (Figure 3B and C). Importantly, following suppression of NQO1 expression, NE regulation of MUC5AC expression was significantly inhibited compared to control siRNA treated cells (Figure 4). These results correlate with the results using dicumarol, a pharmacologic inhibitor of NQO1, implicating NQO1 as a key regulator of NE-mediated MUC5AC expression.
Figure 3.

Knockdown of NQO1 expression by NQO1 siRNA. A549 (70% confluent) were transfected with siRNA for NQO1 (siNQO1, 20 nM), or control siRNA (siControl, 20 nM). 24 h post-transfection, total RNA was collected by Trizol. NQO1 mRNA expression was evaluated by quantitative real-time RT-PCR. The relative NQO1 mRNA level was normalized to 18S rRNA and expressed as percent of control siRNA (siControl) transfected cells. Data are presented as mean ± SEM (n=9, 3 separate experiments). *, significantly different from siControl transfected cells (P<0.0001) (A). Another set of cells with the same treatment were used to collect cell lysate and NQO1 protein expression was evaluated by Western blot (B). The graph summarizes the densitometry data of NQO1 protein level normalized to β-actin and expressed as a percentage of siControl (C). Data are presented as mean ±SEM (n=4, 2 separate experiments). *, significantly different from siControl (P < 0.03).
Figure 4.
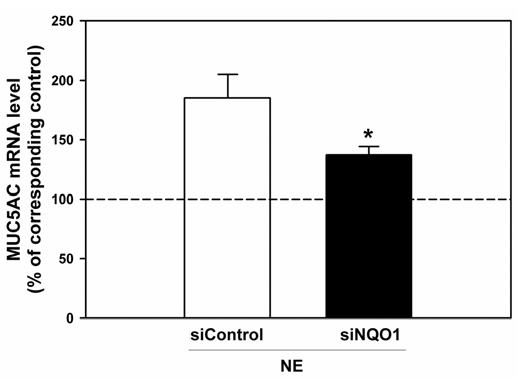
NQO1-knockdown by siRNA transfection abrogates the NE-induced upregulation of MUC5AC mRNA expression. A549 cells (70% confluent) were transfected with siRNA for NQO1 (siNQO1, 20 nM), or control siRNA (siControl, 20 nM). 22h post-transfection, cells were treated with NE (100nM) or control vehicle for 6h and total RNA was collected by Trizol. MUC5AC mRNA expression was evaluated by quantitative real-time RT-PCR and normalized to 18s rRNA expression. The relative MUC5AC mRNA levels from NE-treated cells were expressed as a percentage of their corresponding control-treated cells. Data are presented as mean ± SEM (n=9, 3 separate experiments). The dashed line at 100% represents control levels. *, significantly different from siControl transfected cells (P<0.04).
NE-induced oxidative injury is inhibited by dicumarol
We have previously shown that NE increases MUC5AC expression in A549 cells and primary airway epithelial cells via a pathway involving reactive oxygen species (ROS) [14]. To determine the source of NE induced ROS in A549 cells, cells were treated with NE (100 nM, 2 h) or control vehicle, with or without pre-incubation with dicumarol (30 μM, 1h). Cell lysates were then collected to evaluate for lipid peroxidation as an indicator of reactive oxygen species. NE significantly increased A549 cell lipid peroxidation compared to control-treated samples (Figure 5). Importantly, dicumarol inhibited NE-induced lipid peroxidation (Figure 5). These results suggest that NQO1 plays a role in NE-mediated oxidant stress in epithelial cells, and further link the processes of intracellular oxidant stress and regulation of MUC5AC expression.
Figure 5.

Dicumarol inhibited NE-induced lipid peroxidation in A549 cells. A549 cells were preincubated with dicumarol (30μM, 1h) or 0.1M NaOH and then treated with NE (100nM) or control vehicle for 2h. Cell lysate was collected and evaluated for lipid peroxidation by HPLC. Results are expressed as percent of control. D, dicumarol; NE, neutrophil elastase; NE+D, neutrophil elastase plus dicumarol. Data are presented as mean ± SEM (n=10, 2 separate experiments). The dashed line at 100% represents control levels. *, significantly different from control, P<0.006. #, significantly different from NE (P=0.021).
