

Abstract
Many disease-related processes occur via protein complexes that are considered undruggable with small molecules. An example is RAS, which is frequently mutated in cancer and contributes to initiation and maintenance of the disease by constitutive signal transduction through protein interaction with effector proteins, like PI3K, RAF and RALGDS. Such protein interactions are therefore significant targets for therapy. We describe a single immunoglobulin variable region domain that specifically binds to activated GTP-bound RAS and prevents RAS-dependent tumorigenesis in a mouse model. The crystal structure of the immunoglobulin–RAS complex shows that the variable region competitively binds to the conformationally variant regions of RAS, where its signalling effector molecules interact. This allows the plasma membrane targeted single domain intrabody to inhibit signalling by mutant RAS. This mode of action is a novel advance to directly interfere with oncogenic RAS function in human cancer and shows a universally applicable approach to develop macromolecules to combat cancer. In addition, this method illustrates a general means for interfering with protein interactions that are commonly considered intractable as conventional drug targets.
Keywords: human disease, intrabody, protein–protein interaction, RAS, structure
Introduction
The aetiology of many human diseases such as cancer, neural degeneration and inflammation involves abnormal proteins participating in macromolecular complexes to elicit a biologically relevant effect. As such, protein–protein interactions represent a major potential drug target for manifold human disease indications. Nonetheless, there are currently few small-molecule drugs in clinical trials that are capable of impeding protein interactions, since these generally require clefts in a protein into which a small molecule can fit (Blundell et al, 2006). The development of novel approaches to target protein–protein interactions is therefore a major goal of experimental therapeutics. The most promising reagents currently are macromolecules that have the capability of high affinity and specificity for binding to targets inside a cell. In cancer, a plethora of protein interaction targets offer the possibility of therapeutic benefit. For instance in leukaemias, the chromosomal translocation proteins such as LMO2 form multiprotein complexes that are functionally relevant to cancer (Wadman et al, 1997), and in epithelial tumours the RAS signal transduction pathway is subject to mutation of the RAS proteins themselves or downstream effectors like RAF, RALGDS and PI3K (Downward, 2003). Molecules targeting the protein interactions have a major potential in the long term to be fundamentally important novel drugs. In addition, such molecules will be proof-of-principle for a future generation of drugs aimed specially at the abnormal interactome of human disease cells.
The RAS proteins are guanine nucleotide binding molecules that play key roles in signal transduction as molecular switches, mediated through two switch regions displaying conformational differences between active (GTP bound) and inactive (GDP bound) states (Vetter and Wittinghofer, 2001). Most of the RAS effectors bind to these RAS switch regions (Downward, 2003). RAS is the most important target in cell transformation, being involved in cell proliferation and differentiation through the RAF-MEK-ERK cascade (Marshall, 1995; Kolch, 2005) and cell survival through activation of PI3K (Downward, 2003). The RAS effector, RALGDS, is also involved in RAS-dependent tumorigenesis in vivo (Gonzalez-Garcia et al, 2005) and cell transformation in human cells (Rangarajan et al, 2004). Activating RAS gene mutations are found in as many as 30% of humans, with the highest frequencies in pancreas, colon and lung adenocarcinoma. Mutations of the RAS proteins (K, H or NRAS) create constitutively activated GTP-bound forms that promote cell transformation in a signal-independent manner (Adjei, 2001). In addition, secondary RAS-associated aberrations such as mutation or overexpression of receptor tyrosine kinases (e.g. EGFR, ERBB2) have been indicated in many cancers that lack RAS mutation (Mendelsohn and Baselga, 2000). Therefore, RAS pathways are involved in a higher percentage of human cancers than those simply with RAS mutation. Thus, inhibiting aberrant RAS function has been an exciting possible mode of human cancer therapy. This notion has been supported by observations in mouse models in which oncogenic RAS has been shown to be essential for early onset of tumours and necessary for maintenance of tumour viability (Johnson et al, 2001), as tumours harbouring mutant RAS can regress when mutant RAS expression is stopped (Chin et al, 1999; Fisher et al, 2001).
These facts highlight activated RAS proteins as attractive targets for cancer therapy. Despite this, anti-RAS therapies have not therefore been particularly effective (Friday and Adjei, 2005). Farnesyltransferase inhibitors (FTIs) can inhibit membrane localisation of RAS proteins by preventing post-translational modification, and thus blocking downstream RAS signalling. However, the antitumour activity of FTIs may only partly be due to targeting RAS and may also affect farnesylation of other proteins (Friday and Adjei, 2005). An ideal RAS-based anticancer therapy would involve reagents that can specifically inhibit oncogenic RAS. Antibodies have such qualities of specificity and affinity that can easily be manipulated. However, most oncogenic proteins, including RAS, are located inside cells and not available for antibody-mediated targeting. Over the last decade, antibody engineering has led to development of fragments that can be expressed intracellularly (intrabodies) (Cattaneo and Biocca, 1997), but there are still few intrabodies that work efficiently in the reducing environment of cells due to the usual need for disulphide bonds for correct folding. To overcome this limitation, we developed intracellular antibody capture (IAC), based on in vivo yeast two-hybrid screening (Visintin et al, 1999; Tse et al, 2002; Tanaka and Rabbitts, 2003), and showed that single variable region (V) domains (iDabs) are highly efficient as intrabodies (Tanaka et al, 2003). In the present study, we derived a single domain VH intrabody binding specifically to activated GTP-bound RAS with high affinity and neutralising the oncogenicity in cancerous cells harbouring a RAS mutation. The crystal structure of the intrabody bound to mutant RAS, solved to 2 Å, shows that the intrabody specifically recognises the conformational structure of oncogenic RAS and inhibits RAS-effector protein interactions with RAS. This novel protein provides a gateway for anti-mutant RAS-based human cancer therapy and illustrates a capacity for small proteins to target the ‘undruggable' protein interaction surface, presenting a new spectrum of drug targets in the human disease interactome.
