

Abstract
In humans, complex I of the respiratory chain is composed of seven mitochondrial DNA (mtDNA)-encoded and 38 nuclear-encoded subunits that assemble together in a process that is poorly defined. To date, only two complex I assembly factors have been identified and how each functions is not clear. Here, we show that the human complex I assembly factor CIA30 (complex I intermediate associated protein) associates with newly translated mtDNA-encoded complex I subunits at early stages in their assembly before dissociating at a later stage. Using antibodies we identified a CIA30-deficient patient who presented with cardioencephalomyopathy and reduced levels and activity of complex I. Genetic analysis revealed the patient had mutations in both alleles of the NDUFAF1 gene that encodes CIA30. Complex I assembly in patient cells was defective at early stages with subunits being degraded. Complementing the deficiency in patient fibroblasts with normal CIA30 using a novel lentiviral system restored steady-state complex I levels. Our results indicate that CIA30 is a crucial component in the early assembly of complex I and mutations in its gene can cause mitochondrial disease.
Keywords: blue native PAGE, CIA30, complex I, lentivirus, mitochondria
Introduction
Complex I (NADH-ubiquinone oxidoreductase) is the major entry point of electrons into the mitochondrial respiratory chain and contributes to the establishment of a proton gradient required for ATP synthesis (Brandt, 2006). Complex I is the most complicated of the respiratory chain complexes. It contains 45 different subunits in mammals forming a complex of ∼1 MDa (Carroll et al, 2006). Of the complex I subunits, seven are encoded by mitochondrial DNA (mtDNA) whereas the remainder are encoded by nuclear genes, translated in the cytosol and imported into the organelle via the outer and inner membrane translocases, TOM and TIM (Wiedemann et al, 2004). Electron microscopy indicates that complex I has a bipartite L-shaped configuration consisting of a peripheral matrix arm and a membrane arm (Grigorieff, 1998). The matrix arm contains the flavoproteins and iron sulfur proteins involved in electron transfer, while the membrane arm includes all of the mtDNA-encoded subunits and is involved in ubiquinone reduction and proton pumping (Brandt, 2006). Complex I further assembles into higher ordered supercomplexes or ‘respirasomes' with respiratory chain complexes III and IV (Schagger and Pfeiffer, 2000; Schagger, 2001).
Defects in complex I activity are the most common diagnosis in patients with mitochondrial-based disease (Kirby et al, 1999; Triepels et al, 2001) and they have also been implicated in more common neurological disorders such as Parkinson's disease (Schapira, 2006). Complex I enzyme defects in patients are often associated with decreased levels of holoenzyme (Kirby et al, 2004a, 2004b; Ugalde et al, 2004a) and in some cases, the accumulation of stalled complex I intermediates, pointing to assembly defects (Antonicka et al, 2003; Kirby et al, 2004b). Since ∼50% of patients with complex I defects do not have mutations identified in subunits of complex I (Janssen et al, 2002; Thorburn et al, 2004), it is likely that mutations in genes encoding proteins involved in complex I biogenesis are causative in mitochondrial disease.
Characterization of complex I biogenesis has largely come from studies using Neurospora crassa, mainly because genetic analyses can be performed in this fungus whereas fermentative yeasts such as Saccharomyces cerevisiae lack a true complex I. Analysis of N. crassa complex I assembly using pulse labeling with radioactive amino acids in conjunction with gene disruption has revealed that the membrane and matrix arms assemble independently via separate pathways (Tuschen et al, 1990; Nehls et al, 1992). Furthermore, two additional proteins of 84 and 30 kDa, later termed complex I intermediate associated proteins, CIA84 and CIA30, were isolated with an assembly intermediate of the membrane arm (Kuffner et al, 1998). These proteins were suggested to function as molecular chaperones in the assembly of complex I as they are not components of the mature holoenzyme and mutating them blocked complex assembly (Kuffner et al, 1998). More recently, a human candidate gene (termed NDUFAF1) was identified, with its translation product sharing 28% homology with N. crassa CIA30 (Janssen et al, 2002). Although knockdown of human CIA30 using RNA interference causes reduced levels and activity of complex I (Vogel et al, 2005), the way CIA30 functions is not understood.
Besides CIA30, B17.2L (Ogilvie et al, 2005) has been identified as a mammalian complex I assembly factor while a third protein, AIF, has also been implicated (Vahsen et al, 2004). While mutations in the B17.2L gene have been shown to cause complex I deficiency and encephalomyopathy (Ogilvie et al, 2005), genetic screening of patients has not detected any mutations in the NDUFAF1 gene encoding CIA30 (Janssen et al, 2002). In this report, we show that human CIA30 is peripherally associated with the mitochondrial inner membrane in a number of steady-state complexes, distinct from complex I and other respiratory chain complexes. We show that CIA30 associates with assembly intermediates of complex I that contain both nuclear and mtDNA-encoded subunits. We also describe the first patient to be identified with a CIA30 deficiency, which results in the reduction of the steady-state levels of the protein to only ∼10%. Levels of mature complex I were also significantly reduced due to impaired assembly of the complex. These levels were restored in patient fibroblasts by complementing the deficiency with wild-type CIA30, confirming the defective CIA30 protein as the cause of the complex I deficiency.
Results
Submitochondrial location of CIA30
The human ortholog of the N. crassa complex I chaperone CIA30, has previously been shown to localize to mitochondria (Vogel et al, 2005). To determine its submitochondrial location, human CIA30 was in vitro translated in the presence of 35S-methionine/35S-cysteine and subsequently incubated with isolated mitochondria. As can be seen (Figure 1A, top left panel), the precursor form of the protein (pCIA30) not only associated with mitochondria but over time was processed to the mature form (CIA30). The matrix-targeted precursor of ornithine transcarbamylase (pOTC) (Lingelbach et al, 1986) was also imported as a control (Figure 1A, bottom panels). Unlike the precursor species, the processed forms of CIA30 and OTC were resistant to externally added proteinase K, indicating that they had been imported into mitochondria (Figure 1A, right panels). Furthermore, since processing of CIA30 was dependent on the presence of a membrane potential (Δψm), it can be concluded that CIA30 is synthesized with a presequence that, upon import, is proteolytically removed by matrix proteases (Ryan et al, 2001).
Figure 1.
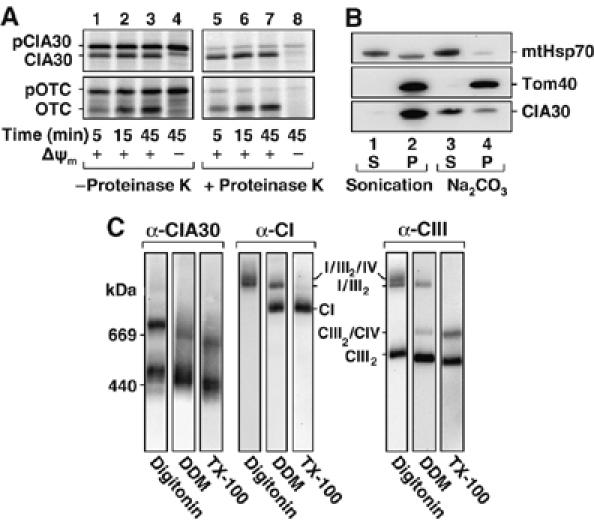
Characterization of CIA30. (A) [35S]-labeled precursor forms of CIA30 (pCIA30) and OTC (pOTC) were incubated with mitochondria isolated from HEK-293T at 37°C for the time points indicated in the presence and absence of a membrane potential (Δψm). Samples were treated with and without proteinase K (50 μg/ml) before SDS–PAGE and phosphorimage analysis. (B) Isolated mitochondria were subjected to either sonication or alkaline (carbonate, Na2CO3) extraction. Following treatments, soluble (S) and insoluble (P) fractions were subjected to SDS–PAGE and Western blot analysis using antibodies against CIA30, mtHsp70 and Tom40. (C) Mitochondria (60 μg protein) isolated from primary fibroblasts were solubilized in 1% (w/v) digitonin, 1% (w/v) DDM or 1% (w/v) Triton X-100 (TX-100) before BN-PAGE and Western blotting with antibodies against CIA30 or subunits of complexes I (CI), or III (CIII). The relative positions of supercomplexes or holoenzymes are indicated.