NE Regulation of NQO1
To determine whether NE regulates NQO1 expression as part of the mechanism of activation, we evaluated NQO1 mRNA expression by real-time RT-PCR and protein levels by Western analysis following NE treatment. Quantitative real-time RT-PCR revealed that in A549 cells, NQO1 mRNA expression increased 20% following NE exposure (30 min) compared to control treated cells. In NHBE cells, NQO1 mRNA expression did not change following NE exposure (1h) compared to control treated cells. On western analysis, NQO1 was detected as a single band at the expected size of 35 kD. NE treatment increased NQO1 protein levels in A549 whole cell lysates by 45 ± 10 % compared to control-treated cells (Figure 6A and B), and increased NQO1 protein levels in NHBE whole cell lysates by 400 ±79% (Figure 7A and B). There were no bands detected when non-immune goat IgG was used in place of primary antibody (data not shown). NE also increased NQO1 activity by 33.4 ± 9 % in A549 cells (Figure 6C) and by 22.5 ± 5% in NHBE cells (Figure 7C) as measured by dicumarol-inhibitable DCPIP reduction. The increase in activity correlated with the increase in NQO1 protein levels. Thus NE increased NQO1 protein levels resulting in increased NQO1 activity as part of the mechanism of MUC5AC mRNA regulation.
Figure 6.
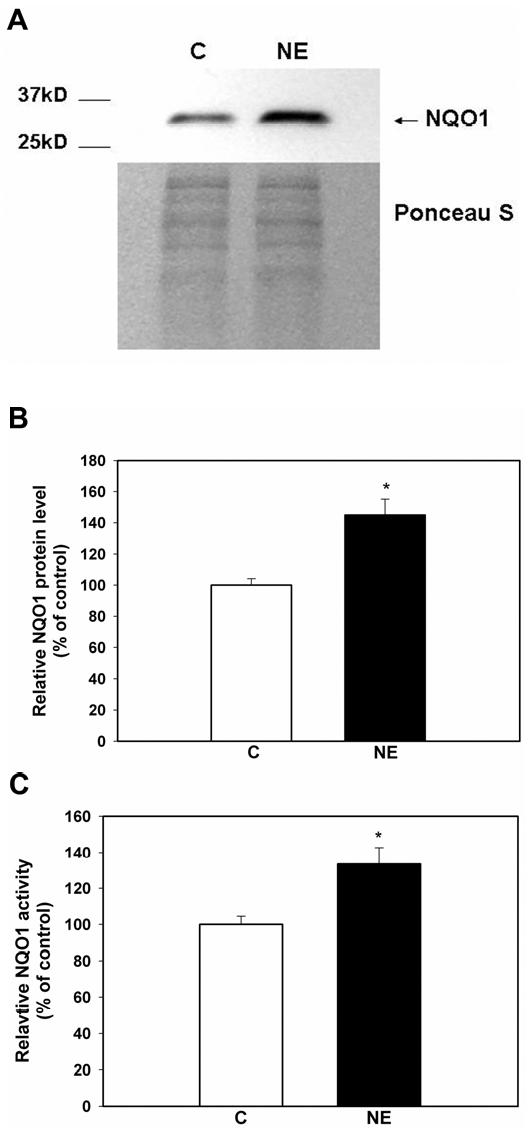
NE increased NQO1 protein and activity level in A549 whole cell lysates. Whole cell lysates of A549 cells following treatment with NE (100nM, 1h) or control vehicle (C) were evaluated for NQO1 protein levels by western blot (A). The graph summarizes the densitometry data of NQO1 protein level normalized to Ponceau S staining. Results are expressed as a percent of control, mean ± SEM (n=10, 4 separate experiments). *, significantly different from control (P= 0.0002) (B). NQO1 activity in whole cell lysate was evaluated using a spectrophotometric assay measuring the reduction of DCPIP at 600 nm. Relative NQO1 activity was expressed as a percent of control, mean ± SEM (n=10, 4 separate experiments). *, significantly different from control (P = 0.0022) (C).
Figure 7.