Results
A single domain antibody fragment that binds mutant RAS
We produced two synthetic single VH domain libraries with fully randomised complementarity determining regions (CDR) within a single stable intrabody framework (Tanaka and Rabbitts, 2003) (the amino-acid sequences are shown in Supplementary Figure 1A). The libraries were screened in yeast with an HRAS(G12V) mutant bait and iDab#6 was isolated (Tanaka et al, 2003) (the VH CDR sequences of iDab#6 are shown in Supplementary Figure 1B, together with a mutant form engineered with CDR1 changes (iDabm#6) and a non-relevant iDab isolated from a library screen using an ATF-2 bait (iDab#27). The comparative binding of RAS by these three iDabs using a COS-7 cell luciferase reporter assay is shown in Supplementary Figure 1C. Only iDab#6 binds to the RAS bait, and it does so through the CDRs because the mutant iDabm#6 has lost binding ability.
The ability of iDab#6 to bind oncogenic HRAS with mutations at either amino acid 12 or 61 was tested in the COS-7 cell-based two-hybrid assay (Figure 1A). Interaction was found with all the oncogenic mutant HRAS proteins, except the (G12P) mutant (a non-transforming protein; Seeburg et al, 1984), and with at least 10 times higher binding than with wild-type HRAS. Furthermore, iDab#6 binds oncogenic mutant RAS belonging to all of the RAS family members (H, K, N, M, and RRAS; Figure 1B). iDab#6 binds to wild-type RAS and RAP mutants (RAP1A, RAP1B and RAP2A) with two orders of magnitude less than mutant RAS and does not significantly bind to RAL mutants (RALA and RALB) (Figure 1B).
Figure 1.
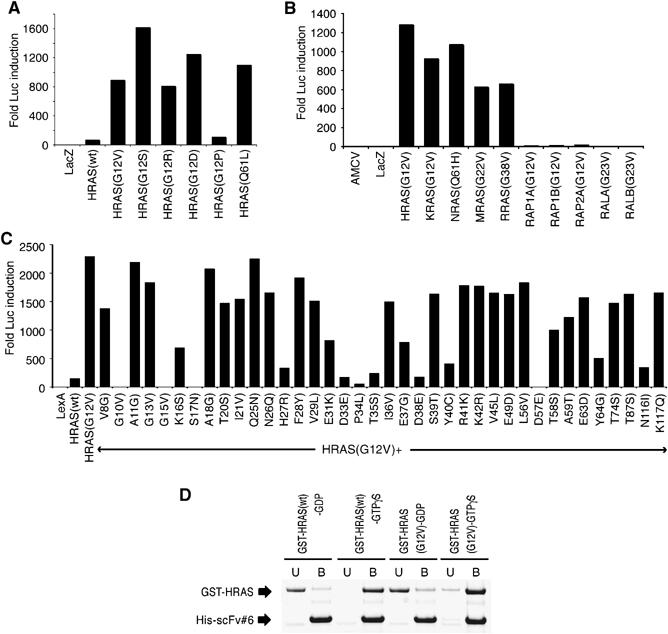
Binding of the anti-RAS single domain with RAS proteins. The binding of the single VH domain to RAS was characterised using luciferase assays in COS-7 cells (A–C) or with bacterially expressed proteins (D). (A–C) The luciferase assays were conducted with a vector expressing iDab#6-VP16 fusion and various mutant forms of RAS, RAP1 or RAL as baits (The sequence alignments of RAS, RAP and RAL are shown in Supplementary Figure 2). (A) Assays with HRAS mutants, (B) RAS family mutants and (C) various mutants of the HRAS(G12V) backbone. (D) Interaction of RAS-GTPγS or -GDP and anti-RAS intrabody in vitro. Purified GST-RAS (wt) or GST-RAS(G12V) were loaded as GDP or GTPγS, as described in Materials and methods, and diluted into an Ni–agarose resin carrying purified His-tagged scFv#6. Following binding, the unbound (U) and bound (B) fractions were sampled and fractioned by SDS–PAGE.
Alignment of the protein sequences of the RAS family (Supplementary Figure 2) shows homology, especially in N-terminal half, suggesting that iDab#6 might bind to this part of RAS (see below). Further binding site characterisation was performed using different point mutants of HRAS, using the (G12V) mutant protein as the backbone (Figure 1C). iDab#6 does not bind to RAS with mutations of amino acids 10, 15, 17 and 57, that are critically conserved at the nucleotide binding site (these mutations cause the low affinity nucleotide and Mg2+ ion binding). Mutations of amino acid 33, 34, 35, 38, 40 or 64 (within the switch I and II regions) significantly decreased interaction of iDab#6, further suggesting that iDab#6 binds to activated RAS in the switch I and II regions.
The specificity of anti-RAS intrabody for activated RAS was examined using in vitro binding assays. Recombinant GST-fusion proteins (GST-HRAS or GST-HRAS(G12V)) were loaded with either GTPγS (GTP analogue) or GDP, mixed with an equimolar amount of His-tagged scFv#6 (the intrabody was made in bacteria as an scFv form, because of improved solubility), as described below, and complexed protein was selected using Ni–agarose resin. We found that scFv#6 bound preferentially to both wild-type HRAS and HRAS(G12V) proteins that were loaded with the GTPγS but not to those loaded with GDP (Figure 1D). This means that the intrabody binds specifically to the activated form of RAS, rather than to particular RAS mutants.
The anti-RAS single domain rescues the untransformed phenotype in cancer cells
The ability of the anti-RAS intrabody to affect the RAS transformation phenotype was examined using mouse NIH3T3-EJ cells (EJ cells), which express mutant HRAS(G12V) (Shih and Weinberg, 1982). Retroviral vectors expressing FLAG-tagged-iDab#6 with subcellular localisation signal peptides specifying nuclear or plasma membrane location (Figure 2A) were expressed in EJ cells. These viral vectors encode soluble proteins (Supplementary Figure 3A) and infect cells with high efficiency (Supplementary Figure 3B). Immunofluorescence data using the FLAG tag for detection showed that the iDab protein predominantly localises according to the signal peptide attached (Supplementary Figure 3C), or in the cytoplasm if no signal peptide was used (Supplementary Figure 3C, iDab#6-cyto).
Figure 2.