CIA30 contains no predicted transmembrane domains and given its N-terminally processed targeting signal, it is likely to reside in the mitochondrial matrix (Ryan et al, 2001). To investigate its possible membrane association, mitochondria were subjected to sonication or alkaline extraction followed by ultracentrifugation to separate membrane and soluble fractions. Samples were subjected to SDS–PAGE and Western blot analysis using antibodies raised against CIA30 (Figure 1B). CIA30 was found in the membrane fraction after sonication, but was predominantly released into the supernatant following alkaline extraction (labeled ‘Na2CO3'; Figure 1B). As controls, the integral outer membrane protein Tom40 was found in the pellet following both conditions as expected (Humphries et al, 2005) while the predominantly matrix-soluble protein mtHsp70 was found in the supernatant. A small amount of mtHsp70 was also in the pellet fraction after sonication since a population is bound to the inner membrane translocase (Ryan and Pfanner, 2001). The import results coupled with Western blot analysis therefore suggest that CIA30 peripherally associates with the matrix face of the mitochondrial inner membrane.
The association of CIA30 with the inner membrane is compatible with CIA30 interacting with inner membrane proteins. Indeed, Vogel et al (2005) previously showed that CIA30 (NDUFAF1) is found in two complexes of 600 and 700 kDa. In that case however, mitochondrial membranes were only solubilized using n-dodecyl-β-D-maltoside (DDM) as detergent. Solubilization with different detergents can result in dissociation of subunits and/or lipids from membrane protein complexes, thereby leading to changes in mobility on blue native (BN)-PAGE (Meisinger et al, 2001; McKenzie et al, 2006). To test whether CIA30 forms other complexes, mitochondria were isolated and solubilized in digitonin, DDM or Triton X-100 and subjected to BN-PAGE followed by Western blot analysis (Figure 1C, left panels). As can be seen, the antibody against CIA30 detected two major complexes under all conditions, with slightly different mobilities for each detergent. The lower complex resolved at 440–500 kDa (sometimes as a doublet), while the higher complex migrated in the 600–700 kDa range. In digitonin-solubilized mitochondria, faint complexes at ∼960 kDa and ∼1.3 MDa were also observed. The mobility of CIA30-containing complexes was compared to those of complexes I and III (and their supercomplex forms; Figure 1C, right panels). Solubilization of mitochondria with DDM results in the release of complex IV from the supercomplex (labeled ‘I/III2/IV' in Figure 1C) and also causes some complex I and complex III to be liberated, whereas Triton X-100 completely resolves complexes into their holoenzyme forms (McKenzie et al, 2007). In all cases, the respiratory chain complexes did not migrate at the same sizes as the CIA30-associated complexes, suggesting that CIA30 is not associated with these mature complexes.
CIA30 and complex I assembly
CIA30 has been implicated in the assembly of complex I in N. crassa by virtue of its association with a stalled assembly intermediate (Kuffner et al, 1998). We asked whether the human CIA30 ortholog associates with complex I subunits in mitochondria. Co-immunoprecipitations of mitochondrial lysates were performed using CIA30 antibodies coupled to protein A-Sepharose (Figure 2). Antibodies from preimmune sera coupled to protein A-Sepharose served as a negative control. Specifically bound proteins were eluted from the beads and subjected to SDS–PAGE followed by Western blot analysis using antibodies against a number of mitochondrial proteins. A sample of the unbound mitochondrial fraction (representing 2% of total) was also analyzed. As can be seen, CIA30 had been completely extracted from the mitochondrial lysate (Figure 2, top panel, compare lanes 2 and 4) while antibodies from the preimmune sera did not pull down CIA30 (Figure 2, top panel, lanes 1 and 3). We next determined the presence of various complex I subunits including the mtDNA-encoded subunit ND1 and the nuclear-encoded subunits NDUFA9, NDUFS3, NDUFS7, NDUFB6, NDUFS5 and NDUFA6. All subunits co-immunoprecipitated with CIA30, except NDUFS5 (Figure 2), which appears to assemble directly into complex I (Ugalde et al, 2004b). Little, if any, protein was detected when preimmune antibodies were used in the co-immunoprecipitations. Subunits from complexes III (core 1) and II (70 kDa) were not immunoprecipitated with CIA30, suggesting that binding is specific for complex I subunits. Unlike CIA30 however, the complex I subunits were not significantly immunodepleted from the mitochondrial extracts since they were all found in the unbound fraction (Figure 2, lane 4). This suggests that CIA30 associates with complex I subunits found only in intermediate subcomplexes and not those within mature complex I. It has recently been shown that NDUFS5 (15 kDa subunit) is not found in any detectable assembly intermediate and may be incorporated directly into the holocomplex at a late stage (Ugalde et al, 2004b). The finding that NDUFS5 does not stably interact with CIA30 in our experiments supports these findings.
Figure 2.
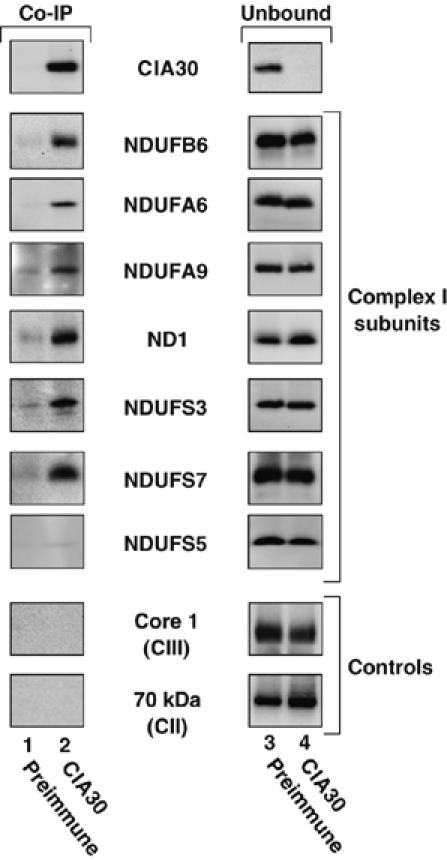
Co-immunoprecipitation of complex I subunits with CIA30. Mitochondria from HEK-293T cells were solubilized in 1% (w/v) Triton X-100 before immunoprecipitation with antibodies against CIA30 or preimmune serum coupled to protein-A Sepharose. The eluted fraction and a sample of the unbound fraction (2% of total) were then applied to Tris–Tricine SDS–PAGE followed by Western transfer and immunodecoration with antibodies as indicated.
We have recently found that newly translated mtDNA-encoded complex I subunits form detectable assembly intermediate complexes at ∼460 and ∼830 kDa on BN-PAGE (Lazarou et al, 2007). The latter complex is most likely the same as that found by Ogilvie et al (2005). After longer incubation times, these assembly intermediates disappear and the subunits are integrated into the fully assembled complex I. We determined whether CIA30 might be associated with intermediate complexes. MtDNA-encoded subunits were specifically radiolabeled in cultured human lymphoblasts for 2 h following a chloramphenicol pretreatment for 16 h. Mitochondria were then solubilized in Triton X-100 and subjected to immunoprecipitations using antibodies against CIA30 or from preimmune serum. The protein complexes not bound to these antibodies were analyzed by BN-PAGE. The preimmune sample shows the presence of a number of respiratory chain complexes containing newly incorporated radiolabeled mtDNA-encoded subunits (Figure 3A, lane 1). The most prominent species is complex V while complexes III, IV and a complex IV intermediate species labeled ‘CIVi' can also be seen (McKenzie et al, 2007). In addition, the ∼460 and ∼830 kDa intermediate complexes containing complex I subunits were present in the unbound fraction following incubation with the preimmune sera (Figure 3A, lane 1). However, these intermediates were specifically depleted following incubation with immobilized CIA30 antibodies (Figure 3A, lane 2). These results indicate that CIA30 associates with these early subcomplexes that contain newly translated mtDNA-encoded subunits. The same radiolabeled samples were subsequently analyzed for the presence of CIA30 complexes using Western blotting (Figure 3A, lanes 3 and 4). With Triton X-100 solubilization, the major CIA30 complexes are at ∼460 kDa (running as a doublet), with minor complexes at ∼600 and ∼830 kDa. These complexes were still present in the unbound fraction following incubation with preimmune antibodies, but they were specifically depleted with the CIA30 antibodies (Figure 3, compare lanes 3 and 4). Subsequent probing for complex I revealed that depletion of CIA30 had no effect on steady-state complex I levels, consistent with CIA30 not associating with the mature holoenzyme (Figure 3, lane 6). The levels of complex II were also assessed as a negative control to demonstrate the specificity of the immunodepletion and to serve as a loading control.