NE increased NQO1 protein and activity levels in NHBE whole cell lysates. Whole cell lysates of NHBE following treatment with NE (500 nM, 1 h) or control vehicle (Ctrl) were evaluated for NQO1 protein levels by western analysis (A). The graph summarizes the densitometry data of NQO1 protein levels normalized to β-actin. Results are expressed as a percent of control (C), mean ± SEM (n=9-12, 4 separate experiments). *, significantly different from control, (p=0.0006) (B). NQO1 activity in the whole cell lysate was evaluated using a spectrophotometric assay measuring the reduction of DCPIP at 600 nm in the presence or absence of dicumarol. Relative NQO1 activity was expressed as a percent of control, mean ±SEM (n=5, 2 separate experiments). *, significantly different from control (P = 0.016) (C).
Previous studies in vivo in rat liver following oxidant stress, have demonstrated that NQO1 translocates to the plasma membrane as part of the mechanism of activation [28, 29] . We performed cell fractionation studies to investigate whether NE caused a similar translocation of NQO1 from the cytosol to the membrane. NQO1 protein was predominantly restricted to the cytosolic compartment, with no change in localization to the plasma membrane following NE treatment (Figure 8A). E-cadherin protein was only detected in the plasma membrane fraction and not the cytosolic fraction; this immunoblot serves as a positive control for detection of proteins in the plasma membrane fraction (Figure 8B). Consistent with western and activity data from whole cell lysates, following NE treatment, NQO1 protein expression significantly increased in the cytosolic fraction by 120 ± 38 % (Figure 8C). The NE-induced increase in NQO1 expression in the cytosol correlated with the 50 ± 3 % increase in NQO1 oxidoreductase activity in the cytosolic fractions (Figure 8D). In contrast, the plasma membrane fraction had no dicumarol-inhibitable NQO1 activity and no increase in activity after NE treatment (data not shown). These data reveal that NE did not activate NQO1 by altering cellular localization, but instead NE increased NQO1 activity by increasing NQO1 protein expression in at least two separate respiratory epithelial cell culture systems.
Figure 8.

NE increased NQO1 protein and activity level in the cytosolic fraction, not in the membrane fraction. Cytosolic and membrane fractions of A549 cells treated with NE (100nM, 1h) or control vehicle (C) were evaluated for NQO1 protein level by western blot (A). E-cadherin was used as a positive control for the plasma membrane fraction (B). The graph summarizes the densitometry data of NQO1 protein level in the cytosol normalized to Ponceau S staining. Results are expressed as a percent of control, mean ±SEM (n=6, 4 separate experiments). *, significantly different from control (P= 0.01) (C). NQO1 activity in the cytosolic fraction was evaluated using a spectrophotometric assay measuring the reduction of DCPIP at 600 nm. Relative NQO1 activity was expressed as a percent of control, mean ±SEM (n=6, 4 separate experiments). *, significantly different from control (P = 0.002)(D).
Discussion
We [14] and others [30] have previously described oxidant stress as a mechanism for NE-induced MUC5AC expression. In this report, we have focused on the role of NQO1 to mediate intracellular oxidant signaling required for MUC5AC regulation. We demonstrate that treatment with dicumarol blocks both NE-induced MUC5AC expression and lipid peroxidation. Although dicumarol has several other pharmacologic functions including inhibition of ATP phosphoribosyltransferase (at >50 μM concentration) [31], microtubule stabilization (at 0.1-10 μM concentration) [32], inhibition of UDP glucuronosyltransferase (IC50 1 μM ) [33], and inhibition of mouse glutathione transferases and glutathione peroxidase II (IC50 ∼10 μM) [34], our results suggest that the effect of dicumarol on inhibition of NE-regulated MUC5AC expression is due to its inhibition of NQO1. First, in the presence of dicumarol, there was little residual NQO1 activity by a specific spectrophotometric assay (≤10% residual activity). Further, dicumarol blocked NE-induced lipid peroxidation (Figure 5); dicumarol inhibition of glutathione transferase or peroxidase should increase oxidant stress which is inconsistent with our results. We further support the hypothesis that NQO1 mediates NE-regulated MUC5AC expression by demonstrating that suppression of NQO1 by siRNA inhibited NE-induced MUC5AC expression, indicating that NQO1 is required for this process. Thus, we propose a novel role for NQO1 as an important signaling molecule in the NE-triggered regulation of MUC5AC expression.