The anti-RAS single domain reverts the RAS-transformed phenotype of mouse and human tumour cells. Retroviruses encoding iDab#6 or mutant (iDabm#6) iDab were infected into RAS-transformed mouse NIH3T3-EJ or DLD-1 and HT-1080 human cells. (A) A schematic diagram of retroviral constructs. To express anti-RAS single domain in mammalian cells, the cells were infected with ecotropic retrovirus (Costa et al, 2000). To localise the intrabody in the cells, a signal peptide was fused to the C-terminal of the anti-RAS intrabodies (either a nuclear localisation signal, nls or a plasma membrane targeting element, memb.). LTR, long terminal repeat; FLAG, antibody tag; IRES, internal ribosome entry site; SP, signal peptide (the sequences shown are for the nls or memb.). (B) The morphology of uninfected NIH3T3-D4 and NIH3T3-EJ are shown in top left and right panels and of NIH3T3-EJ cells infected with retrovirus encoding iDab#6-memb or mutant iDab#6 (iDabm#6-memb) in bottom left and right panels; images were obtained 48 h after infection. (C, D) Anchorage-independent growth of DLD-1 (C) and HT-1080 (D). The cells were infected with retrovirus expressing iDab#6 or retrovirus only. Forty-eight hours after infection, the EGFP expressing cells were sorted, seeded in soft agar and colonies (>0.1 mm) were counted at 3 weeks. Colony forming efficiency was the number of colonies per seeded cells. (E) Phalloidin staining of HT-1080 cells. Uninfected cells or cells infected with retrovirus encoding iDab#6 or iDabm#6 (left, middle or right) were stained 48 h after infection to show F-actin stress fibre formation.
Untransformed NIH3T3 cells (NIH3T3-D4) grow as a monolayer with flat morphology (Figure 2B, top left panel), whereas EJ cells exhibit small round cell bodies and form foci due to lack of contact inhibition effect (Figure 2B, top right). EJ cells also have reduced F-actin stress fibre formation and increased accumulation of actin filaments (Supplementary Figure 3D, top left panel). Expression of iDab#6 with membrane localisation signal (iDab#6-memb) reverted EJ cells to an untransformed phenotype (Figure 2B, bottom left panel) and restored F-actin stress fibres (Supplementary Figure 3D, bottom right). When iDab#6 was expressed in the cytoplasm or nucleus, no change in EJ cell morphology was observed (Supplementary Figure 3D, top right and bottom left panels show iDab#6-cyto and iDab#6-nuc, respectively). This neutralisation effect of iDab#6-memb is not simply because the iDab competes with RAS for sites on the plasma membrane, since the mutant iDabm#6-memb had no effect on transformation (Figure 2B, bottom right panel).
The effect of anti-RAS intrabody on human cancer cells was also studied, using HT-1080 (a fibrosarcoma with NRAS(Q61K) mutation) and DLD-1 (a colorectal adenocarcinoma with KRAS (G13D) and P53(S241F) mutations). These lines were infected with retrovirus expressing iDab#6-memb, and the ability of infected cells to show anchorage-independent growth in soft agar assay was assessed (Figure 2C and D). Inhibition was observed in both HT-1080 and DLD-1 cells infected with retrovirus expressing iDab#6-memb, but not in cells infected with retrovirus only. Expression of intrabody causes a morphological change in HT-1080 with reorganised F-actin stress fibres (Figure 2E, middle panel compared with uninfected cells or cells infected with retrovirus expressing the mutated iDabm#6-memb; Figure 2E, left and right panels, respectively). This is a similar to the phenotypic change reported in MCH603c8 cells (a derivative of HT-1080 without the mutant NRAS(Q61K); Plattner et al, 1996).
Inhibition of tumorigenesis and metastasis by anti-RAS iDab in a mouse model
The in vivo efficacy of the anti-RAS intrabody was evaluated by injecting retrovirally infected EJ cells into athymic nude mice. All mice subcutaneously injected with EJ cells infected with retrovirus only or retrovirus expressing the mutant iDabm#6-memb formed subcutaneous tumours up to ∼18 days, whereas tumours did not develop in the mice injected with EJ cells expressing iDab#6-memb up to 18 days (Figure 3A). However, 29 days after injection, half of these mice did have small tumours, but cells isolated from these did not express the iDab#6-memb, as observed by Western analysis or EGFP expression (the viral vector carries an IRES-EGFP segment), while the cells from tumours infected with iDabm#6-memb did (Supplementary Figure 4B and C, respectively). In addition, we assessed the ability of anti-RAS intrabody to inhibit tumorigenesis of the human HT-1080 and DLD-1 cells (Figure 3B and C). The cells infected with retrovirus only or with virus encoding the mutant iDabm#6-memb produced to tumours in almost all mice within ∼5 weeks, whereas none of the mice expressing iDab#6-memb developed tumours throughout our observation period.
Figure 3.

The anti-RAS single domain inhibits tumorigenesis and metastasis in a mouse model. The effect of the anti-RAS single domain iDab#6 on the growth of mouse (NIH3T3-EJ; A, D) or human (HT-1080; B, or DLD-1; C) tumour cells was examined using grafts in immunodeficient nude mice. (A–C) Cells were infected with retroviral vectors expressing iDab#6 or the mutant iDabm#6, each with a membrane (memb.) localisation signal. Forty-eight hours after infection, the EGFP expressing cells were flow sorted, propagated in culture and injected subcutaneously into nude mice (5 × 104 cells for NIH3T3-EJ, 2.5 × 106 for HT-1080 or 5 × 105 cells for DLD-1). If tumour sizes reached 17 mm diameter, the experiment was terminated, otherwise growth was observed for 42 days (for NIH3T3 cells) or 13 weeks (for HT-1080 or DLD-1). (D) Cells (105) were injected intravenously into nude mice. Three weeks after injection, the mice were killed, and lungs were dissected for examination. Representative images are shown from mice injected with either NIH3T3-D4 cells (top left), NIH3T3-EJ (top right), NIH3T3-EJ infected with retrovirus expressing iDab#6-memb (bottom left) or mutant iDabm#6-memb (bottom right).
An experimental lung metastasis assay in nude mice was used to measure the effects of iDab#6 on extravasation and colonisation of tumour cells within the lung. EJ cells infected with retrovirus only or virus encoding iDabm#6-memb caused lung metastasis with multiple foci at ∼3 weeks after intravenous injection (Figure 3D, upper and lower right panels, respectively), whereas none of the mice infected with EJ cells expressing iDab#6 formed lung foci (Figure 3D, lower left). We conclude that the anti-RAS intrabody iDab#6 inhibits RAS-dependent tumorigenicity and metastasis in these models.