Figure 3.
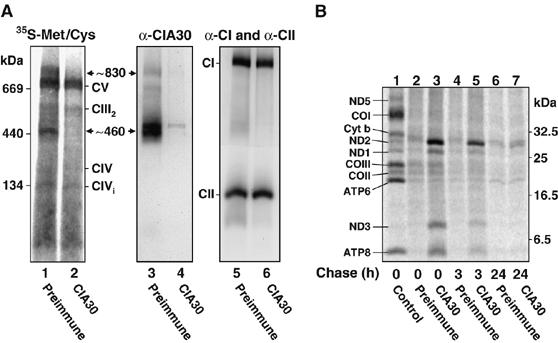
mtDNA-encoded complex I (CI) subunits associate with CIA30 at early stages in their assembly. (A) BN-PAGE analysis of mitochondria containing radiolabeled mtDNA-encoded subunits (0 h chase) immunodepleted with the αCIA30 antibody (lanes 2, 4 and 6). Preimmune serum was also used for control depletion (lanes 1, 3 and 5). Protein complexes containing radiolabeled mtDNA-encoded subunits were detected by autoradiography (lanes 1 and 2). After this, the lanes were subjected to Western blot analysis using antibodies against CIA30 (lanes 3 and 4) followed by antibodies against CI and complex II (CII) (lanes 5 and 6). (B) After radiolabeling, cells were chased for various times as indicated. Mitochondria were isolated, solubilized in Triton X-100 and incubated with protein A-Sepharose coupled to CIA30 antibodies or preimmune sera. Bound proteins were analyzed by SDS–PAGE and phosphorimaging (lanes 2–7). A labeled subunit profile (control) is shown in lane 1 and represents 16.5% of material used in each co-immunoprecipitation. The mtDNA-encoded subunits are indicated.
Our results suggest that CIA30 interacts with newly synthesized mtDNA subunits in complex I intermediates but not with the mature holoenzyme. To determine which mtDNA-encoded subunits associate with CIA30, a co-immunoprecipitation analysis was again undertaken. Mitochondrial extracts were used from cells that had been chased for various times after mtDNA subunit labeling. In this case, radiolabeled mtDNA-encoded subunits are present in intermediate assemblies at early time points while at later times (e.g. 24 h—see Figure 4C and McKenzie et al, 2007), they will have largely assembled into the complex I holoenzyme. Immunoprecipitated radiolabeled proteins were separated on SDS–PAGE and detected by phosphorimage analysis. Owing to the lack of available antibodies, only the ND1 subunit could be definitively identified based on its exact migration with endogenous ND1 (data not shown). Nevertheless, based on the mobilities of other subunits (COX1, 2, 3, cytochrome b, ATP6, ATP8 and ND5) and previous assignments (Chomyn, 1996), we could also confidently assign spots corresponding to subunits ND2 and ND3. At 0 and 3 h chase, the complex I subunits corresponding to ND1, ND2 and ND3 were co-immunoprecipitated with CIA30 (Figure 3B, lanes 3 and 5). The subunit corresponding to the mobility of ND5 was not specifically co-immunoprecipitated while ND4, ND4L and ND6 were not detected. The ATP8 subunit of complex V was also seen in the co-immunoprecipitates, but unlike the complex I subunits it was not specifically enriched against the total sample (lane 1), suggesting that it may represent a contamination. By 24 h chase, the radiolabeled subunits were no longer co-precipitated with CIA30 (Figure 3B, lane 7), consistent with these subunits integrating into the mature holoenzyme. Our results indicate that at early stages of complex I assembly, CIA30 is found in complexes of ∼460 and ∼830 kDa that contain (among others) the mtDNA-encoded subunits ND1, ND2 and ND3.
Figure 4.
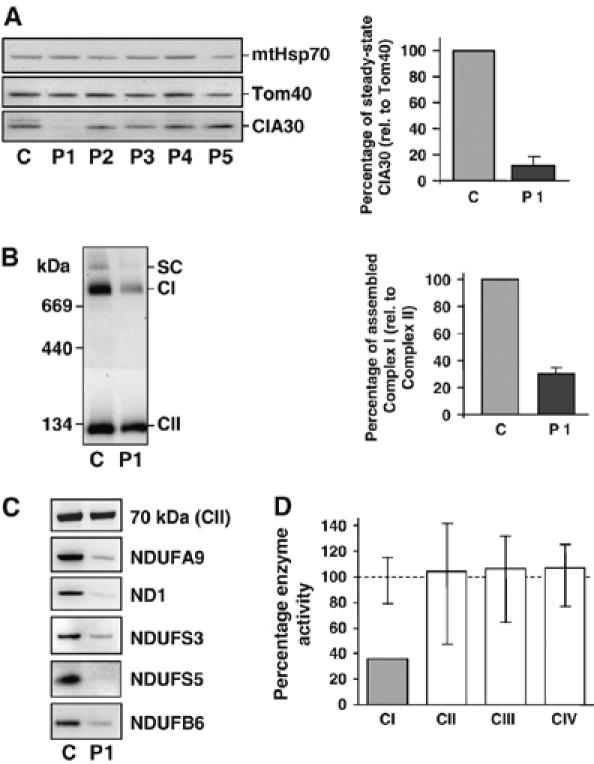
Identification of a complex I-deficient patient cell line with decreased CIA30. (A) Mitochondria isolated from fibroblasts of control (C) and patients (P1–P5) with complex I defects were applied to SDS–PAGE, Western blotted and probed with antibodies against CIA30, Tom40 and mtHsp70 (left panel). Patients had fibroblast complex I activities ranging from 10 to 50% of controls with unknown genetic cause (P2) or with two pathogenic mutations in the complex I subunit genes NDUFV1 (P3), NDUFS4 (P4) or NDUFS6 (P5). The steady-state levels of CIA30 in patient 1 (P1) were quantified against Tom40. Data are mean±s.e.m., n=3 (right panel). (B) Steady-state levels of complex I from patient 1 (P1) fibroblasts were analyzed by BN-PAGE and immunoblotting using antibodies against complex I (CI) and complex II (CII; left panel). SC=complex I/complex III2 supercomplex. The steady-state levels of complex I were quantified against complex II. Data are mean±s.e.m., n=3 (right panel). (C) Mitochondrial extracts from control (C) and patient 1 (P1) fibroblasts were subjected to SDS–PAGE and Western blot analysis against complex I subunits and the complex II 70 kDa subunit (as control). (D) Respiratory chain enzyme activities in patient 1 (P1) lymphoblasts were measured and standardized against the mitochondrial marker enzyme citrate synthase and shown as a percentage of the control mean. Vertical lines represent the observed range for 6 control lymphoblast cell lines.