NQO1 is highly expressed in normal human respiratory epithelium, particularly in ciliated columnar epithelial cells and lung cancers [18]. NQO1 expression and activity are increased by hyperoxia [35], and oxidative stress induced by vitamin depletion [28] via activation of oxidant-sensitive promoter elements in the 5' flanking region of the gene. There are two regulatory elements in the promoter of the NQO1 gene: the antioxidant response element (ARE) and the xenobiotic response element [36]. The ARE sequence is similar to the AP-1 binding site, thus several transcription factors that recognize both elements including AP-1 and other basic leucine zipper proteins may regulate NQO1 expression. One of these factors, NF-E2-related factor 2 is activated by oxidant stress and upregulates NQO1 expression following hyperoxic lung injury [37], cigarette smoke [38], or diesel particle exposure [39]. Extracellular signal regulated protein kinase has been reported to participate in NQO1 ARE activation [36]. Given that NE increases oxidant stress in epithelial cells by several different mechanisms, it is possible that NE regulation of NQO1 expression in A549 cells may be secondary to generation of reactive oxygen species via another mechanism.
In addition to transcriptional regulation, we conclude that NE regulates NQO1 at the translational level in A549 cells based on the discrepancy between a 20% increase in NQO1 mRNA compared to a much greater increase (120%) in NQO1 protein in the cytosol following NE exposure. Interestingly, in NHBE, NE does not alter NQO1 mRNA expression, but instead significantly increases NQO1 protein levels suggesting a regulatory mechanism involving translation or post-translational stability. The mechanism(s) regulating NQO1 expression as well as the differential regulation between cancer-derived and primary airway epithelial cells have not yet been clearly delineated. To our knowledge, this is the first report of NQO1 translational or post-translational regulation and future studies will focus on the molecular mechanisms required for up-regulation following NE exposure.
We propose a new function for NQO1, as a central regulator of intracellular oxidant signaling resulting in MUC5AC gene regulation. Most of the literature on NQO1 to date focuses on its role as a detoxification and an antioxidant enzyme [36]. NQO1 has also been a target for enzyme-directed anticancer pharmacotherapy due to its high level of expression in many human solid tumors [40]; NQO1 detoxifies carcinogens and stabilizes the tumor suppressor p53 [36]. However, some of the hydroquinones produced by NQO1 reduction are less stable than their quinone precursors. These less stable products are prone to both auto-oxidation with resulting ROS production, and rearrangements that lead to the production of alkylating or arylating species [27, 41, 42]. In airway epithelial cells, the prooxidant function of NQO1 may be beneficial or harmful. Although increased mucin production may initially augment host defenses, subsequent mucin overproduction and the resulting airway obstruction may impair pulmonary function.
There is an increasing body of epidemiologic literature which suggests a linkage between wild type NQO1 and susceptibility to pollution-induced pulmonary disease. When subjects with the genotype of wild-type NQO1 plus Glutathione-S-Transferase M1 (GSTM1)-null are exposed to ozone during exercise, they have decreased pulmonary function including FEV1, MEF50 and MEF 75 [21], or increased biomarkers of inflammation including 8-isoprostane, thiobarbituric reactive substances, and leukotriene B4 [43]. Furthermore, in a GSTM1-null population, the presence of an inactive NQO1 polymorphism (Pro187Ser) has been shown to have a protective effect against the development of asthma in children with high lifetime ozone exposure [22]. This work lends support to our concept of NQO1 as a source of oxidant stress, and may help to explain the pulmonary morbidity associated with air pollution.
Given the epidemiologic data linking wild-type NQO1 genotype with asthma and pulmonary susceptibility to ozone, we speculate that NQO1 may be activated/ upregulated in response to air pollutants, leading to airway obstruction by increasing the expression and secretion of mucins. There are several published reports that residual oil fly ash [44], and tobacco smoke, in the absence [45, 46] or presence [47] of inflammatory mediators, activate intracellular ROS and induce mucin expression. Residual oil fly ash [48] and other sources of reactive oxygen and reactive nitrogen species [49] also increase mucin secretion. The link between endogenous ROS and mucin gene regulation is further strengthened by the observation that antioxidants block the pollutant-induced regulation of mucin expression. There is limited information to date in these model systems firmly establishing the source of oxidant stress responsible for regulation of mucin expression. We speculate that NQO1 would be an important airway epithelial target to evaluate following air pollutant exposures.