Structure of the RAS single domain complex
Details of the intrabody binding site on GTP-bound HRAS were obtained by solving the X-ray crystal structure of HRAS–intrabody complex. Protein was prepared after coexpressing HRAS(G12V) and anti-RAS intrabody in an Fv format in the cytoplasm of bacteria and purifying the complex by chromatography. The anti-RAS Fv comprised the VH#6 plus an anti-RAS VL isolated from a synthetic VL intrabody library, using a yeast three-hybrid-based screening method (manuscript in preparation). These V domains bind to same antigenic region to form a functional Fv, and the Fv was used to facilitate consistent protein production and the yield of purified complex was up to 6 mg/l culture. The crystal structure was solved at 2 Å and determined by the molecular replacement method using the coordinates of anti-HEL Fv (PDB, 1A2Y) (Dall'Acqua et al, 1998) and mutant HRAS(G12D) with GTP analogue (HRAS/GPPNP) (Franken et al, 1993) as search models. Ribbon representation and molecular surface models of the RAS-Fv complex are shown in Figure 4.
Figure 4.

Crystal structure of the RAS–anti-RAS single domains complex. HRAS(G12V) protein complexed with the anti-RAS #6 in an Fv format is shown in ribbon form (A) or space filling (B), where HRAS(G12V) is shown in green and the Fv proteins VH and VL are shown in cyan and orange, respectively. The CDRs of VH and VL are in yellow and lemon and the RAS switch I and II regions are in red and purple, respectively. The GTP and Mg2+ ion in RAS are in blue and magenta, respectively.
The structure of HRAS(G12V)-GTP complexed with Fv was similar to the previously reported isolated HRAS(G12D)/GPPNP (PDB, 1AGP) (Franken et al, 1993) and HRAS(G12V)/GPPNP (complexed with PI3Kγ, PDB, 1HE8) (Pacold et al, 2000), showing that no significant alteration results from binding with the single domain (Supplementary Figure 5C–E). Similarly, the anti-RAS Fv structure is very similar to several reported structures of isolated Fvs, except within the CDR regions (data not shown). Both VH and VL domains have disulphide bonds between the conserved cysteines on the β strands B and F, consistent with proper folding of these domains. The intra-domain disulphide bonds formed presumably during purification by auto-oxidation. However, we have also found that the cysteine residues can be mutated to alanine or valine without losing the binding ability with RAS in vitro and in vivo, and that the disulphide bond-free mutation does not affect overall Fv structure (manuscript in preparation). The CDRs of VH domain are mainly in contact with the conformationally flexible parts of GTP-bound RAS, namely the switch I and the switch II regions (detailed in Figure 5A and B). Only a small part of VL CDR3 contacts the switch II region, and thus interaction of the Fv with RAS predominantly involves the VH segment (as expected, since the single VH domain, without the VL domain, has a 6 nM affinity for RAS (see below), and it functions intracellularly. Most of the interaction between RAS and VH is via hydrogen bonds derived from main or side chains of respective residues (Figure 5A), consistent with the finding that the interaction of Fv with RAS is inversely related to the salt concentration and pH (data not shown). Residues of all three VH CDR regions are involved in binding, with two to four residues from each CDR contributing equally to the interaction with the region including switch I of RAS.
Figure 5.

The binding site of the single domains on the RAS molecule. (A) A stereo diagram of the HRAS(G12V)-GTP-Fv binding interface. HRAS(G12V) is in green and the VH and VL chains are in cyan and orange, respectively. The CDRs of VH and VL are in yellow and lemon and the RAS switch I and II regions are in red and purple, respectively. Residues involved in the interface are shown in cylinder configuration. Specific residues of RAS are shown in blue, VH in red and VL in brown. Putative hydrogen bonds are indicated by dashed lines. (B) Schematic representation of the interacting residues in HRAS (green) and in the anti-RAS antibody (VH, yellow; VL, lemon). Putative hydrogen bonds are indicated by dotted lines. (C) The structures of HRAS(G12V)-GTP (green, red and purple) bound to anti-RAS Fv and of HRAS-GDP (blue) (PDB, 4Q21) (Milburn et al, 1990) are superimposed to illustrate the selectivity of iDab#6 single VH domain binding to activated GTP-bound RAS.
This structural analysis shows why iDab#6 is specific for activated RAS. Affinity determination of the anti-RAS intrabody binding to HRAS was performed using surface plasmon resonance analysis, employing iDab#6, or its scFv format, with GST-HRAS (loaded in vitro with GDP or GTPγS). Complexes formed selectively with HRAS(GTPγS) in a concentration-dependent manner (Figure 6A and B) and only background levels of association/dissociation were observed with HRAS(GDP) (Figure 6C). Kinetic measurements were used to calculate an equilibrium dissociation constant (Kd) of approximately 0.35 nM for scFv#6-RAS(GTPγS) and 6.2 nM for iDab#6-RAS(GTPγS). Superimposition the HRAS(G12V)-GTP with HRAS-GDP structures (PDB, 4Q21) shows that a major conformational difference exists in the switch I and II region of GTP-bound compared to GDP-bound RAS (Supplementary Figure 5A), and the region of GDP-bound RAS cannot form hydrogen bonds with the CDRs of the VH of iDab#6 (Supplementary Figure 6B, E, F and G). Therefore, the intrabody specifically binds to activated, GTP-bound RAS, because the hydrogen bonds needed for the nanomolar affinity do not form with GDP-bound RAS.
Figure 6.
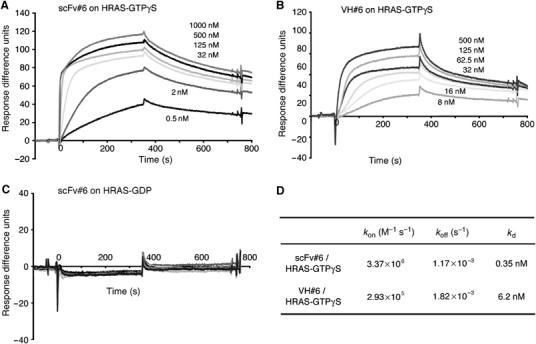
Affinity of anti-RAS intrabody for RAS. The binding affinity of the anti-RAS intrabody was measured using purified protein by surface resonance plasmon method measured using a BIAcore 2000. (A–C) Representative sensogrammes of scFv#6 bound to GST-HRAS(G12V)-GTPγS, bound to GST-HRAS(wt)-GDP (C), or of VH#6 bound to GST-RAS(G12V)-GTPγS are shown. The response difference units of sensogrammes, were normalised to the response of the channel trapping GST protein. (D) The table summarises values for the association (kon) (M−1 s−1) or dissociation rates (koff) (s−1), and the calculated equilibrium dissociation constants (Kd), using the BIAevaluation 2.1 software.