Identification of a complex I-deficient patient cell line with reduced levels of CIA30
A previous genetic screen of 13 patients with complex I deficiency but containing no known mutation in genes encoding any of its subunits, did not reveal any mutations in the NDUFAF1 gene encoding CIA30 (Janssen et al, 2002). Since this screening methodology did not focus on steady-state CIA30 protein levels, we undertook a Western blot analysis of fibroblast cell lines from patients diagnosed with enzymatically deficient complex I (Figure 4A, left panel). Although CIA30 varied in its levels between control and patient cell lines, it was severely reduced in patient 1 (Figure 4A), who had presented with cardioencephalomyopathy. Antibodies against mtHsp70 and Tom40 served as loading controls. Quantification against Tom40 revealed that the levels of CIA30 were ∼10% compared to control (Figure 4A, right panel). CIA30 levels in patient 1 lymphoblasts were also decreased to ∼10% of control values (data not shown). The level of steady-state complex I in patient 1 fibroblasts, as measured on BN-PAGE, was also reduced to ∼30% of control values (Figure 4B). SDS–PAGE and Western blot analysis of mitochondrial extracts from patient 1 fibroblasts showed that the levels of all five complex I subunits tested were reduced compared to control mitochondria (Figure 4C). Consistent with the reduction of complex I steady-state levels, enzymatic analysis of patient 1 lymphoblasts showed a complex I deficiency of 36% residual activity relative to the marker enzymes citrate synthase or complex II. The activities of complexes II–IV were all normal (Figure 4D). Like lymphoblasts, patient 1 fibroblasts also showed a specific reduction in complex I activity (see Supplementary Figure 1).
Complex I assembly defects are found in patient 1 cells
The decreased levels of complex I and its subunits in patient 1 cells may be due to a defect in subunit assembly and/or increased turnover. To test this, we analyzed the assembly of newly synthesized mtDNA-encoded subunits into complex I. As can be seen (Figure 5A), at early chase times the ∼830 kDa intermediate (marked *) is detectable in control fibroblasts (lanes 1 and 2), but is not visible in patient 1 fibroblasts (lanes 5 and 6). The ∼460 kDa band is also reduced in patient 1 fibroblasts, although a faint band is visible at ∼400 kDa (Figure 5A, lanes 5 and 6). By 24 h chase, mature complex I is detectable in control fibroblasts (Figure 5A, lane 4), but is not present in patient 1 fibroblasts (lane 8). The assembly of mtDNA-encoded subunits into the other respiratory chain complexes is not altered, demonstrating that the defect lies specifically in complex I assembly.
Figure 5.
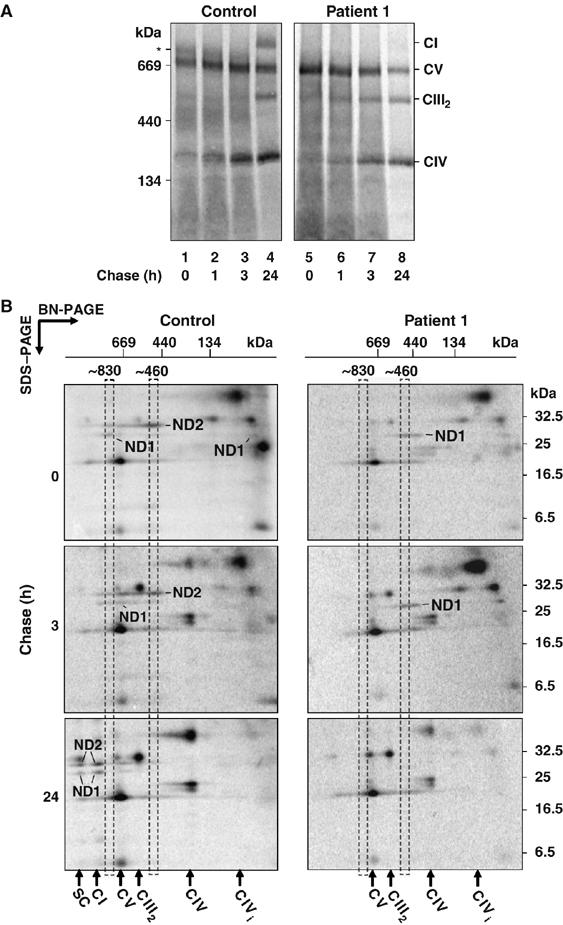
Complex I assembly is impaired in patient 1 cells. (A) After radiolabeling of mtDNA-encoded subunits, control or patient 1 fibroblasts were chased for various times as indicated. Mitochondria were isolated, solubilized in Triton X-100 and subjected to BN-PAGE followed by phosphorimage analysis. CI, CIII2, CIV and CV are indicated. The asterisk (*) denotes the ∼830 kDa intermediate complex. (B) 2D-PAGE analysis of radiolabeled mtDNA-encoded subunits from both control (left panels) and patient 1 (right panels) fibroblasts at different chase times. Mitochondria were isolated, solubilized in Triton X-100 and subjected to BN-PAGE in the first dimension, followed by SDS–PAGE in the second dimension and phosphorimager analysis. Positions of the ∼460 and ∼830 kDa assembly intermediates and the respiratory chain complexes are shown. The positions of ND1 and ND2 are indicated. CI=complex I, CV=complex V, CIII2=complex III dimer, CIV=complex IV, CIVi=complex CIV intermediate, SC=CI/CIII2 supercomplex.
Radiolabeled mtDNA-encoded translation products from both control and patient 1 fibroblasts were also analyzed after various chase times using two dimensional (2D)-PAGE analysis (Figure 5B). ND2 assembled into the ∼460 kDa intermediate complex in control mitochondria but in patient 1 mitochondria, ND2 was absent (Figure 5B, top panels). Before the chase (‘0 h chase'), ND1 was found in the ∼830 kDa intermediate complex in control mitochondria but in patient 1 it appeared to stall in an ∼400 kDa complex (Figure 5B, top panels). This stalled complex was still present in patient 1 mitochondria at 3 h chase (Figure 5B, middle panels). After 24 h chase, ND1 in control mitochondria had almost fully assembled into complex I or its supercomplex whereas in patient 1 mitochondria, it was completely absent (Figure 5B, bottom panels). SDS–PAGE analysis was also performed, confirming the loss of ND2 between 0 and 1 h chase and the loss of ND1 after 24 h chase in patient 1 mitochondria (see Supplementary Figure 2). These results suggest that the complex I defect in patient 1 cells is due to an impaired assembly of complex I subunits into early intermediates.
Genetic analysis of the CIA30-deficient patient
Genomic DNA sequencing analysis of the patient with CIA30 deficiency revealed two heterozygous mutations in exon 3 of the NDUFAF1 gene, c.1001A>C and c.1140A>G, named according to their cDNA nomenclature (Figure 6A). The c.1001A>C mutation was maternally inherited and predicts a Thr to Pro substitution (T207P) while the c.1140A>G mutation was paternally inherited and predicts a Lys to Arg substitution (K253R). Both Thr 207 and Lys 253 are highly conserved across different species (Figure 6B). The c.1001A>C and c.1140A>G mutations are not known SNPs and were absent from 106 control alleles sequenced (data not shown).
Figure 6.