Oxidant stress may regulate gene expression at both the transcriptional and post-transcriptional level. Oxidant stress regulates MUC5AC mRNA expression via activation of the epidermal growth factor receptor [12, 50] and by an alternative mechanism, post-transcriptional regulation [11]. Several mammalian genes are post-transcriptionally regulated by oxidant stress. Expression of vascular endothelial growth factor [51], nitric oxide synthase [52], macrophage inflammatory protein 2 [53], and p21waf1/cip1 [54] are all increased as a result of oxidant-induced stabilization of mRNA. The mechanisms of oxidant-regulated mRNA stability are not well understood but may involve post-translational modification of RNA-binding proteins [55] or altered interaction of RNA-binding proteins with mRNA stability domains [56]. These reports emphasize that intracellular oxidant signals regulate airway gene expression at multiple levels. Particularly in the case of MUC5AC, oxidant signaling may result in activation of several regulatory pathways and a greatly augmented effect on gene expression.
In summary, our results demonstrate that NQO1 is a source for the ROS responsible for NE-induced MUC5AC expression and suggest a new role for NQO1 in airway epithelium in the pathophysiology of chronic lung diseases and susceptibility to air pollutants such as ozone.
Acknowledgments
We thank Jacob G. Cuellar, Lingxiang Zhu and Vera B. Jaffe for technical assistance. We thank Thomas P. Kennedy for helpful discussions. This work was supported by NIH grants: HL65611 (JAV), HL082504 (JAV) and HL081763 (BMF). Angela S. Byrd was supported by a Howard Hughes Medical Institute Medical Student Fellowship.
Abbreviations
- ALI
air-liquid interface
- ARE
antioxidant response element
- BSA
bovine serum albumin
- CB
chronic bronchitis
- CF
cystic fibrosis
- DMEM
Dulbecco's Minimum essential medium
- ECL
enhanced chemiluminescence
- FBS
fetal bovine serum
- GSTM1
glutathione-S-transferase μ1
- NE
neutrophil elastase
- NHBE
normal human bronchial epithelia
- NQO1
NAD(P)H quinone oxidoreductase 1
- PBS
phosphate buffered saline
- RA
retinoic acid
- ROS
reactive oxygen species
- RT-PCR
reverse transcriptase-polymerase chain reaction
- siRNA
short interfering RNA
- SSC
saline, sodium citrate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Diseases. III. McGraw-Hill; New York: 2001. pp. 5121–5188. [Google Scholar]
- 2.Senior RM, Shapiro SD. Chronic obstructive pulmonary disease: epidemiology, pathophysiology, and pathogenesis. In: Fishman AP, editor. Fishman's Pulmonary Diseases and Disorders. Vol. 1. McGraw-Hill; New York: 1998. pp. 659–681. [Google Scholar]
- 3.Voynow JA. What does mucin have to do with lung disease? Paediatr Respir Rev. 2002;3:98–103. doi: 10.1016/s1526-0550(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 4.Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am. J. Respir. Crit. Care Med. 1994;150:448–454. doi: 10.1164/ajrccm.150.2.8049828. [DOI] [PubMed] [Google Scholar]
- 5.Stockley RA, Burnett D. Alpha1-antitrypsin and leukocyte elastase in infected and noninfected sputum. Am Rev Respir Dis. 1979;120:1081–1086. doi: 10.1164/arrd.1979.120.5.1081. [DOI] [PubMed] [Google Scholar]
- 6.Christensen TG, Breuer R, Hornstra LJ, Lucey EC, Stone PJ, Snider GL. An ultrastructural study of the response of hamster bronchial epithelium to human neutrophil elastase. Exp Lung Res. 1987;13:279–297. doi: 10.3109/01902148709069594. [DOI] [PubMed] [Google Scholar]
- 7.Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, Ho SB, Foster WM. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1293–1302. doi: 10.1152/ajplung.00140.2004. [DOI] [PubMed] [Google Scholar]