The anti-RAS single domain prevents effector molecules binding to RAS
Most RAS effector molecules, including RAF, RALGDS and PI3K, only interact with GTP-bound RAS and in the switch I conformation (Vetter and Wittinghofer, 2001), binding to a similar location as the VH of iDab#6. The crystal structures of respective RAS binding domains (RBD) complexed with RAS, or the RAS-like protein RAP1A, have been resolved to high resolution and could be superimposed our RAS–anti-RAS VH iDab#6 structure (Supplementary Figure 6). The intrabody binds to RAS with a high degree of overlap to the region of RAS bound by the RBD segments. Furthermore, the interface accessible surface areas (ASA) of RAS for iDab (688.2 Å2) or Fv (852.8 Å2) are larger that of the RAS-RBD complex (Supplementary Table I). The Kd of the RAS–intrabody complexes is approximately 0.4 nM for scFv#6-RAS(GTPγS) and 6 nM for iDab#6-RAS(GTPγS) (Figure 6D). These dissociation constants are lower than the 160 nM Kd of RAS-RAF-RBD (Sydor et al, 1998), 1 μM Kd for RAS-RALGDS-RBD (Linnemann et al, 2002) and 2.8 μM for RAS-PI3Kγ complexes (Pacold et al, 2000). These findings provide compelling evidence that the anti-RAS intrabody functions by inhibiting the RAS-effector protein interactions, thereby interfering with RAS-associated signal transduction pathways.
The ability of the intrabody to block the RAS-effector interactions was confirmed using biochemical studies in vitro (Figure 7). GST-RALGDS-RBD or GST-RAF-RBD fusion proteins were expressed in bacteria and incubated with radioactive HRAS loaded with GTPγS, in the presence or absence of purified, bacterially synthesised anti-RAS intrabody (in the scFv format, mutant scFvm#6 or scFv#6). Protein complexes were isolated (pulled down) using glutathione–Sepahrose resin, followed by gel separation for detection of HRAS by autoradiography and of RBD or single domain by Coomassie blue staining. Each GST-RBD protein quantitatively pulled down HRAS either in the absence of intrabody protein (Figure 7) or in the presence of the mutant intrabody scFvm#6 (Figure 7C). However, adding scFv#6 protein in increasing amounts reduced binding of the RAF-RBD or RALGDS-RBD to HRAS(GTPγS) in a dose-dependent manner (Figure 7A and B). These interactions between RAS and RBD proteins were almost completely abolished with equimolar amounts of GST-RBD and scFv#6.
Figure 7.
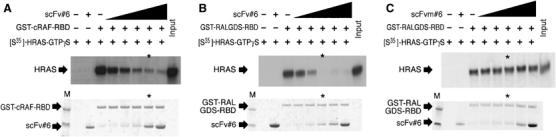
The anti-RAS single domain competes for binding of RAS effector molecules. (A–C) Competition of RAS-effector protein interaction by the anti-RAS intrabody in vitro. Biochemical studies of RAS-RAF or RAS-RALGDS interaction were performed using GST-fusion protein pull downs the presence of increasing amounts of anti-RAS scFv#6 (indicated by black shaded area)+presence, −absence. [35S-]HRAS(G12V) was loaded with GTPγS and mixed with differing concentrations of purified scFv#6 (A, B), mutant scFv#6 (C) plus by glutathione–Sepharose carrying GST-cRAF-RBD (A) or GST-RALGDS-RBD (B, C). The pulled down samples were eluted and fractionated by SDS–PAGE, followed by exposure to X-ray film, to detect [35S]-HRAS(G12V)-GTPγS (middle panel). The same amount of scFv and GST-RBD mixture without [35S]-HRAS(G12V)-GTPγS was fractioned on SDS–PAGE staining with CBB (lower panel). Asterisks indicate the lane in which scFv and GST-RBD were mixed in a 1:1 ratio. First lane (M) in lower panel is loaded with prestained SDS–PAGE standard molecular weight markers (estimated size: high, 53 kDa and low, 35.5 kDa).
Discussion
RAS-mediated cancer prevention by avoidance of protein–protein interaction
One of the most frequent mutations in human cancer is either directly RAS family mutations or aberrant signalling by RAS caused by secondary mutations, or amplification such as in receptor tyrosine kinases. Remarkable progress in the understanding of RAS function in normal and cancerous cells suggests targeting RAS for cancer therapy would block or reverse its aberrant functions. We have isolated a soluble, stably expressed single VH domain intrabody that specifically binds to active GTP-bound RAS with high affinity. The intrabody functionally distinguishes between the oncogenic properties of mutant RAS and the cell division/proliferation properties of normal RAS, presumably as there are alternative, normal pathways for signal transduction (Downward, 2003). By expressing the intrabody in cells harbouring mutant RAS, we have been able to prevent tumour formation at the site of transplantation and also in a metastatic tumour model. A previously described scFv intrabody, made from a rat monoclonal anti-RAS neutralising antibody, showed efficacy as an RAS inhibitor in mammalian cells (Canevari et al, 2002), but this scFv forms insoluble aggregates (Cardinale et al, 2003).
The crystal structure of the antigen–antibody complex shows that the single VH domain specifically binds to the switch I region on the active form RAS. This structural study shows the precision with which the CDR regions of the VH forms hydrogen binds with RAS residues, defining the binding site and demonstrating why the intrabody blocks RAS function, since it covers the surface of RAS where most of effectors (RAF, RALGDS and PI3K) interact. This binding competes RAS-effector interactions and prevents RAS-dependent signal transduction, and thus represents a canonical example of the type of inhibition.