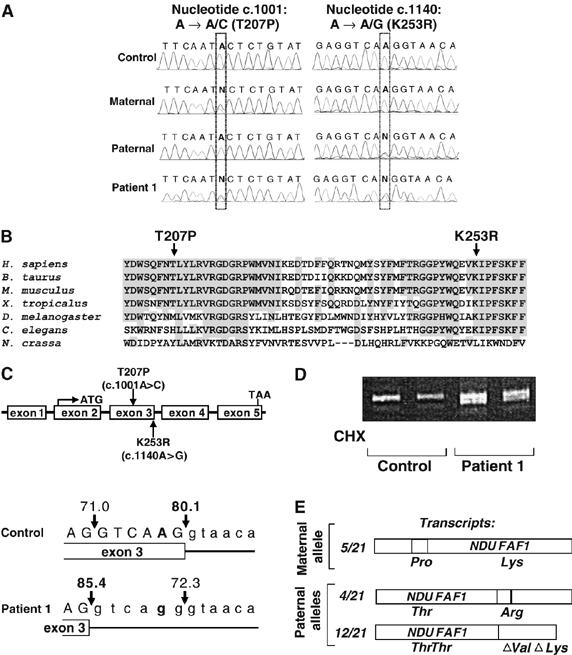
Genetic screening of NDUFAF1 in patient 1. (A) Chromatographic trace of relevant NDUFAF1 sequence from control, patient 1 and the patient's parents. The patient is compound heterozygous for a c.1001A>C and a c.1140A>G mutation, the patient's mother was heterozygous for the c.1001A>C mutation while the patient's father was heterozygous for the c.1140A>G mutation. (B) Amino-acid sequence alignment of the mutated region of CIA30 from Homo sapiens (human, accession #Q9Y375), Bos taurus (bovine, accession #XP_615202), Mus musculus (mouse, accession #NP_081451), Xenopus tropicalis (xenopus, accession #NP_001017153.1), Drosophila melanogaster (accession #NP_651718.1), Caenorhabditis elegans (accession #NP_506361.1) and N. crassa (accession #O42636). Shading represents identical residues in four or more species. (C) Predicted NDUFAF1 gene structure showing the location of the mutations within exon 3 (top) and the predicted donor splice sites in control and patient 1 exon 3 (bottom). Predicted percentage scores for splice site probability are shown. SpliceSiteFinder predicts that the c.1140A>G mutation moves the preferred donor splice site 6 bp upstream in patient 1 resulting in the deletion of 6 bp of coding sequence. (D) PCR analysis using primers located in NDUFAF1 exons 2 and 4 of cDNA from control and patient 1 fibroblasts grown without and with cycloheximide (CHX). Patient cDNA shows a second smaller fragment in addition to the expected 333 bp fragment, consistent with the splicing defect predicted by SpliceSiteFinder. (E) Diagrammatic representation of the three sequences identified by cloning the NDUFAF1 cDNA fragment spanning exons 2–4 from patient 1 cDNA. Of 21 sequenced clones, five cDNA sequences represented the expected maternal allele carrying the c.1001A>C (Thr to Pro) mutation. Of 16 paternally derived cDNA sequences (containing c.1001A), four contained the c.1140A>G (Lys to Arg) mutation while 12 lacked the final 6 bp encoded by exon 3 (loss of Val and Lys), consistent with the predicted splicing defect.
cDNA sequencing confirmed the maternally inherited heterozygous mutation at c.1001 and the presence of double sequence suggested a deletion/insertion event had occurred 6 bp upstream of the exon 3/4 boundary (not shown). The c.1140A>G mutation is positioned in the splice site consensus sequence at the end of exon 3 and was suspected to be causing a splicing defect. The NDUFAF1 exon 3 and intron 3 sequence was analyzed using the SpliceSiteFinder prediction program (http://violin.genet.sickkids.on.ca/~ali/splicesitefinder.html), which predicts the presence and relative efficiencies of donor and acceptor sites and assigns them a probability value from 0 to 100 (Shapiro and Senapathy, 1987). The c.1140A>G mutation is predicted to give a preferred donor splice site 6 bp upstream of the wild-type site (Figure 6C). This alternative splice site would lead to a 6 bp deletion of the cDNA, predicting the loss of Val252 and Lys253, which appear evolutionarily conserved in higher eukaryotes (Figure 6B).
cDNA was prepared from control and patient cells and PCR-amplified using primers located in exons 2 and 4. Control cDNA showed a single amplicon of the predicted size but patient cDNA also showed an additional smaller amplicon, consistent with the predicted small deletion being present (Figure 6D). The smaller amplicon was present in cells grown with and without cycloheximide, suggesting the apparent splicing defect did not cause nonsense-mediated mRNA decay (Amrani et al, 2006). To confirm that these results reflected the predicted splicing defect, 21 individual cDNAs were cloned and sequenced. Three different cDNA sequences were identified and these are depicted schematically in Figure 6E. Five cDNA sequences represented the expected maternal allele and contained the c.1001A>C mutation. Two different paternally derived cDNA sequences (containing c.1001A) were identified; four of these contained the c.1140A>G mutation whereas 12 lacked the final 6 bp encoded by exon 3, consistent with the predicted 6 bp deletion encompassing c.1136 to c.1141 (Figure 6E).
Complementation with wild-type CIA30 in cells from patient 1 restores complex I levels
To verify that the reduced level of complex I observed in patient 1 is in fact due to a CIA30 deficiency, complementation analysis was performed (Figure 7). Patient fibroblasts were stably transfected with wild-type NDUFAF1 using a lentiviral transfection system. In this case, NDUFAF1 was placed under the control of the estrogen promoter and can be transcriptionally induced with 4-hydroxy-tamoxifen (4HT). As previously shown, CIA30 is virtually undetectable in fibroblasts from patient 1 (Figure 7A) and the levels of complex I are also low (Figure 7B) in comparison to control fibroblasts. Induction of wild-type NDUFAF1 in transfected patient 1 fibroblasts resulted in increased levels of CIA30 protein and exceeded control levels following 74 h incubation with 4HT (Figure 7A, compare lanes 1 and 7). The expression of wild-type CIA30 in patient cells also correlated with the restoration of assembled complex I (Figure 7B). Induction of CIA30 into control cells had no effect on complex I levels (data not shown). We conclude that the genetic defect in NDUFAF1 is responsible for reduced levels of complex I in this patient and that CIA30 is a critical factor in complex I assembly.
Figure 7.
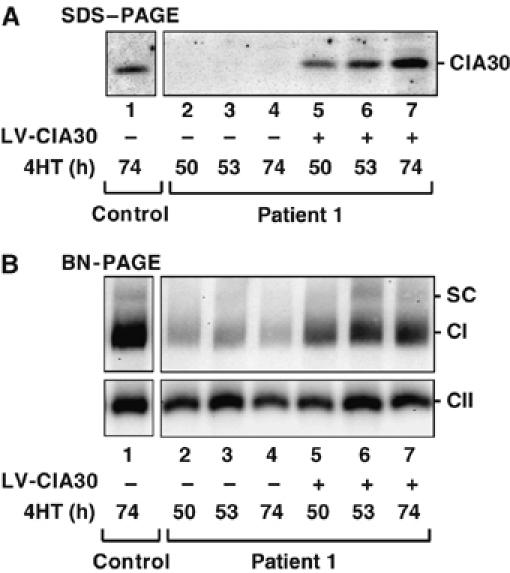
Expression of wild-type CIA30 in the patient cell line increases complex I levels. Patient 1 fibroblasts (lanes 2–4) and those transfected with wild-type CIA30 using lentiviral (LV) vectors (lanes 5–7) were incubated with 4HT for the indicated times. Untransfected control cells are also shown (lane 1). (A) Mitochondria were isolated and subjected to SDS–PAGE and Western blotting using antibodies against CIA30. (B) The same samples as in A were solubilized in Triton X-100 and subjected to BN-PAGE and immunoblot analysis using antibodies against complex I and complex II subunits. SC=complex I/complex III2 supercomplex.
Discussion
Involvement of CIA30 in complex I assembly
CIA84 and CIA30 were the first factors identified in the assembly of fungal complex I. This complex differs somewhat to its mammalian counterpart and contains only 39 subunits, some of which are fungal-specific (Marques et al, 2005). Kuffner et al (1998) found that disruption of the 21.3 kDa subunit of N. crassa complex I lead to assembly defects and the subsequent accumulation of two membrane intermediates of 350 and 200 kDa. The 350 kDa intermediate was found to be composed of a number of mtDNA-encoded subunits and at least six nuclear-encoded subunits, as well as the chaperones CIA30 and CIA84. Both CIA84 and CIA30 appeared to bind independently to the 350 kDa intermediate and neither interacted with fully assembled complex I, leading to the proposition that they act as molecular chaperones (Kuffner et al, 1998). While no further characterization of N. crassa CIA30 and CIA84 has been reported to date, studies of mammalian complex I have identified a number of mitochondrial proteins involved in complex I biogenesis. These include human CIA30 (NDUFAF1) (Vogel et al, 2005), B17.2L (Ogilvie et al, 2005), AIF (Vahsen et al, 2004), complex III (Acin-Perez et al, 2004) and complex IV (Diaz et al, 2006). While the human ortholog of CIA84 has been identified at the sequence level, no characterization has been reported (Gabaldon et al, 2005).