- 8.Kim KC, Wasano K, Niles RM, Schuster JE, Stone PJ, Brody JS. Human neutrophil elastase releases cell surface mucins from primary cultures of hamster tracheal epithelial cells. Proc. Natl. Acad. Sci. USA. 1987;84:9304–9308. doi: 10.1073/pnas.84.24.9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fahy JV, Schuster A, Ueki I, Boushey HA, Nadel JA. Mucus hypersecretion in bronchiectasis. The role of neutrophil proteases. Am. Rev. Respir. Dis. 1992;146:1430–1433. doi: 10.1164/ajrccm/146.6.1430. [DOI] [PubMed] [Google Scholar]
- 10.Park JA, He F, Martin LD, Li Y, Chorley BN, Adler KB. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase C{delta}-mediated mechanism. Am J Pathol. 2005;167:651–661. doi: 10.1016/s0002-9440(10)62040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voynow JA, Young LR, Wang Y, Horger T, Rose MC, Fischer BM. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am J Physiol. 1999;276:L835–L843. doi: 10.1152/ajplung.1999.276.5.L835. [DOI] [PubMed] [Google Scholar]
- 12.Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;283:L531–L540. doi: 10.1152/ajplung.00455.2001. [DOI] [PubMed] [Google Scholar]
- 13.Fischer BM, Cuellar JG, Diehl ML, deFreytas AM, Zhang J, Carraway KL, Voynow JA. Neutrophil elastase increases MUC4 expression in normal human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L671–L679. doi: 10.1152/ajplung.00220.2002. [DOI] [PubMed] [Google Scholar]
- 14.Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol. 2002;26:447–452. doi: 10.1165/ajrcmb.26.4.4473. [DOI] [PubMed] [Google Scholar]
- 15.Phan SH, Gannon DE, Ward PA, Karmiol S. Mechanism of neutrophil-induced xanthine dehydrogenase to xanthine oxidase conversion in endothelial cells: evidence of a role for elastase. Am J Respir Cell Mol Biol. 1992;6:270–278. doi: 10.1165/ajrcmb/6.3.270. [DOI] [PubMed] [Google Scholar]
- 16.Aoshiba K, Yasuda K, Yahui S, Tamaoki J, Nagai A. Serine proteases increase oxidative stress in lung cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L556–L564. doi: 10.1152/ajplung.2001.281.3.L556. [DOI] [PubMed] [Google Scholar]
- 17.Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- 18.Siegel D, Franklin WA, Ross D. Immunohistochemical detection of NADPH:Quinone oxidoreductase in human lung and lung tumors. Clin. Can. Res. 1998;4:2065–2070. [PubMed] [Google Scholar]
- 19.Ernster L. DT Diaphorase. Methods Enzymol. 1967;10:309–317. [Google Scholar]
- 20.Cadenas E. Antioxidant and prooxidant functions of DT-diaphorase in quinone metabolism. Biochem Pharmacol. 1995;49:127–140. doi: 10.1016/s0006-2952(94)00333-5. [DOI] [PubMed] [Google Scholar]
- 21.Bergamaschi E, De Palma G, Mozzoni P, Vanni S, Vettori MV, Broeckaert F, Bernard A, Mutti A. Polymorphism of quinone-metabolizing enzymes and susceptibility to ozone-induced acute effects. Am. J. Respir. Crit. Care Med. 2001;163:1426–1431. doi: 10.1164/ajrccm.163.6.2006056. [DOI] [PubMed] [Google Scholar]
- 22.David G, Romieu I, Sienra-Monge JJ, Collins WJ, Ramirez-Auilar M, del Rio-Navarro BE, Reyes-Ruiz NI, Morris RW, Marzec JM, London SJ. Nicotinamide adenine dinucleotide (Phosphate) reduced:quinone oxidoreductase and glutathione s-transferase M1 polymorphisms and childhood asthma. Am. J. Respir. Crit. Care Med. 2003;168:1199–1204. doi: 10.1164/rccm.200305-684OC. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Hamati E, Lee PK, Lee WM, Wachi S, Schnurr D, Yagi S, Dolganov G, Boushey H, Avila P, Wu R. Rhinovirus induces airway epithelial gene expression through double-stranded RNA and IFN-dependent pathways. Am J Respir Cell Mol Biol. 2006;34:192–203. doi: 10.1165/rcmb.2004-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho S, Niehans G, Lyftogt C, Yan P, Cherwitz D, Gum E, Dahiya R, Kim Y. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res. 1993;53:641–651. [PubMed] [Google Scholar]