A general approach to the undruggable protein interface
Our data show that a single domain shows great promise as a macromolecular drug (macrodrug; Forster et al, 2005) against RAS-mediated cancers. A possible use of this intrabody would be in combination with chemotherapeutic agents, to exert a potent antitumour cytotoxic-like effect in vivo. To achieve this goal, powerful methods for macrodrug delivery will need to be implemented (a problem that also besets RNAi therapeutic approaches), such as delivery of expression vectors via viral particles (Verma and Weitzman, 2005) or nanoparticles (Torchilin, 2005), or the use of protein transduction (Wadia and Dowdy, 2002). For therapy, the iDab is a RAS-specific antagonist that should be more effective in interfering with RAS-dependent cancer than cascade-specific blockers such as FTI, antisense, RNAi or other anti-RAS therapeutic reagents in clinical trials. In support of this option, we have also found that the intrabody inhibits growth of a human cancer cell line, with mutations in addition to RAS mutation, in a mouse model.
Finally, the single domain described here functions inside cells by blocking protein–protein interaction. This is generally considered to be an undruggable protein property (Blundell et al, 2006), because unlike the interaction of enzymes with non-protein substrates (small-molecule drugs), protein–protein interactions usually occur across large, flat surfaces, and not in tight binding pockets (Lo Conte et al, 1999). Our results show that single domain antibody fragments transform this into a druggable protein property. It should be noted that methods such as RNAi are incapable of discriminating between functional domains of proteins, as the approach works by downregulating mRNA and thus offers no subtlety in therapeutic use. Single domain intrabodies, on the other hand, can easily be derived, using IAC (Tse et al, 2002; Tanaka and Rabbitts, 2003), to any part of a protein considered to have a protein interaction partner, whose inhibition is of possible therapeutic importance. In addition, using mouse models as a preclinical test, as we have done in the present study, can validate the protein partnership as being relevant to disease. Therefore, these small proteins will be valuable not only to interfere with protein–protein interactions in diseases but also as potential research tools to validate disease-relevant interactomes of disease cells.
Materials and methods
Mammalian two-hybrid assay and retroviral infection of mammalian cells
Mammalian luciferase reporter assays were performed as described in Supplementary data. The method of retroviral infection is also described in Supplementary data. Briefly, recombinant retroviruses were generated by transfecting ecotropic packaging cells (Phoenix-E; Costa et al, 2000) cells with pGC-IRES-iDab plasmids, according to the online protocol (http://www.stanford.edu/group/nolan/protocols/pro_helper_free.html). Cells used were mouse NIH3T3-EJ cells (Shih and Weinberg, 1982) (this line has an exogenous human HRAS(G12V) mutant gene) or low-passage NIH3T3-D4 cells DLD-1 (ATCC, CCL-221, a human colorectal adenocarcinoma with KRAS(G13D) mutation) and HT-1080 (ATCC, CCL-121, a human fibrosarcoma with NRAS(Q61K)).
For infection of recombinant retroviruses, cells were plated at ∼2–5 × 105 in six-well plates and overlaid with 1 ml of recombinant retroviral supernatant using 4 μg/ml polybrene (Sigma). At 48 h post-infection, the cells were used for assays. For fluorescence staining, the cells were fixed and stained for F-actin staining with 0.1 μg/ml TRITC-phalloidin (Sigma), or for immunofluorescence staining with anti-FLAG mouse monoclonal antibody (M2, Sigma) and Cy3-linked anti-mouse IgG antibody (GE Healthcare). For soft agar assays, 5 × 103 cells were seeded in the medium containing agar. Cells were grown at 37°C for 3 weeks and the colonies (>0.1 mm in size) were counted.
Tumorigenicity and lung metastasis assays in mice
For determining effects of the anti-RAS intrabody on RAS-dependent tumorigenicity and metastasis in vivo, mouse and human cell lines expressing intrabodies after retroviral transduction were injected subcutaneously or intravenously into nude mice, as described in Supplementary data. A total of 5 × 104 NIH3T3-D4, NIH3T3-EJ cells, 5 × 105 of DLD-1 or 2.5 × 106 HT-1080 EGFP expressing cells were injected subcutaneously into 6 to 8-week-old female athymic MF1 nu/nu mice. Subcutaneous tumour growth was measured five times per week using a calliper. The assays were terminated when the tumour size reached maximum of 17 mm diameter, or at 42 days after injection (for NIH3T3 cells), or 13 weeks (for DLD-1 and HT-1080 cells) if no tumours were apparent. For lung metastasis assays, 105 NIH3T3-D4 or NIH3T3-EJ cells were injected intravenously (tail vein) into nude mice. At 3 weeks after injection, the mice were dissected and pathological and biochemical examinations were performed.
Protein expression and purification
For pull-down and affinity measurement assays, recombinant GST-HRAS(wt), GST-HRAS(G12V), GST-cRAF-RBD and GST-RALGDS-RBD fusion proteins, and His-tagged anti-RAS scFv and VH, were expressed in bacteria and purified, as described in Supplementary data.
For crystallography, the HRAS-anti-RAS Fv heterotrimer complex was prepared by coexpression of the three chains in Escherichia coli. The plasmid pRK-HISTEV-VH-RAS-VL was transformed into C41 (DE3). The transformed bacterial cell were cultured to an OD600 of 0.6 and induced with IPTG (final 0.5 mM) at 37°C for 4 h. The purification procedure of the complex was the same as for the scFv described in Supplementary data, except the extraction buffer (25 mM Na phosphate, pH 7.4, 300 mM NaCl, 20 mM imidazole) and gel filtration buffer (20 mM Tris–HCl pH 8.0 at room temperature (RT), 150 mM NaCl). The purified proteins were concentrated to 40 mg/ml, snap frozen and stored at –70°C.