In this report, we have further characterized the role of CIA30 in the biogenesis of human complex I. Through in vitro import analysis and studies on the endogenous protein, we show that CIA30 is a nuclear-encoded protein that is imported into the matrix where it presumably associates with components on the inner membrane. Vogel et al (2005) reported that in digitonin-solubilized mitochondrial extracts, CIA30 is present in two complexes of 600 and 700 kDa. We also observed CIA30 in a number of complexes, but these resolved slightly differently with major steady-state complexes between 440–500 kDa and 600–700 kDa. The migration of these complexes varied slightly, depending on the detergent employed. A recent publication clarified the sizes of the CIA30 complexes as being in the 500–850 kDa range (Vogel et al, 2007b), which is more consistent with our data. Regardless, we showed through co-immunoprecipitation studies and immunodepletion analysis that CIA30 associates with newly translated mtDNA-encoded subunits corresponding to ND1, ND2 and ND3 of the membrane arm, as well as many nuclear-encoded subunits of complex I, including a number found in the matrix arm. Indeed, after Triton X-100 solubilization complex I assembly intermediates of ∼460 and ∼830 kDa comigrate with CIA30-containing complexes and are immunodepleted with the CIA30 antibody (Figure 3). The ∼830 kDa complex represents a late assembly intermediate of complex I and appears to lack subunits of the tip of the matrix arm (Kirby et al, 2004b; Ogilvie et al, 2005). Importantly, the NDUFS5 subunit did not coprecipitate with CIA30. Ugalde et al (2004b) showed that this subunit assembles at a late step and does not form stable intermediates. This suggests that CIA30 does not associate with all subunits and is consistent with results showing that it is not stably bound to fully assembled complex I.
CIA30 and mitochondrial disease
The identification of N. crassa CIA30 lead to the screening for genetic defects in the orthologous human NDUFAF1 gene in 13 complex I-deficient patients without identified mutations in genes encoding complex I subunits; however, none were found (Janssen et al, 2002). By screening patient cell extracts with CIA30 antibodies using Western blot analysis, we were able to detect a patient with significantly reduced levels of CIA30 protein. Subsequent sequence analysis of NDUFAF1 transcripts in patient cells indicate that a c.1140A>G mutation leads to mis-splicing, resulting in a 6 bp deletion in ∼75% of paternally derived transcripts (deletion of V252 and K253). The remaining ∼25% of paternal transcripts encode CIA30 with a K253R mutation. In contrast, the maternally derived c.1001A>C mutation results in a T207P change in CIA30. As patient 1 fibroblasts are deficient in CIA30, the T207P and deletion mutations are likely to cause increased turnover of the protein. The K253R variant may also be unstable but given the relatively mild substitution of one positively charged amino acid for another, it is plausible that this variant may account for the residual protein and complex I assembly seen in patient cells. Introduction of normal CIA30 into patient cells through a lentiviral-mediated approach restored complex I to levels similar to control. In contrast, overexpression of CIA30 in control cells did not lead to increased steady-state complex I levels (data not shown). Thus, our results indicate that the mutations seen in NDUFAF1 are responsible for the defect in complex I and the resulting disease pathology. Our results are consistent with those of Vogel et al (2005) who found complex I levels can be reduced following knockdown of NDUFAF1 using RNA interference.
Most patients with nuclear gene mutations impacting on complex I present in infancy or early childhood and have severe disease often leading to death in early childhood (Kirby et al, 1999). At the time of this report, patient 1 is alive and medically stable at 20 years of age although he displays myopathy, visual impairment, kyphoscoliosis and mild intellectual handicap (a full patient history is provided as Supplementary material). Presumably the presence of some CIA30 is sufficient to promote the assembly of residual complex I seen in the patient cells thereby leading to the ‘relatively' mild phenotype displayed. In addition, since we only assayed CIA30 levels in fibroblasts and lymphoblasts, we cannot exclude that other cells have different splicing and/or protease activities that result in different CIA30 levels in other tissues.
Role of CIA30 in complex I assembly
Reduced CIA30 levels in patient mitochondria correlate with the loss of a ∼460 kDa complex I intermediate that contains ND2. SDS–PAGE analysis indicates that ND2 is still expressed in these cells but is rapidly turned over. In the absence of this intermediate, ND1 (and other subunits) stall at ∼400 kDa and are lost over time. These results would be consistent with much of the ∼400 kDa complex containing ND1 being degraded as it cannot assemble with the missing ∼460 kDa intermediate. This prevents formation of the ∼830 kDa complex and hence complete assembly of the holoenzyme. Given that there is some steady-state level of complex I still present in these cells, the assembly process cannot be completely defective. However, the steady-state levels of those complex I subunits tested were significantly reduced in patient mitochondria implying that there must also be increased turnover of unassembled subunits. This is consistent with other examples where reduced levels of mature complex I lead to decreased steady-state levels of its subunits (Acin-Perez et al, 2004; McKenzie et al, 2006).
Studies of complex I assembly in humans, either in patients with complex I defects (Antonicka et al, 2003) or in a model system whereby complex I was firstly depleted to monitor de novo synthesis (Ugalde et al, 2004b), have resulted in two different models for complex I assembly. This discrepancy highlights the fact that the assembly process is yet to be fully elucidated. We propose that CIA30 is involved in the initial assembly of a membrane subcomplex that most likely contains ND2 and ND3. The presence of other ND subunits within these complexes was not clarified due to their insufficient labeling. ND1 and other subunits associate with this subcomplex through separate module(s) forming a stable ∼830 kDa intermediate. The presence of both ND1 and ND2 could be seen in this intermediate in Figure 5B. The assembly of subunits into the matrix arm of complex I correlate with the release of CIA30.
While we have uncovered that CIA30 is indeed a complex I assembly factor and its defects cause disease, we are yet to understand precisely how this protein functions in this process. This is the focus of future investigations. It is also probable that other factors involved in complex I assembly will be identified, perhaps in association with CIA30. As a comparative example, complex IV (cytochrome c oxidase) of the respiratory chain comprises only 13 subunits but requires more than 15 different components for its coordinated assembly into a mature complex (Fontanesi et al, 2006). As CIA30 can be observed in multiple large molecular weight complexes, all of which could not be attributed to its association with complex I assembly intermediates, it may be in complex with novel partner proteins. Recently, Vogel et al (2007a) identified a protein termed Ecsit that interacts with CIA30 and is involved in complex I biogenesis. The identification of Ecsit and other potential partner proteins may lead to uncovering the mechanism of CIA30 action and hence lead to a greater understanding of complex I biogenesis and its defects in disease.
Materials and methods
Patient details
Patient 1 was first investigated at 11 months for failure to thrive. He presented with severe cardiac failure in association with a viral illness at 15 months and hypertrophic cardiomyopathy was diagnosed. At that time, he was noted to have developmental delay, hypotonia and lactic acidosis, with blood lactate levels typically in the range of 5–10 mM (normal <2.5 mM) in the first few years of life and 2.5–3.5 mM subsequently. He was diagnosed with Wolff-Parkinson-White syndrome at 3 years of age, had cortical visual dysfunction at 7 years, pigmentary retinopathy at 11 years, kyphoscoliosis developed in his teenage years and osteoporosis was diagnosed at 16 years of age. At 20 years of age, he has mild to moderate intellectual disability. Full details are provided as Supplementary material.
Cloning
The cDNA encoding CIA30 (accession number NM_016013) was obtained from the IMAGE Consortium. For bacterial expression of CIA30, the sequence encoding the predicted mature protein (residues 25–327) was cloned into pQE30 (Qiagen) at BamHI and SmaI sites. For construction of pF5UAS-CIA30, the entire CIA30 open reading frame was cloned into pF5UAS at BamHI and XbaI sites.