- 25.Madden MC, Thomas MJ, Ghio AJ. Acetaldehyde (CH3CHO) production in rodent lung after exposure to metal-rich particles. Free Radical Biology & Medicine. 1999;26:1569–1577. doi: 10.1016/s0891-5849(99)00027-1. [DOI] [PubMed] [Google Scholar]
- 26.Snedecor GW, Cochran WG. Statistical Methods. Iowa State University Press; Ames: 1980. [Google Scholar]
- 27.Ross D, Siegel D. NADPH:Quinone Oxidoreductase 1 (NQO1, Dt-Diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004;382:115–145. doi: 10.1016/S0076-6879(04)82008-1. [DOI] [PubMed] [Google Scholar]
- 28.Navarro F, Navas P, Burgess JR, Bello RI, De Cabo R, Arroyo A, Villalba JM. Vitamin E and selenium deficiency induces expression of the ubiquinone-dependent antioxidant system at the plasma membrane. Faseb J. 1998;12:1665–1673. doi: 10.1096/fasebj.12.15.1665. [DOI] [PubMed] [Google Scholar]
- 29.Navarro F, Arroyo A, Martin SF, Bello RI, de Cabo R, Burgess JR, Navas P, Villalba JM. Protective role of ubiquinone in vitamin E and selenium-deficient plasma membranes. Biofactors. 1999;9:163–170. doi: 10.1002/biof.5520090211. [DOI] [PubMed] [Google Scholar]
- 30.Takeyama K, Dabbagh K, Jeong Shim J, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J Immunol. 2000;164:1546–1552. doi: 10.4049/jimmunol.164.3.1546. [DOI] [PubMed] [Google Scholar]
- 31.Dall-Larsen T, Kryvi H, Klungsoyr L. Dinitrophenol, dicoumarol and pentachlorophenol as inhibitors and parasite substrates in the ATP phosphoribosyltransferase reaction. Eur J Biochem. 1976;66:443–446. doi: 10.1111/j.1432-1033.1976.tb10568.x. [DOI] [PubMed] [Google Scholar]
- 32.Madari H, Panda D, Wilson L, Jacobs RS. Dicoumarol: a unique microtubule stabilizing natural product that is synergistic with Taxol. Cancer Res. 2003;63:1214–1220. [PubMed] [Google Scholar]
- 33.Segura-Aguilar JE, Barreiro V, Lind C. Dicoumarol-sensitive glucuronidation of benzo(a)pyrene metabolites in rat liver microsomes. Arch Biochem Biophys. 1986;251:266–275. doi: 10.1016/0003-9861(86)90074-3. [DOI] [PubMed] [Google Scholar]
- 34.Mays JB, Benson AM. Inhibition of mouse glutathione transferases and glutathione peroxidase II by dicumarol and other ligands. Biochem Pharmacol. 1992;44:921–925. doi: 10.1016/0006-2952(92)90124-2. [DOI] [PubMed] [Google Scholar]
- 35.Merker MP, Audi SH, Bongard RD, Lindemer BJ, Krenz GS. Influence of pulmonary arterial endothelial cells on quinone redox status: effect of hyperoxia-induced NAD(P)H:quinone oxidoreductase 1. Am J Physiol Lung Cell Mol Physiol. 2006;290:L607–619. doi: 10.1152/ajplung.00302.2005. [DOI] [PubMed] [Google Scholar]
- 36.Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NADPH:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Inter. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 37.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 38.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baulig A, Garlatti M, Bonvallot V, Marchand A, Barouki R, Marano F, Baeza-Squiban A. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L671–679. doi: 10.1152/ajplung.00419.2002. [DOI] [PubMed] [Google Scholar]
- 40.Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat Res. 2004;555:149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe N, Forman HJ. Autooxidation of extracellular hydroquinones is a causative event for the cytotoxicity of menadione and DMNQ in A549-S cells. Arch Biochem. Biophys. 2003;411:145–157. doi: 10.1016/s0003-9861(02)00716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brar S, Kennedy TP, Whorton AR, Sturrock AB, Huecksteadt TP, Ghio AJ, Hoidal JR. Reactive oxygen species from NAD(P)H: quinone oxidoreductase constitutively activate NF-kB in malignant melanoma cells. Am. J. Physiol. Cell Physiol. 2001;280:C659–C676. doi: 10.1152/ajpcell.2001.280.3.C659. [DOI] [PubMed] [Google Scholar]