Crystallography, structure determination and refinement
Crystals were grown using the sitting-drop vapour diffusion method. A 2 μl volume of purified protein was mixed with an equal volume of reservoir solution (17–18% PEG3350, 400 mM zinc acetate, 100 mM sodium cacodylate, pH 5.8, 0.03% dichloromethane) and set in 24-well Cryschem plates (Hampton Research), with 400 μl of reservoir solution. Crystals grew to a maximum size within 3 days at 19°C. SDS–PAGE analysis of the crystals showed the presence of the VH, VL and RAS components. The larger crystals were transferred to cryoprotectant (reservoir with 25% PEG3350) and frozen by immersion in liquid N2. Final data sets were collected with beamlines ID14-4 at the European Synchrotron Radiation Facility in Grenoble, France. Data collection statistics are shown in Table I. Autoindexing indicated that the crystals belonged to point group P2,2,2 with cell dimensions a=75.528 Å, b=84.632 Å, c=62.592 Å and a Matthews coefficient (Vm) of 2.15 Å3/Da (corresponding to a solvent content of 42.2% vol/vol) for three molecules (one complex) in the asymmetric unit. The reflections were indexed and integrated with MOSFLM (Leslie, 1992), merged and scaled with SCALA (CCP4, 1994). Molecular replacement was carried out with AMoRe (Navaza, 1994). The initial model consisted of the structure of HRAS(G12D) with GPPNP (PDB, 1AGP) (Franken et al, 1993). A second rotation/translation search with the structure of Fv of the anti-hen egg white lysozyme (HEL) monoclonal antibody D1.3 (PDB, 1A2Y) (Dall'Acqua et al, 1998) located the Fv molecule. The programme O (Jones et al, 1991) was used to build the model into the 2Fo−Fc and Fo−Fc maps with iterative rounds of refinement, using REFMAC (Murshudov et al, 1997). Figures were prepared with Pymol (http://www.pymol.org) software.
Table 1.
Structure data collection and refinement statistics
| Data collection and processing | |
| X-ray source | ESRF, ID14-4 |
| Wavelength (Å) | 0.934 |
| Space group | P2,2,2 |
| Unit cell dimensions | |
| a (Å) | 75.53 |
| b (Å) | 84.63 |
| c (Å) | 62.59 |
| Resolution range (Å)a | 50.32–2.00 (2.11–2.00) |
| Number of complexes/asymmetric unit | 1 |
| Observationsa | 92071 (12969) |
| Unique reflectionsa | 27214 (3867) |
| Completeness (%)a | 98.2 (97.2) |
| Rmergea,b | 0.097 (0.36) |
| Means I/σ (I)a | 10.4 (3.0) |
| Multiplicitya | 3.4 (3.4) |
| Refinement | |
| Reflections | 25816 |
| Number of atoms | 3330 |
| Protein atoms | 3003 |
| GTP atoms | 32 |
| Mg atom | 1 |
| Zn atoms | 5 |
| Water atoms | 289 |
| Rcryst (%)c | 19.4 |
| Rfree (%)c (% data used) | 27.0 (5.0) |
| Ramachandran plot statistics | |
| Core (%) | 90.8 |
| Allowed (%) | 8.3 |
| Generous (%) | 0.9 |
| Disallowed (%) | 0 |
| R.m.s. deviations from ideality | |
| Bond length (Å) | 0.019 |
| Bond angle (deg) | 1.812 |
| Values in parentheses are for the highest-resolution shell (2.11–2.00Å). | |
| Rmerge=∑hlk(∑i∣Ihlk−〈Ihlk〉∣)/∑hlk∣〈Ihlk〉∣. | |
| Rcryst and Rfree=∑∣∣Fobs∣−∣Fcalc∣∣/∑∣Fobs∣; Rfree calculated with the percentage of the data shown in parentheses. | |
In vitro pull-down and competition assays
For pull-down assays, equal molar amounts of GDP- or GTPγS-loaded GST-RAS and His-tagged anti-RAS scFv were mixed at RT for 1 h, and added to Ni-NTA agarose (for scFv pull down) or glutathione–Sepharose (for RAS pull down) and mixed by rotation for a further 1 h at RT. The mixture was centrifuged briefly and the supernatant was collected (unbound fraction). The agarose/sepharose pellets were washed five times with PBS and resuspended in SDS–PAGE buffer (bound fraction). The samples were analysed by SDS–PAGE and visualised by staining Coomassie brilliant blue (CBB). For in vitro competition assays, [35S]-HRAS(G12V) was prepared using TNT transcription/translation system (Promega), according to the manufacturer's instruction. After in vitro translation, [35S]-HRAS(G12V) was loaded with GTPγS and MgCl2. The GST-cRAF-RBD or GST-RALGDS-RBD was mixed with [35S]-HRAS(G12V)-GTPγS and varying amounts of anti-RAS scFv. Complexed proteins were pulled down by glutathione–Sepharose and analysed as described above. The respective protein ratios of reaction were assessed by SDS–PAGE staining with CBB and pulled down [35S]-HRAS(G12V) was fractionated by SDS–PAGE and visualised by autoradiography.
Affinity measurement of intrabodies
The binding kinetics of antibody with antigen were measured using a BIAcore 2000 (Biacore), as described elsewhere (Tanaka and Rabbitts, 2003), with modification. Briefly, a polyclonal goat anti-GST antibody (GE Healthcare) was immobilised on a CM5 sensor chip (Biacore) by amine coupling. GST or GDP- or GTPγS-loaded GST-RAS was injected for trapping on the sensor chip through the anti-GST antibody. Antibody fragment binding experiments were performed by injecting purified scFv or VH (0.5–1000 nM) in buffer containing 20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 5 mM MgCl2 and 0.005% Tween 20. Evaluation and calculation of the binding parameters was carried out according to the BIA evaluation 2.1 software.
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Table 1
Acknowledgments
We thank Drs RA Weinberg for NIH3T3 cells EJ clone, CJ Marshall for NIH3T3-D4 cells, J Avruch for cDNAs of M-RAS, R-RAS, RALA and RALB1, JH Camonis for cDNAs of RALA and RALB, M Matsuda for cDNAs of RAP1 and RAP2, J Downward for cDNAs of cRAF-RBD, GL Costa for pGCIRES, LM Albritton for pcDNA3-ATRC-1, V Smith, G King and R Berks for animal experiments, E Obayashi for pRKHISTEV and TEV protease, and R Grenfell for cell sorting, and also thank Elena Micossi for assistance with ESRF beamline ID14-4. This work was supported by the Medical Research Council and TT was supported by the National Foundation For Cancer Research.
The coordinates of the RAS–anti-RAS Fv complex have been deposited in the Protein Data Bank with the entry code 2uzi
References
- Adjei AA (2001) Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 93: 1062–1074 [DOI] [PubMed] [Google Scholar]
- Blundell TL, Sibanda BL, Montalvao RW, Brewerton S, Chelliah V, Worth CL, Harmer NJ, Davies O, Burke D (2006) Structural biology and bioinformatics in drug design: opportunities and challenges for target identification and lead discovery. Philos Trans R Soc Lond B Biol Sci 361: 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canevari S, Biocca S, Figini M (2002) Re: blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 94: 1031–1032, author reply 1032 [DOI] [PubMed] [Google Scholar]
- Cardinale A, Filesi I, Mattei S, Biocca S (2003) Evidence for proteasome dysfunction in cytotoxicity mediated by anti-Ras intracellular antibodies. Eur J Biochem 270: 3389–3397 [DOI] [PubMed] [Google Scholar]
- Cattaneo A, Biocca S (1997) Intracellular Antibodies: Development and Applications. Springer: New York, USA [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, Horner II JW, Cordon-Cardo C, Yancopoulos GD, DePinho RA (1999) Essential role for oncogenic Ras in tumour maintenance. Nature 400: 468–472 [DOI] [PubMed] [Google Scholar]
- Costa GL, Benson JM, Seroogy CM, Achacoso P, Fathman CG, Nolan GP (2000) Targeting rare populations of murine antigen-specific T lymphocytes by retroviral transduction for potential application in gene therapy for autoimmune disease. J Immunol 164: 3581–3589 [DOI] [PubMed] [Google Scholar]
- Dall'Acqua W, Goldman ER, Lin W, Teng C, Tsuchiya D, Li H, Ysern X, Braden BC, Li Y, Smith-Gill SJ, Mariuzza RA (1998) A mutational analysis of binding interactions in an antigen–antibody protein–protein complex. Biochemistry 37: 7981–7991 [DOI] [PubMed] [Google Scholar]
- Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3: 11–22 [DOI] [PubMed] [Google Scholar]
- Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, Whitsett JA, Koretsky A, Varmus HE (2001) Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 15: 3249–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster A, Pannell R, Drynan L, Cano F, Chan N, Codrington R, Daser A, Lobato N, Metzler M, Nam CH, Rodriguez S, Tanaka T, Rabbitts T (2005) Chromosomal translocation engineering to recapitulate primary events of human cancer. Cold Spring Harb Symp Quant Biol 70: 275–282 [DOI] [PubMed] [Google Scholar]
- Franken SM, Scheidig AJ, Krengel U, Rensland H, Lautwein A, Geyer M, Scheffzek K, Goody RS, Kalbitzer HR, Pai EF, Wittinghofer A (1993) Three-dimensional structures and properties of a transforming and a nontransforming glycine-12 mutant of p21H-ras. Biochemistry 32: 8411–8420 [DOI] [PubMed] [Google Scholar]
- Friday BB, Adjei AA (2005) K-ras as a target for cancer therapy. Biochim Biophys Acta 1756: 127–144 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ (2005) RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell 7: 219–226 [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T (2001) Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410: 1111–1116 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Kolch W (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 6: 827–837 [DOI] [PubMed] [Google Scholar]
- Leslie AGW (1992) Recent changes to the MOSFLM package for processing film and image plate data. In Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography. Warrington, UK: Daresbury Laboratory [Google Scholar]
- Linnemann T, Kiel C, Herter P, Herrmann C (2002) The activation of RalGDS can be achieved independently of its Ras binding domain. Implications of an activation mechanism in Ras effector specificity and signal distribution. J Biol Chem 277: 7831–7837 [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein–protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179–185 [DOI] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J (2000) The EGF receptor family as targets for cancer therapy. Oncogene 19: 6550–6565 [DOI] [PubMed] [Google Scholar]
- Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, Kim SH (1990) Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247: 939–945 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) A MoRe: an automated package for molecular replacement. Acta Cryst A 50: 157–163 [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103: 931–943 [DOI] [PubMed] [Google Scholar]
- Plattner R, Anderson MJ, Sato KY, Fasching CL, Der CJ, Stanbridge EJ (1996) Loss of oncogenic ras expression does not correlate with loss of tumorigenicity in human cells. Proc Natl Acad Sci USA 93: 6665–6670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan A, Hong SJ, Gifford A, Weinberg RA (2004) Species- and cell type-specific requirements for cellular transformation. Cancer Cell 6: 171–183 [DOI] [PubMed] [Google Scholar]
- Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD (1984) Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature 312: 71–75 [DOI] [PubMed] [Google Scholar]
- Shih C, Weinberg RA (1982) Isolation of a transforming sequence from a human bladder carcinoma cell line. Cell 29: 161–169 [DOI] [PubMed] [Google Scholar]
- Sydor JR, Engelhard M, Wittinghofer A, Goody RS, Herrmann C (1998) Transient kinetic studies on the interaction of Ras and the Ras-binding domain of c-Raf-1 reveal rapid equilibration of the complex. Biochemistry 37: 14292–14299 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Lobato MN, Rabbitts TH (2003) Single domain intracellular antibodies: a minimal fragment for direct in vivo selection of antigen-specific intrabodies. J Mol Biol 331: 1109–1120 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Rabbitts TH (2003) Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. EMBO J 22: 1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin VP (2005) Recent advances with liposomes as pharmaceutical carrriers. Nature Rev Drug Discov 4: 145–160 [DOI] [PubMed] [Google Scholar]
- Tse E, Lobato MN, Forster A, Tanaka T, Chung GTY, Rabbitts TH (2002) Intracellular antibody capture technology: application to selection of single chain Fv recognising the BCR-ABL oncogenic protein. J Mol Biol 317: 85–94 [DOI] [PubMed] [Google Scholar]
- Verma IM, Weitzman MD (2005) Gene therapy: twenty-first century medicine. Annu Rev Biochem 74: 711–738 [DOI] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294: 1299–1304 [DOI] [PubMed] [Google Scholar]
- Visintin M, Tse E, Axelson H, Rabbitts TH, Cattaneo A (1999) Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc Natl Acad Sci USA 96: 11723–11728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia JS, Dowdy SF (2002) Protein transduction technology. Curr Opin Biotechnol 13: 52–56 [DOI] [PubMed] [Google Scholar]
- Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, Rabbitts TH (1997) The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J 16: 3145–3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Table 1