Cell culture
HEK-293T cells were cultured in DMEM (Invitrogen) supplemented with 5% (v/v) fetal calf serum (FCS) and penicillin/streptomycin. Patient and control primary fibroblasts and Epstein–Barr virus-transformed lymphoblasts were cultured in DMEM, containing 10% (v/v) FCS supplemented with penicillin/streptomycin and 50 μg/ml uridine. All cells were incubated at 37°C under an atmosphere of 5% CO2 and 95% air.
In vitro protein import into isolated mitochondria
Plasmid DNA or PCR products were used for in vitro transcription using SP6 RNA polymerase (Promega). In vitro translation of the RNA transcripts was performed according to Ryan et al (2001). CIA30 and pOTC (Peralta et al, 1993) were translated in rabbit reticulocyte lysate in the presence of 35S-methionine/35S-cysteine (EXPRE35S35S Protein Labeling Mix; Perkin Elmer Life Sciences, USA) and incubated with isolated mitochondria at 37°C for various times as indicated. Proteinase K treatment and dissipation of Δψm were performed according to Ryan et al (2001).
Blue-native PAGE
BN gel electrophoresis was performed as described previously (Schagger and von Jagow, 1991) with minor modifications (McKenzie et al, 2007). Mitochondrial pellets (50–100 μg protein) were solubilized for 30 min on ice in 50 μl of 20 mM bis–Tris pH 7.4, 50 mM NaCl, 10% (v/v) glycerol containing either 1% (w/v) digitonin (Merck), 1% (w/v) Triton X-100 (Sigma) or 1% (w/v) DDM (or lauryl maltoside; Sigma). After removal of insoluble material by centrifugation, BN-PAGE loading dye (final concentrations: 0.5% (w/v) Coomassie Blue G, 50 mM ɛ-amino n-caproic acid (Sigma), 10 mM bis–Tris pH 7.0) was added and protein complexes separated on a 4–13% acrylamide–bisacrylamide BN-PAGE gel. Thyroglobulin (669 kDa), ferritin (440 kDa) and bovine serum albumin (140 and 67 kDa) were used as markers.
Antibodies
Monoclonal antibodies against the complex I subunits NDUFS3 (30 kDa), NDUFS7 (20 kDa/PSST), NDUFB6 (17 kDa), NDUFS5 (15 kDa) and NDUFA6 (14 kDa), the complex III subunit core 1 and the complex II subunit 70 kDa were from Molecular Probes. Antibodies against CIA30, Tom40, NDUFA9 and NDUFB6 were raised in rabbits as described previously (Johnston et al, 2002). The ND1 antibody was a gift from A Lombes (Paris, France) and the mtHsp70 antibody from Nick Hoogenraad (Melbourne, Australia). For detection of complex I on BN-PAGE, antibodies against NDUFA9 were used.
Radiolabeling of mtDNA-encoded translation products
Mitochondrial translation products were labeled as described previously (Chomyn, 1996; McKenzie et al, 2007). The individual mtDNA-encoded subunits were identified based on their size estimates, previous assignments (Chomyn, 1996), 2D-PAGE analysis and their migration with endogenous forms seen with Western blot analysis.
Construction of lentiviral vectors and infection
Inducible lentiviral vectors were based on the pF lentiviral vectors (Naldini et al, 1996) that were a kind gift of I Verma (San Diego, USA). pFUW was modified by introducing a PGK hygromycin-resistance cassette generated by PCR. The ERT2 tamoxifen regulatable ER (Casanova et al, 2002) was a kind gift of T Mantamadiotis (Melbourne, Australia) and was cloned as a fusion with Gal4 VP16 into pFU-PGK-HygroW to generate pFUGEV16. pFUW was also modified by introducing an SV40 puromycin-resistance cassette. This vector was further modified by replacing the ubiquitin promoter with a 5 × multimerized Gal4-binding site to generate pF5UAS. Full sequence details can be obtained upon request.
HEK-293T cells were grown on 10 cm plates to a confluency of 60% and transfected using LipoFectamine2000 (Invitrogen) according to the manufacturer's instructions. One plate was transfected with 5 μg pCMV-δR8.2 (structural gene expression), 2 μg pCAG4-E20 and 3 μg of pF5UAS-CIA30 while the other plate was transfected with 5 μg pCMV δR8.2, 2 μg pCAG4-E20 and 3 μg of pFUGEV16 (expression vector containing transcriptional activator). The supernatants containing packaged virus were harvested 48 h after transfection, combined and filtered through a 0.45 μM membrane filter. Hexabromide (Sigma) was then added to a final concentration of 5 μg/ml and the supernatant was overlaid onto patient fibroblasts for infection. At 72 h later, double infected cells were selected for by the addition of 1 μg/ml puromycin (Sigma) and 0.1 mg/ml hygromycin B (Sigma). Cells were grown in selection media for a period of 72–96 h with media replenished every 24 h. CIA30 expression was induced by addition of 100 nM 4HT to the media. Media containing 4HT was replenished every 24 h.
Genetic analysis
The five exons of NDUFAF1 were amplified from genomic DNA using primers that incorporated at least 30 bp of 5′ and 3′ intronic sequence. In addition, the entire coding region of NDUFAF1 was amplified from cDNA. All amplicons were sequenced using BigDye Terminator Cycle Sequencing kit (Applied Biosystems). For cloning of patient 1 partial cDNA spanning the exon 3/4 boundary, a 333 bp PCR fragment was generated using a forward primer in exon 2 (5′ CGAAGTGGGTACTGTGCAAT 3′) and a reverse primer in exon 4 (5′ TTTATCAGCCAAGGTGAATCC 3′.
Miscellaneous
Mitochondria were isolated from cultured cells according to Pallotti and Lenaz (2001). Sonication and carbonate treatments of mitochondria were performed as described previously (Ryan et al, 2001). Respiratory chain and control enzymes were measured as described (Rahman et al, 1996; Kirby et al, 1999). Tris–Tricine SDS–PAGE was performed as described previously (Schagger and von Jagow, 1987) but using a linear 10–16% polyacrylamide gradient. Western blotting was performed using a semi-dry transfer method (Harlow and Lane, 1999). Immunoreactive proteins from blots were detected using a ChemiGenius chemiluminescence system (SynGene) using horseradish peroxidase-coupled secondary antibodies and SuperSignal West Pico chemiluminescent substrate (Pierce Co.). 2D-PAGE was performed as described previously (McKenzie et al, 2007).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary material
Acknowledgments
We are grateful to the family for participation in this study. We thank J Hoogenraad for technical expertise, A Ohtake for helpful suggestions and A Lombes, T Mantamadiotis, I Verma for reagents This work was supported by grants (MTR, DRT, JS), Postdoctoral Fellowships (MMcK, DMK), RD Wright Fellowship (JS) and a Principal Research Fellowship (DRT) from the Australian National Health and Medical Research Council and a grant from the Australian Research Council (MTR). CJRD and ML are supported by Australian Postgraduate Research Scholarships and CS by a University of Melbourne Postgraduate Research Scholarship.