- 43.Corrradi M, Alinovi R, Goldoni M, Vettori MV, Folesani G, Mozzoni P, Cavazzini S, Bergamaschi E, Rossi L, Mutti A. Biomarkers of oxidative stress after controlled human exposure to ozone. Toxicol. Lett. 2002;134:219–225. doi: 10.1016/s0378-4274(02)00169-8. [DOI] [PubMed] [Google Scholar]
- 44.Longphre M, Li D, Li J, Matovinovic E, Gallup M, Samet JM, Basbaum CB. Lung Mucin Production Is Stimulated by the Air Pollutant Residual Oil Fly Ash. Tox Appl Pharmacol. 2000;162:86–92. doi: 10.1006/taap.1999.8838. [DOI] [PubMed] [Google Scholar]
- 45.Gensch E, Gallup M, Sucher A, Li D, Gebremichael A, Lemjabbar H, Mengistab A, Dasari V, Hotchkiss J, Harkema J, Basbaum C. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J. Biol. Chem. 2004;279:39085–39093. doi: 10.1074/jbc.M406866200. [DOI] [PubMed] [Google Scholar]
- 46.Shao M, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am. J. Phyiol. Lung Cell Mol. Physiol. 2004;287:L420–L427. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 47.Baginski TK, Dabbagh K, Satjawatcharaphong C, Swinney DC. Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. Am J Respir Cell Mol Biol. 2006;35:165–174. doi: 10.1165/rcmb.2005-0259OC. [DOI] [PubMed] [Google Scholar]
- 48.Jiang N, Dreher KL, Dye JA, Li Y, Richards JH, Martin LD, Adler KB. Residual oil fly ash induces cytotoxicity and mucin secretion by guinea pig tracheal epithelial cells via an oxidant-mediated mechanism. Toxicol Appl Pharmacol. 2000;163:221–230. doi: 10.1006/taap.1999.8886. [DOI] [PubMed] [Google Scholar]
- 49.Martin LD, Krunkosky TM, Voynow JA, Adler KB. The role of reactive oxygen and nitrogen species in airway epithelial gene expression. Environ Health Perspect. 1998;106(Suppl 5):1197–1203. doi: 10.1289/ehp.98106s51197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casalino-Matsuda S, Monzon ME, Conner GE, Salathe M, Forteza RM. Role of hyaluronan and reactive oxygen species in tissue kallikrein-mediated epidermal growth factor receptor activation in human airways. J. Biol. Chem. 2004;279:21606–21616. doi: 10.1074/jbc.M309950200. [DOI] [PubMed] [Google Scholar]
- 51.Kuroki M, Voest EE, Amano S, Beerepoot LV, Takashima S, Tolentino M, Kim RY, Rohan RM, Colby KA, Yeo K-T, Adamis AP. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J. Clin. Invest. 1996;98:1667–1675. doi: 10.1172/JCI118962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 2000;86:347–354. doi: 10.1161/01.res.86.3.347. [DOI] [PubMed] [Google Scholar]
- 53.Shi MM, Chong I-W, Godleski JJ, Paulauskis JD. Regulation of macrophage inflammatory protein-2 gene expression by oxidative stress in rat alveolar macrophages. Immunology. 1999;97:309–315. doi: 10.1046/j.1365-2567.1999.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esposito F, Cuccovillo F, Vanoni M, Cimino F, Anderson CW, Appella E, Russo T. Redox-mediated regulation of p21waf1/cip1 expression involves a post-transcriptional mechanism and activation of the mitogen-activated protein kinase pathway. Eur. J. Biochem. 1997;245:730–737. doi: 10.1111/j.1432-1033.1997.00730.x. [DOI] [PubMed] [Google Scholar]
- 55.Clerch LB, Massaro D. Oxidation-reduction-sensitive binding of lung protein to rat catalase mRNA. J. Biol. Chem. 1992;267:2853–2855. [PubMed] [Google Scholar]
- 56.Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell. 1993;72:19–28. doi: 10.1016/0092-8674(93)90046-s. [DOI] [PubMed] [Google Scholar]