References
- Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, Moraes CT, Enriquez JA (2004) Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13: 805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Sachs MS, Jacobson A (2006) Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 7: 415–425 [DOI] [PubMed] [Google Scholar]
- Antonicka H, Ogilvie I, Taivassalo T, Anitori RP, Haller RG, Vissing J, Kennaway NG, Shoubridge EA (2003) Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J Biol Chem 278: 43081–43088 [DOI] [PubMed] [Google Scholar]
- Brandt U (2006) Energy converting NADH:quinone oxidoreductase (complex I). Annu Rev Biochem 75: 69–92 [DOI] [PubMed] [Google Scholar]
- Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE (2006) Bovine complex I is a complex of 45 different subunits. J Biol Chem 281: 32724–32727 [DOI] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Lemberger T, Shimshek DR, Sprengel R, Mantamadiotis T (2002) ER-based double iCre fusion protein allows partial recombination in forebrain. Genesis 34: 208–214 [DOI] [PubMed] [Google Scholar]
- Chomyn A (1996) In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol 264: 197–211 [DOI] [PubMed] [Google Scholar]
- Diaz F, Fukui H, Garcia S, Moraes CT (2006) Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol Cell Biol 26: 4872–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi F, Soto IC, Horn D, Barrientos A (2006) Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am J Physiol Cell Physiol 291: C1129–C1147 [DOI] [PubMed] [Google Scholar]
- Gabaldon T, Rainey D, Huynen MA (2005) Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (complex I). J Mol Biol 348: 857–870 [DOI] [PubMed] [Google Scholar]
- Grigorieff N (1998) Three-dimensional structure of bovine NADH:ubiquinone oxidoreductase (complex I) at 22 A in ice. J Mol Biol 277: 1033–1046 [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D (1999) Using Antibodies: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Humphries AD, Streimann IC, Stojanovski D, Johnston AJ, Yano M, Hoogenraad NJ, Ryan MT (2005) Dissection of the mitochondrial import and assembly pathway for human Tom40. J Biol Chem 280: 11535–11543 [DOI] [PubMed] [Google Scholar]
- Janssen R, Smeitink J, Smeets R, van Den Heuvel L (2002) CIA30 complex I assembly factor: a candidate for human complex I deficiency? Hum Genet 110: 264–270 [DOI] [PubMed] [Google Scholar]
- Johnston AJ, Hoogenraad J, Dougan DA, Truscott KN, Yano M, Mori M, Hoogenraad NJ, Ryan MT (2002) Insertion and assembly of human tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J Biol Chem 277: 42197–42204 [DOI] [PubMed] [Google Scholar]
- Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR (1999) Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology 52: 1255–1264 [DOI] [PubMed] [Google Scholar]
- Kirby DM, McFarland R, Ohtake A, Dunning C, Ryan MT, Wilson C, Ketteridge D, Turnbull DM, Thorburn DR, Taylor RW (2004a) Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet 41: 784–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, Kirk EP, Boneh A, Taylor RW, Dahl HH, Ryan MT, Thorburn DR (2004b) NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest 114: 837–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffner R, Rohr A, Schmiede A, Krull C, Schulte U (1998) Involvement of two novel chaperones in the assembly of mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Mol Biol 283: 409–417 [DOI] [PubMed] [Google Scholar]
- Lazarou M, McKenzie M, Ohtake A, Thorburn DR, Ryan MT (2007) Analysis of the assembly profiles for mitochondrial and nuclear encoded subunits into Complex I. Mol Cell Biol 27: 4228–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingelbach KR, Graf LJ, Dunn AR, Hoogenraad NJ (1986) Effect of deletions within the leader peptide of pre-ornithine transcarbamylase on mitochondrial import. Eur J Biochem 161: 19–23 [DOI] [PubMed] [Google Scholar]
- Marques I, Duarte M, Assuncao J, Ushakova AV, Videira A (2005) Composition of complex I from Neurospora crassa and disruption of two ‘accessory' subunits. Biochim Biophys Acta 1707: 211–220 [DOI] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol 361: 462–469 [DOI] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2007) Analysis of mitochondrial subunit assembly into respiratory chain complexes using blue native polyacrylamide gel electrophoresis. Anal Biochem 364: 128–137 [DOI] [PubMed] [Google Scholar]
- Meisinger C, Ryan MT, Hill K, Model K, Lim JH, Sickmann A, Muller H, Meyer HE, Wagner R, Pfanner N (2001) Protein import channel of the outer mitochondrial membrane: a highly stable Tom40-Tom22 core structure differentially interacts with preproteins, small tom proteins, and import receptors. Mol Cell Biol 21: 2337–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Nehls U, Friedrich T, Schmiede A, Ohnishi T, Weiss H (1992) Characterization of assembly intermediates of NADH:ubiquinone oxidoreductase (complex I) accumulated in Neurospora mitochondria by gene disruption. J Mol Biol 227: 1032–1042 [DOI] [PubMed] [Google Scholar]
- Ogilvie I, Kennaway NG, Shoubridge EA (2005) A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest 115: 2784–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotti F, Lenaz G (2001) Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol 65: 1–35 [DOI] [PubMed] [Google Scholar]
- Peralta D, Lithgow T, Hoogenraad NJ, Hoj PB (1993) Prechaperonin 60 and preornithine transcarbamylase share components of the import apparatus but have distinct maturation pathways in rat liver mitochondria. Eur J Biochem 211: 881–889 [DOI] [PubMed] [Google Scholar]
- Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR (1996) Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 39: 343–351 [DOI] [PubMed] [Google Scholar]
- Ryan MT, Pfanner N (2001) Hsp70 proteins in protein translocation. Adv Protein Chem 59: 223–242 [DOI] [PubMed] [Google Scholar]
- Ryan MT, Voos W, Pfanner N (2001) Assaying protein import into mitochondria. Methods Cell Biol 65: 189–215 [DOI] [PubMed] [Google Scholar]
- Schagger H (2001) Respiratory chain supercomplexes. IUBMB Life 52: 119–128 [DOI] [PubMed] [Google Scholar]
- Schagger H, Pfeiffer K (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19: 1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, von Jagow G (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166: 368–379 [DOI] [PubMed] [Google Scholar]
- Schagger H, von Jagow G (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199: 223–231 [DOI] [PubMed] [Google Scholar]
- Schapira AH (2006) Etiology of Parkinson's disease. Neurology 66: S10–S23 [DOI] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15: 7155–7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn DR, Sugiana C, Salemi R, Kirby DM, Worgan L, Ohtake A, Ryan MT (2004) Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim Biophys Acta 1659: 121–128 [DOI] [PubMed] [Google Scholar]
- Triepels RH, Hanson BJ, van den Heuvel LP, Sundell L, Marusich MF, Smeitink JA, Capaldi RA (2001) Human complex I defects can be resolved by monoclonal antibody analysis into distinct subunit assembly patterns. J Biol Chem 276: 8892–8897 [DOI] [PubMed] [Google Scholar]
- Tuschen G, Sackmann U, Nehls U, Haiker H, Buse G, Weiss H (1990) Assembly of NADH: ubiquinone reductase (complex I) in Neurospora mitochondria. Independent pathways of nuclear-encoded and mitochondrially encoded subunits. J Mol Biol 213: 845–857 [DOI] [PubMed] [Google Scholar]
- Ugalde C, Janssen RJ, van den Heuvel LP, Smeitink JA, Nijtmans LG (2004a) Differences in assembly or stability of complex I and other mitochondrial OXPHOS complexes in inherited complex I deficiency. Hum Mol Genet 13: 659–667 [DOI] [PubMed] [Google Scholar]
- Ugalde C, Vogel R, Huijbens R, Van Den Heuvel B, Smeitink J, Nijtmans L (2004b) Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum Mol Genet 13: 2461–2472 [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G (2004) AIF deficiency compromises oxidative phosphorylation. EMBO J 23: 4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RO, Janssen RJ, Ugalde C, Grovenstein M, Huijbens RJ, Visch HJ, van den Heuvel LP, Willems PH, Zeviani M, Smeitink JA, Nijtmans LG (2005) Human mitochondrial complex I assembly is mediated by NDUFAF1. FEBS J 272: 5317–5326 [DOI] [PubMed] [Google Scholar]
- Vogel RO, Janssen RJ, van den Brand MA, Dieteren CE, Verkaart S, Koopman WJ, Willems PH, Pluk W, van den Heuvel LP, Smeitink JA, Nijtmans LG (2007a) Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev 21: 615–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RO, van den Brand MA, Rodenburg RJ, van den Heuvel LP, Tsuneoka M, Smeitink JA, Nijtmans LG (2007b) Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Mol Genet Metab 91: 176–182 [DOI] [PubMed] [Google Scholar]
- Wiedemann N, Frazier AE, Pfanner N (2004) The protein import machinery of mitochondria. J Biol Chem 279: 14473–14476 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary material