

Abstract
FANCJ also called BACH1/BRIP1 was first linked to hereditary breast cancer through its direct interaction with BRCA1. FANCJ was also recently identified as a Fanconi anemia (FA) gene product, establishing FANCJ as an essential tumor suppressor. Similar to other FA cells, FANCJ-null (FA-J) cells accumulate 4N DNA content in response to DNA interstrand crosslinks (ICLs). This accumulation is corrected by reintroduction of wild-type FANCJ. Here, we show that FANCJ interacts with the mismatch repair complex MutLα, composed of PMS2 and MLH1. Specifically, FANCJ directly interacts with MLH1 independent of BRCA1, through its helicase domain. Genetic studies reveal that FANCJ helicase activity and MLH1 binding, but not BRCA1 binding, are essential to correct the FA-J cells' ICL-induced 4N DNA accumulation and sensitivity to ICLs. These results suggest that the FANCJ/MutLα interaction, but not FANCJ/BRCA1 interaction, is essential for establishment of a normal ICL-induced response. The functional role of the FANCJ/MutLα complex demonstrates a novel link between FA and MMR, and predicts a broader role for FANCJ in DNA damage signaling independent of BRCA1.
Keywords: BRIP1/BACH1, FANCJ, Fanconi anemia, interstrand crosslinks, mismatch repair, MutLα (MLH1/PMS2)
Introduction
In the absence of DNA repair proteins, cell cycle checkpoints and/or DNA damage repair pathways are not properly activated. This inability to actively respond to DNA damage can lead to massive chromosomal damage and even cell death. In some cases, mutations in DNA repair proteins can contribute to multiple cancer syndromes. Studies on the genetic causes of the cancer-prone syndrome Fanconi anemia (FA) revealed that genetic mutations associated with hereditary breast cancer were also associated with FA. For example, the hereditary breast cancer gene, BRCA2 was shown to be the gene defect in the FA-D1 patient complementation group, revealing that BRCA2 was FANCD1 (Howlett et al, 2002). Likewise, FANCJ (also called BACH1/BRIP1) was identified as the gene defective in the FANCJ-null (FA-J) patient complementation group (Levitus et al, 2005; Levran et al, 2005; Litman et al, 2005), and was initially linked to hereditary breast cancer. This link was based on its direct binding to BRCA1 and through the identification of two breast cancer patients with mutations in FANCJ, which also altered its helicase activity in vitro (Cantor et al, 2004, 2001). This connection was furthered by the finding that FANCJ (BRIP1) mutations confer a two-fold increase in the risk of developing breast cancer (Seal et al, 2006).
While other FA genes have not been linked to breast cancer, the network of at least 13 genes (designated FANCA to FANCN) is critical for maintaining chromosomal integrity (Thompson, 2005). Although the molecular function of these proteins is not clear, several gene products, including FANCA, B, C, D, E, F, G, L and M, form a nuclear core complex (the FA core complex), that is required for monoubiquitination of FANCD2. The FA proteins BRCA2/FANCD1, PALB2/FANCN and FANCJ are not required for this event and are considered downstream of FANCD2 monoubiquitination. Nevertheless, all FA proteins contribute to processing interstrand crosslinks (ICLs) (Thompson, 2005). Consequently, in the absence of FA proteins, ICL treatment leads to reduced cell viability and an accumulation of cells with a 4N DNA content representing cells in either late S or G2/M. This ICL-induced cell cycle progression defect and sensitivity to ICLs is restored upon reintroduction of the missing FA gene (Dutrillaux et al, 1982; Kaiser et al, 1982; Kupfer and D'Andrea, 1996; Kupfer et al, 1997; Heinrich et al, 1998; Sala-Trepat et al, 2000; Chandra et al, 2005). However, the FA-related function or associated partners required for a proper ICL response is not known.
Consistent with other FA cells, FA-J cells have an ICL-induced cell cycle progression defect that can be corrected upon re-introduction of wild-type (WT) FANCJ cDNA (Litman et al, 2005). This cell cycle progression defect has also been described as a prolonged G2/M arrest (Miglierina et al, 1991) or 4N DNA content accumulation (Akkari et al, 2001). The cause of this ICL response in FA cells is not presently understood, but is thought to involve delayed repair and/or failure to restart replication (Thompson et al, 2005). Unlike the majority of FA proteins, FANCJ has defined domains. Specifically, FANCJ binds directly to BRCA1 (Cantor et al, 2001) and is a DNA helicase (Cantor et al, 2004). Determining the importance of these domains could further our understanding of how FA proteins function in an ICL-induced response. Attempts to define the functions of FANCJ domains in the ICL response have been limited to chicken DT40 cells, where the FANCJ/BRCA1 interaction is not conserved (Bridge et al, 2005). If FANCJ operates independent of BRCA1 for a particular ICL response function, a remaining question will be whether FANCJ forms a complex with other proteins independent of BRCA1 to perform that function.
Here, we investigated whether FANCJ helicase activity or the FANCJ interaction with two distinct proteins was required for restoring FANCJ's ICL response. Specifically, we identified that FANCJ interacts with the MutLα mismatch repair complex, independent of BRCA1. Our findings demonstrate for the first time that the FANCJ/MLH1 interaction is as critical as FANCJ helicase activity for restoring a normal cell cycle progression and resistance of FA-J cells to ICLs. In contrast, the FANCJ/BRCA1 interaction is dispensable for normalizing the response of FA-J cells to ICLs, suggesting that FANCJ functions in distinct complexes to facilitate multifaceted DNA repair functions.
Results
FANCJ functions independently of BRCA1 to correct FA-J cells
We had previously shown that introduction of WT FANCJ cDNA into FA-J cells corrects the ICL-induced cell cycle progression defect (Litman et al, 2005). However, it is unclear how FANCJ contributes to the ICL response to restore the FA pathway, especially given that FANCJ's role in the FA pathway appears to be independent of BRCA1, at least in chicken cells (Bridge et al, 2005). To verify and extend this finding, we addressed whether FANCJ binding to BRCA1 was required to correct the ICL-induced cell cycle progression defect in FA-J cells. We reconstituted FA-J cells with vector, WT, or the S990A FANCJ construct that is ablated for BRCA1 binding (Yu et al, 2003) (Figure 1C). Both WT and S990A versions of FANCJ corrected the ICL-induced cell cycle progression defect observed in FA-J cells compared to vector alone (Figure 1A and B). These data support the finding that FANCJ operates independent of BRCA1 to correct FA-J cells.
Figure 1.
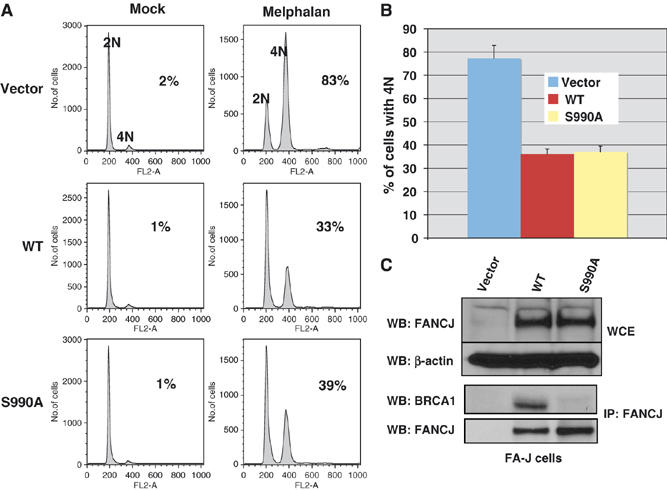
The FANCJ/BRCA1 interaction is dispensable for correction of the 4N DNA accumulation defect in FA-J cells. (A) FA-J cells were reconstituted with vector, WT or S990A, and FANCJ expression was analyzed in whole-cell extracts (WCE) by Western blot. β-Actin served as a loading control for the WCE samples. (B) Immunoprecipitations with FANCJ (E67) were analyzed by Western blot with the indicated Abs. (C) FA-J cells reconstituted with vector, WT, or S990A FANCJ were either left untreated or treated with melphalan, and the percentage of cells with 4N DNA content was analyzed by FACS. The percentage of cells with 4N DNA content after ICL-treatment was averaged for each cell line from four independent experiments, with standard deviation (s.d.) indicated by error bars.
FANCJ is physically linked to the MutLα complex
Since FANCJ binding with BRCA1 was not required to correct the ICL-induced cell cycle progression defect in FA-J cells, we set out to identify additional FANCJ interacting partners that may function with FANCJ in this ICL-induced response. A WT double tagged FANCJ construct was used to create a stable line of HeLa S3 cells. Using a two-step immunoaffinity strategy, the double tagged FANCJ was sequentially immunopurified (Nakatani and Ogryzko, 2003). Interacting proteins co-purifying with the double tagged WT FANCJ were eluted and visualized by silver stain. FANCJ migrated at the expected ∼140 kDa size (Figure 2A). Individual bands were excised from the gel and analyzed by mass spectrometry (LC-MS/MS). As expected, FANCJ copurified with BRCA1 that was identified as the 250 kDa band. Unique partners were identified, including the MMR proteins, MLH1 and PMS2, which form the MutLα heterodimer (Schofield and Hsieh, 2003) (Figure 2A). Western blot analyses using specific antibodies (Abs) confirmed the presence of these proteins (Figure 2B). To determine whether the MutLα complex associated with the native FANCJ protein, MCF7 cell extracts were immunoprecipitated (IP) with FANCJ Abs E67 and E47, and the presence of co-precipitating MLH1, PMS2 and BRCA1 proteins was evaluated by Western blot (Figure 2C). While FANCJ Ab precipitated the MutLα complex in the MCF7 cells, a MutLα complex was not precipitated with preimmune Abs (PI) or FANCJ Abs, in 293T cells, which lack expression of the MutLα complex (Trojan et al, 2002). Moreover, FANCJ was not precipitated with the MLH1 Ab in FA-J cells, which lack expression of FANCJ, unless FANCJ was reintroduced (Figure 2D). In contrast, a FANCJ/MLH1 interaction was readily detected in other FA cell lines, irrespective of gene correction, such as FA-A, FA-D1 and FA-D2 (Supplementary Figure 1). Furthermore, the interaction between FANCJ and the MutLα complex was stable in HeLa cells in the presence or absence of DNA damage (Figure 2E).
Figure 2.
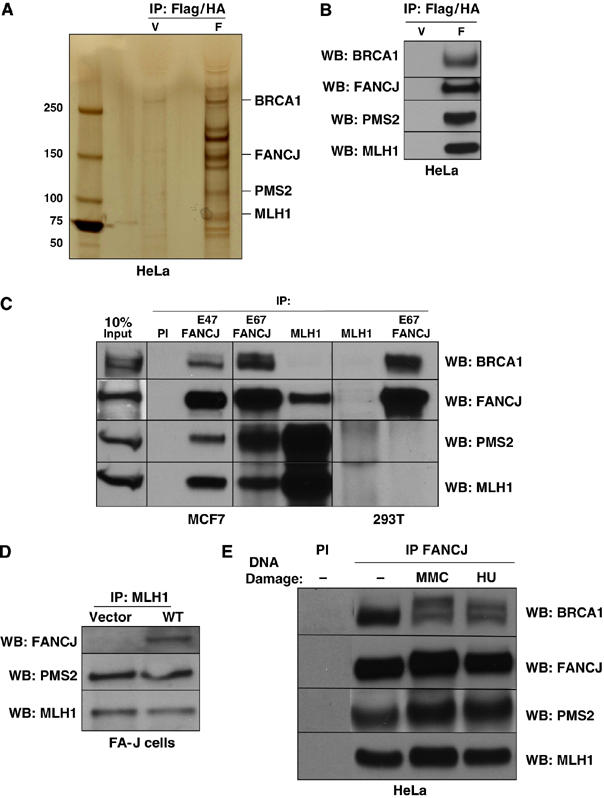
FANCJ interacts with the MMR proteins MLH1 and PMS2. (A) Silver-stained gel of the WT FANCJ (F) compared to vector (V)-purified complexes from HeLa S3 cells by consecutive Flag and HA purification steps (Flag/HA). Identified unique bands are indicated and FANCJ is observed as two species, the 140 kDa band is labeled. (B) Western blot detection of Flag/HA-purified FANCJ complexes. (C) Immunoprecipitations with either FANCJ (E67 or E47) or MLH1 Abs from MCF7 or 293T cells were analyzed by Western blot with the indicated Abs. (D) Western blot shows the presence of the indicated proteins from MLH1 IPs from FA-J cells reconstituted with vector or WT FANCJ. (E) HeLa cells were either left untreated or treated with 1 mM HU for 24 h or 2.4 μg/ml MMC for 1 h. HeLa cell lysates were IPed with PI or FANCJ Abs followed by Western blot analysis with the indicated Abs.
The helicase domain of FANCJ binds directly to MLH1 independent of BRCA1
MLH1 was previously reported to be part of a BRCA1 complex (Wang et al, 2000; Greenberg et al, 2006); therefore, we examined whether BRCA1 mediated the interaction between FANCJ and the MutLα complex. First, we noted that unlike FANCJ, BRCA1 was not readily detected in an MLH1 precipitation (Figure 2C). Next, we addressed whether FANCJ precipitated with the MutLα complex in BRCA1-deficient cells. Expression of BRCA1 was stably suppressed in MCF7 cells by an shRNA vector, as previously demonstrated (Litman et al, 2005). In cells expressing both a control shRNA specific to eGFP or an shRNA specific to BRCA1, FANCJ Abs efficiently co-precipitated the two components of the MutLα complex (Figure 3A), suggesting that FANCJ binds the MutLα complex independent of BRCA1. In support of this finding, the helicase domain of FANCJ was required for MLH1 binding, while a C-terminal region of FANCJ was required for BRCA1 binding (Figure 3B). To further assess the nature of the FANCJ/MLH1 interaction, we incubated recombinant FANCJ or BRCA1 with MLH1 that had been translated in vitro. MLH1 and FANCJ were precipitated by their corresponding Abs, and their interactions were analyzed by Western blot. FANCJ and MLH1 proteins were co-precipitated with both FANCJ and MLH1 IPs, whereas BRCA1 was robustly precipitated only in the FANCJ IP (Figure 3C). A direct interaction between FANCJ and MLH1 was confirmed by ELISA assay using purified recombinant proteins. FANCJ bound MLH1 in a protein concentration-dependent manner (Figure 3D). Furthermore, the interaction of FANCJ and MLH1 was demonstrated to be DNA independent, as evidenced by the similar colorimetric signal observed for FANCJ/MLH1 interaction in the presence of ethidium bromide (EtBr) or DNaseI (Figure 3E). These results suggest that FANCJ makes direct contacts with MLH1, independent of BRCA1 or PMS2.
Figure 3.
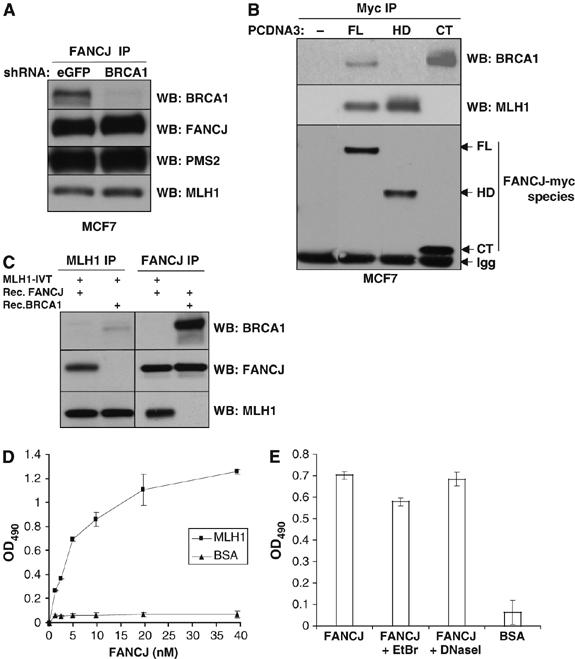
FANCJ helicase domain associates with the MutLα complex independent of BRCA1 and through a direct interaction with MLH1. (A) MCF7 cells were stably infected with a lentivirus encoding shRNA for either eGFP or BRCA1. FANCJ IP was performed followed by Western blot for the indicated proteins. (B) MCF7 cells were transiently transfected with pCDNA3 vectors containing no insert (−), full-length FANCJ (FL), helicase domain including amino-acid residues 1–882 (HD) or C-terminus including residues 882–1249 (CT) of FANCJ, and then IPed with the Myc Ab (9E10). Arrows indicate the respective FANCJ myc-tagged species. Immunoglobulin (IgG) is shown. (C) Western blot of the indicated IP experiments in which in vitro translated MLH1 was incubated with recombinant FANCJ or BRCA1 proteins. (D) Purified recombinant MLH1 or BSA was coated onto ELISA plates. Following blocking with 3% BSA, the wells were incubated with increasing concentrations of purified recombinant FANCJ (0–40 nM) for 1 h at 30°C, and bound FANCJ was detected by ELISA using a rabbit polyclonal Ab against FANCJ, followed by incubation with secondary horseradish peroxidase (HRP)-labeled Abs and OPD substrate. Data points are the mean of three independent experiments performed in duplicate, with s.d. indicated by error bars. (E) ELISA was performed as described in panel D using 4.9 nM FANCJ alone or in the presence of EtBr (50 μg/ml) or DNaseI (2 μg/ml). BSA (3%) was used as a control instead of MLH1 during the coating step.
PMS2 contributes to the FANCJ/MLH1 interaction in vivo
Given that MLH1 forms a heterodimer with PMS2, we next assessed whether PMS2 binding to MLH1 contributed to the MLH1/FANCJ interaction in vivo. To address this possibility, we tested the ability of different MLH1 constructs to precipitate FANCJ in the absence or presence of PMS2. WT full-length MLH1 and several MLH1-myc fusion proteins of varying length were generated and transiently transfected into MutLα-null 293T cells. To determine which of these MLH1 fragments were expressed and/or co-IPed FANCJ, MLH1 was precipitated from cell lysates with either myc or MLH1 Abs. While cotransfecting PMS2 with MLH1 did not alter the expression of MLH1, the ability of FANCJ to form a complex with MLH1 was enhanced. With the addition of PMS2, FANCJ precipitated with the MLH1 constructs N2, C2, and C3, which in the absence of PMS2 had failed to precipitate FANCJ (Figure 4A). Thus, in the presence of PMS2, only one of the two MLH1-FANCJ interacting domains (478–508) (D1) or (736–744) (D2) was required (see Figure 4C), suggesting that PMS2 facilitates the MLH1/FANCJ interaction. PMS2 stability is dependent on the MLH1 C-terminus (Mohd et al, 2006); not surprisingly, we found that ablation of a C-terminal region of MLH1 (703–725) reduced both PMS2 and FANCJ binding (Figure 4B).
Figure 4.
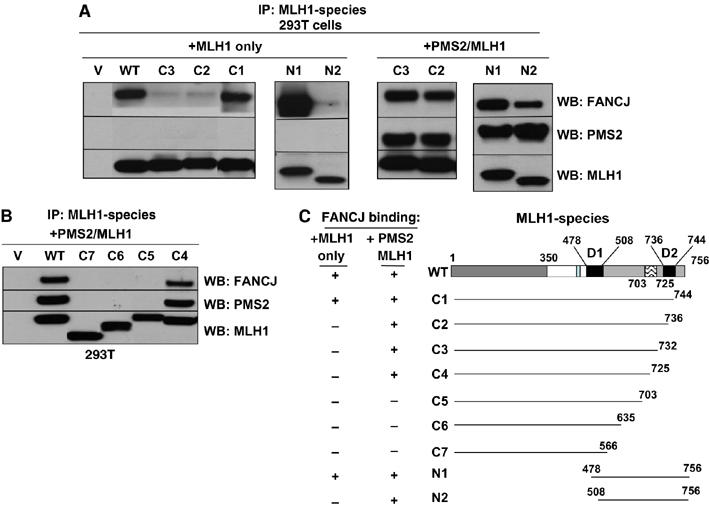
PMS2 facilitates the FANCJ interaction with the MLH1 C-terminus. (A) MLH1 or Myc (9E10) IP experiments were performed from 293T cells that were transfected with vector alone (V), full-length MLH1 (WT) or MLH1 species, alone or in combination with PMS2 (C1–C3, N1, N2). IP products were analyzed by Western blot with FANCJ, PMS2, and MLH1 Abs. (B) MLH1 IP experiments were performed from 293T cells that were transfected with vector alone (V), full-length MLH1 (WT) or MLH1 species, in combination with PMS2 (C4–C7). IP products were analyzed by Western blot with FANCJ, PMS2, and MLH1 Abs. (C) Schematic representation of the MLH1/FANCJ dimer domains (D1, D2), and the region between 703–725 is highlighted as an essential element for maintaining the MLH1/PMS2/FANCJ complex.
MutLα functions downstream of FANCD2 monoubiquitination
To appreciate the physiological significance of a FANCJ/MutLα interaction, we next, addressed whether the MutLα complex functioned with FANCJ in the FA pathway. We had previously shown that in FANCJ-deficient cells, DNA damage-induced FANCD2 monoubuiquitination was intact (Litman et al, 2005). Similarly, we found that incubation of MutLα-deficient cells (HCT116 and HEC-1A) with hydroxyurea (HU) leads to efficient FANCD2 monoubiquitination (Supplementary Figure 2A), suggesting that similar to FANCJ, MutLα functions downstream of FANCD2.
Given that suppression of MMR proteins has been reported to reduce the survival of cells upon ICL-treatment, (Aquilina et al, 1998; Fiumicino et al, 2000), we next asked whether similar to FANCJ deficiency, MutLα deficiency also sensitizes cells to ICLs. First, we suppressed MutLα using siRNA reagents in MCF7 cells versus a luciferase control. Second, we reconstituted HCT116 cells null for MutLα with vector or MutLα expressing cDNAs. In both experiments, there was no measurable change in ICL sensitivity, in the presence or absence of MutLα expression (Supplementary Figure 2B and data not shown). Given that MMR proteins bind and process ICLs (Duckett et al, 1996; Yamada et al, 1997; Zhang et al, 2002), activate multiple DNA damage-induced checkpoints, such as intra S and G2/M (4N) arrest (Brown et al, 2003; Cejka et al, 2003), and participate in the repair of ICLs by promoting recombination (Zheng et al, 2006), we considered that MutLα suppression could bypass ICL sensitivity through loss of checkpoint (Cejka et al, 2003), and/or by activating default non-recombination-based repair pathways, as reported (Zheng et al, 2006). Thus, we considered that to unmask function of MutLα in the ICL response with FANCJ, it would be necessary to selectively ablate the MLH1/FANCJ interaction, while maintaining other MLH1 functional interactions (i.e. PMS2 binding).
Disruption of the native MLH1/FANCJ interaction generates ICL sensitivity
To define the domain on FANCJ required for MLH1 binding, we generated several FANCJ-myc fusion proteins of varying length and expressed them in MCF7 cells (Figure 5A and E). To determine which of these FANCJ fragments were expressed and/or co-IPed MLH1, FANCJ was precipitated from cell lysates with myc Abs. Full-length FANCJ and FANCJ expression constructs including the FANCJ N-terminal amino-acid residues 1–145 precipitated MLH1 (Figure 5A and C). These results suggested that FANCJ N-terminal residues 1–145 were required for binding to MLH1. To assess whether residues in this region were sufficient for MLH1 binding, we inserted FANCJ residues 128–158 within the eGFP gene sequence to create an eGFP-fusion protein. In contrast to eGFP alone, the eGFP-FANCJ fusion protein readily co-precipitated MLH1 (Figure 5B), suggesting that FANCJ 128–158 was sufficient for MLH1 binding. Furthermore, expression of the eGFP-FANCJ fusion protein in cells perturbed the formation of the native FANCJ/MLH1 interaction, as determined by both FANCJ and MLH1 IP and Western blot experiments (Figure 5D), confirming that this region of FANCJ was essential for mediating the MLH1 interaction.
Figure 5.
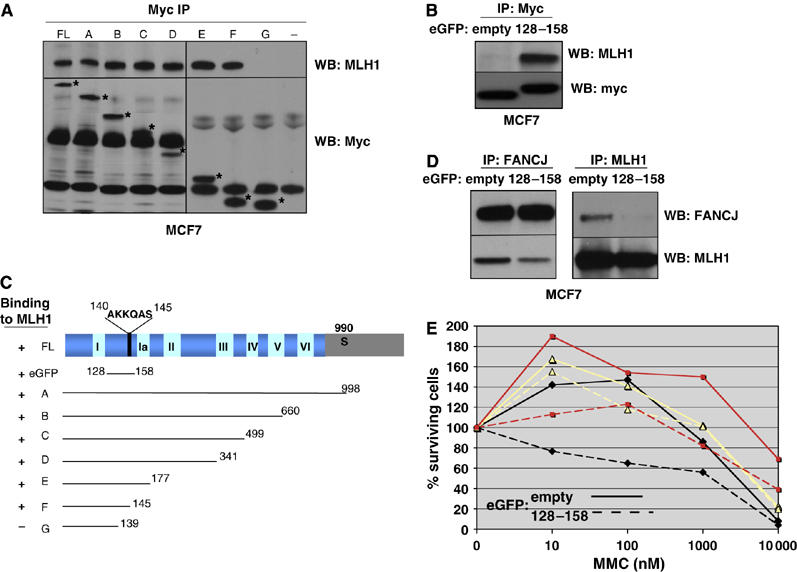
Expression of FANCJ residues 128–158 disrupts the FANCJ/MLH1 interaction to generate ICL sensitivity. (A) Myc (9E10) IP experiments were performed from MCF7 cells that were transfected with vector alone (−), full-length FANCJ (FL) and the different FANCJ constructs (A–G) shown in panel C, followed by Western blot with MLH1, and Myc Abs. The asterisk denotes the migration of the different myc-tagged FANCJ species. (B, D) Myc IP experiments were performed from MCF7 cells that were transfected with either eGFP empty vector or the 128–158 FANCJ-eGFP constructs, followed by Western blot with the indicated Abs. (C) The different FANCJ constructs are indicated with a positive (+) or negative (−) to indicate binding to MLH1. (E) MCF7 cells transfected with vector alone or the 128–158 FANCJ-eGFP construct, treated with increasing concentrations of MMC and incubated for 4–5 days. Cell growth was measured by ATP content. Three independent representative experiments are shown and depicted by lines with squares, triangles, and diamonds. Solid lines represent cells transfected with empty-eGFP vector and hatched lines represent cells transfected with 128–158-eGFP vector (for color figure see online version).
Next, we addressed whether expression of the 128–158 FANCJ-eGFP fusion protein and the resulting perturbation of the native FANCJ/MLH1 interaction would render cells sensitive to ICLs. MCF7 cells were transfected with vectors expressing either the 128–158 FANCJ-eGFP fusion protein or eGFP alone, plated and treated with increasing concentrations of Mitomycin C (MMC). The overall trend upon expression of the 128–158 FANCJ-eGFP fusion protein was reduced cellular survival compared to expression of the eGFP control, despite some variability between experiments (Figure 5E). While the enhanced sensitivity was consistent with the possibility that a FANCJ/MLH1 interaction was required for ICL repair, we considered that binding of the fusion protein to MLH1 might have altered additional MLH1 functions not specific to FANCJ. Thus, we sought to identify a method to ablate the FANCJ/MLH1 interaction without altering native MLH1 protein or being reliant on transfection efficiency to disrupt the native FANCJ/MLH1 interaction.
Lysines 141 and 142 of FANCJ are required for the FANCJ/MLH1 interaction
Given that mutational analysis revealed that FANCJ co-precipitated with MLH1, except when FANCJ residues 140–145 were absent (Figure 5A and C), we assessed the importance of these residues for binding MLH1 within the context of the full-length FANCJ protein. Thus, we generated three independent FANCJ mutant constructs that converted lysine 141 and 142 to alanine (K141/142A), glutamine 143 to a glutamic acid (Q143E), or serine 145 to an alanine (S145A). While the WT FANCJ and all three mutant versions were expressed and efficiently co-precipitated BRCA1, the K141/142A version demonstrated a dramatic reduction in the co-precipitation of MLH1 (Figure 6A), suggesting that these two lysines were required for MLH1 binding.
Figure 6.
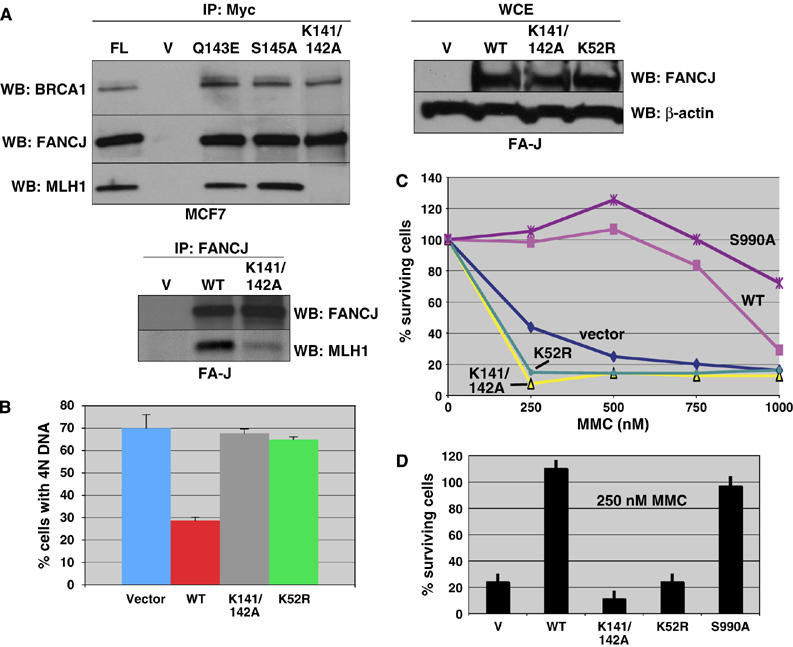
MLH1 binding to FANCJ is essential to correct FA-J cells. (A) Myc IP experiments were performed from MCF7 cells that were transfected with vector alone (−), FL, V, Q143E, S145A, and K141/142A FANCJ constructs, followed by Western blot with the indicated Abs. FA-J cells were reconstituted with empty vector, WT, K141/142A, or K52R FANCJ vectors, and FANCJ expression was analyzed by whole-cell extracts; β-actin served as a loading control for the WCE samples. Western blot shows the presence of the indicated proteins from FANCJ IPs from FA-J cells reconstituted with vector, WT, or K141/142A FANCJ. (B) FA-J cell lines reconstituted with empty vector, WT, K141/142A, or K52R FANCJ were either left untreated or treated with melphalan. The percentage of cells with 4N DNA content after ICL treatment was averaged for each cell line from four independent experiments, with standard deviation (s.d.) indicated by error bars. (C) FA-J cells reconstituted with vector, WT, K141/142A, K52R, or S990A FANCJ were seeded on 24-well plates and incubated overnight under normal growth conditions. The cells were then treated with the indicated doses of MMC and incubated for 8 days. On the final day, the cells were counted and the percentage of live cells was calculated. Experiments were performed in triplicate and a representative plot is shown. (D) The IC50 dose for the FA-J vector (250 nM) was compared for all mutants, and error bars represent the standard deviation.
We considered that the K141/142A mutation in FANCJ could have not only disrupted MLH1 binding, but also FANCJ helicase activity. Thus, we generated recombinant versions of WT (Cantor et al, 2004) and the K141/142A FANCJ proteins to assess whether this mutant version was enzymatically active. The recombinant K141/142 FANCJ protein was detected as a single Coomassie-stained band analyzed by SDS–PAGE, that co-migrated with the WT FANCJ recombinant protein (Supplementary Figure 3A and D). The DNA unwinding activity of K141/142A FANCJ on a forked duplex DNA substrate was compared to unwinding activity of WT FANCJ. Both K141/142A FANCJ and WT FANCJ were found to be proficient in unwinding, whereas K52R FANCJ, as previously demonstrated, failed to unwind the forked duplex substrate (Gupta et al, 2005) (Supplementary Figure 3B). Furthermore, K141/142A FANCJ and WT FANCJ unwound the forked duplex substrate in a protein concentration-dependent manner, achieving 90% of unwound substrate at the highest helicase concentration tested (Supplementary Figure 3C). Thus, the K141/142 mutant only disrupts MLH1 binding, but not FANCJ helicase activity.
FANCJ function depends on MLH1 binding to correct FA-J cells
Next, we tested the ability of K141/142A FANCJ cDNA to correct the cell cycle progression defect in FA-J cells. We used retroviral infection to stably infect FA-J cells with cDNA encoding the vector, WT, K141/142A or K52R Flag/HA-tagged FANCJ constructs, which expressed similarly (Figure 6A). Moreover, an MLH1 IP demonstrated that the MLH1/PMS2 complex was intact in FA-J cells and was able to precipitate the reconstituted FANCJ (Figure 2D). As in MCF7 cells, in FA-J cells MLH1 co-IPed with WT FANCJ, but was dramatically reduced with the K141/142A mutant FANCJ (Figure 6A). FA-J cells containing vector, WT, K52R, or K141/142A FANCJ were treated with melphalan to induce ICLs, as described (Litman et al, 2005). The proportion of vector containing FA-J cells with 4N DNA content increased after melphalan treatment, similar to previous experiments. As before, the proportion of WT FANCJ containing FA-J cells with 4N DNA content (∼30%) was about half that of vector containing cells (∼70%). We found that cells containing the catalytically inactive FANCJ helicase (K52R) failed to correct the 4N accumulation defect (∼66%). Likewise, the proportion of K141/142A FANCJ containing FA-J cells with 4N DNA content (∼68%) resembled that of vector containing FA-J cells, suggesting that introduction of K141/142A FANCJ did not correct the cell cycle progression defect in FA-J cells (Figure 6B). These data suggest that the FANCJ/MLH1 interaction is essential for restoration of the FA pathway in FA-J cells.
To further confirm and examine the importance of FANCJ binding to MLH1 for the ICL-induced response, we assessed whether FANCJ binding to MLH1 was required for FANCJ to correct the ICL-induced sensitivity of FA-J cells. However, the ability of WT FANCJ to correct the ICL sensitivity of FA-J cells had not been previously reported. Thus, we first tested and confirmed that reintroduction of WT FANCJ corrected the ICL sensitivity of FA-J cells treated with MMC. The vector-reconstituted FA-J cells were more sensitive to MMC than the WT FANCJ-reconstituted cells with an IC50 of 250 and ∼900 mM MMC, respectively (Figure 6C). In contrast to WT, both the K141/142A- and K52R FANCJ-reconstituted FA-J cells were sensitive to ICLs with an IC50 of less than 250 mM MMC. Moreover, the K141/142A FANCJ-reconstituted FA-J cells were more sensitive than the vector or K52R FANCJ-reconstituted FA-J cells (P<0.01). As before, the S990A FANCJ corrected the ICL response similar to WT. Although the correction of the ICL sensitivity was greater with the S990A FANCJ than with the WT FANCJ, at 250 mM MMC the values were not significantly different (with P-values between 0.0027 and 0.22) (Figure 6C). These findings clearly demonstrate that both FANCJ helicase activity and FANCJ binding to MLH1 are required for FANCJ to functionally correct the ICL sensitivity of FA-J cells.
Discussion
In this study, we addressed whether FANCJ helicase activity or different FANCJ complexes are essential for FANCJ's function in the ICL response. Specifically, we have shown that FANCJ forms a complex with the MutLα heterodimer, which is composed of the mismatch repair proteins MLH1 and PMS2. FANCJ directly interacts with MLH1 independent of BRCA1, and this DNA-independent interaction is within the FANCJ helicase domain, C-terminal to nucleotide binding box 1, and includes lysines 141 and 142. This is the first report that demonstrates that a direct interaction between FANCJ and MLH1 is as essential for the ICL-induced response as the FANCJ helicase activity. Furthermore, our data suggest that formation of a FANCJ/BRCA1 complex is not required for normalization of the ICL-induced response in FA-J cells.
The question remains as to how FANCJ or mismatch repair proteins function in the ICL response. Following exposure to ICLs, ATR is activated and initiates a signal cascade through the phosphorylation of downstream substrates, ultimately leading to checkpoint activation, DNA damage repair and/or apoptosis. Intriguingly, MMR proteins were recently proposed to act as direct sensors of DNA methylation and initiate the intra S-phase checkpoint, by helping to recruit ATR-ATRIP to sites of DNA damage (Yoshioka et al, 2006). Furthermore, mismatch repair proteins have been implicated in sensing and processing ICLs. In particular, the MutSβ complex was shown to bind to intrastrand crosslinks produced by cisplatin (Duckett et al, 1996; Yamada et al, 1997), and the MutSβ complex was shown to be involved with the removal of ICLs produced by psoralen (Zhang et al, 2002). Furthermore, in the absence of MMR signaling, ICL repair proceeded in an alternate pathway promoting a non-recombination-dependent mechanism (Zheng et al, 2006). Our study now suggests that MLH1 binding to FANCJ is functionally important for the ICL-induced response, as disruption of the native FANCJ/MLH1 interaction reduced cell survival following ICL treatment. Moreover, FANCJ K141/142A mutant, defective in MLH1 binding, fails to correct FA-J cells. The finding that MLH1 deficiency did not generate gross changes in the cellular survival following ICLs may stem from the loss of both MMR DNA damage repair and signaling. In fact, the loss of DNA damage signaling and the subsequent repair is essential in establishing the resistance of MMR-deficient cells to DNA methylation (O'Brien and Brown, 2006). The multiplicity of MMR functions in the DNA damage response has been recently uncovered through separation-of-function mutations (O'Brien and Brown, 2006).
Here, we attempted a similar approach to dissect the role of MLH1 and FANCJ in the ICL response. Three approaches were attempted to selectively ablate the FANCJ/MLH1 interaction in vivo. In one of these approaches, we attempted to generate an MLH1 mutant that lacked FANCJ binding. While MLH1 and FANCJ bind directly in vitro, the interaction is facilitated by PMS2 in vivo. In the absence of PMS2, FANCJ binding to MLH1 requires two MLH1 C-terminal domains D1 (478–508) and D2 (736–744). However, in the presence of PMS2, only one of these domains was required. Moreover, both the regions on MLH1 (703–725) required for FANCJ binding in the presence of PMS2 are also essential for PMS2 stabilization. The complexity of the MLH1/PMS2/FANCJ interaction confounded attempts to selectively ablate the FANCJ/MLH1 interaction through MLH1 mutagenesis. Fortunately, the binding of MLH1 to FANCJ was less complex and therefore, the FANCJ/MLH1 interaction could be selectively disrupted by both mutagenesis and peptide disruption. We found that disruption of this complex caused defects in the ICL response, consistent with the MLH1/FANCJ interaction being required for ICL repair.
Conceivably, for ICL repair, mismatch repair complexes including MutLα, mobilize or regulate FANCJ helicase activity to unwind DNA in the vicinity of the DNA damage to facilitate repair processes. Thus, disruption of FANCJ helicase activity or MLH1 binding could interfere with ICL response. In support of this possibility, the MLH1 homologue in Escherichia coli, MutL binds the DNA helicase UvrD gene product Helicase II (Hall et al, 1998; Spampinato and Modrich, 2000) and stimulates its helicase activity (Dao and Modrich, 1998; Yamaguchi et al, 1998). However, we did not detect an effect of the MutLα complex, inhibitory or stimulatory, on FANCJ catalyzed unwinding of a forked duplex, 5′ flap, or Holliday Junction substrate (data not shown). It is possible that regulation of FANCJ helicase activity by MLH1 may require additional MMR proteins, and/or that the physical interaction between FANCJ and MLH1 serves a non-catalytic role in mediating the ICL response.
Alternatively, the catalytic activity of FANCJ may serve an entirely different purpose (see Figure 7). For example, the FANCJ helicase activity could serve to displace MutLα from DNA. This type of model has also been proposed for other helicases. For example, the Srs2 helicase is proposed to displace a checkpoint protein to facilitate checkpoint exit (Vaze et al, 2002). If true, in the absence of FANCJ (FA-J cells), or in FA-J cells with a catalytically inactive FANCJ and/or a FANCJ mutant that lacks MLH1 binding, the MutLα complex would fail to be displaced from DNA. As such, the MutLα complex would be stuck or take longer to be displaced from DNA, leading to a prolonged G2/M arrest and/or delay in the completion of repair. In support of this model, ICL treatment of FA-J cells reconstituted with vector, K52R or K141/142A FANCJ constructs demonstrates both hyper-G2/M arrest and sensitivity to ICLs. Furthermore, peptide perturbation of the MLH1/FANCJ interaction in MCF7 cells lead to enhanced ICL sensitivity.
Figure 7.
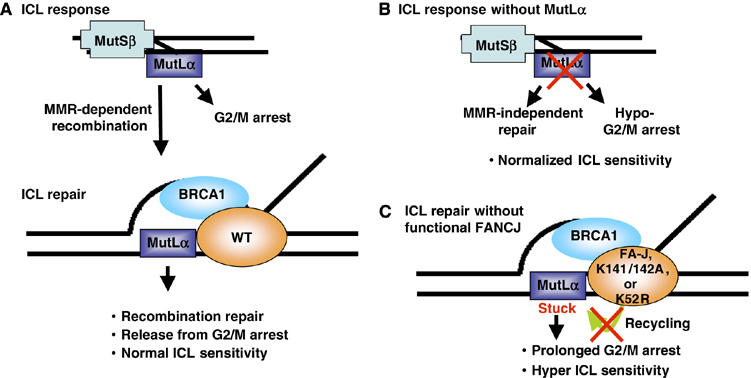
Model depicting how FANCJ and MutLα proteins function to mediate the ICL response. (A) The normal ICL response is proposed to include MMR proteins. This is supported by the findings that the MMR machinery has been shown to specifically bind crosslinked DNA adducts (Duckett et al, 1996; Yamada et al, 1997; Zhang et al, 2002), mediate ICL recombination repair (Zheng et al, 2006) and induce a G2/M arrest (Cejka et al, 2003). Furthermore, MMR proteins including the MutLα complex, similar to BRCA1 and FANCJ are essential for recombination processes (de Wind et al, 1995; Jasin, 2002; Mohindra et al, 2002; Litman et al, 2005). (B) The ICL response without MutLα is predicted to lead to a MMR-independent non-recombination-based mechanism with a minimal G2/M arrest, so that ICL sensitivity is normalized. (C) ICL repair without functional FANCJ is predicted to be directed to recombination as in panel A, but generates a prolonged G2/M arrest, due to absent or dysfunctional FANCJ protein. In the absence of FANCJ protein, helicase activity or MLH1 binding, FANCJ is unable to displace MutLα from recombination intermediates, and consequently, the MutLα complex remains stuck or tethered to DNA for a longer time period, delaying the exit from the G2/M arrest and enhancing ICL sensitivity.
This proposed role for FANCJ in displacing MutLα is also not at odds with the finding that suppression of the MutLα complex in MCF7 cells did not lead to MMC sensitivity (Supplementary Figure 2). It has been reported that in the absence of MMR signaling there is a reduced G2/M arrest following DNA damage (O'Brien and Brown, 2006), and compensating non-recombination repair pathways are engaged (Zheng et al, 2006). The expectation from these findings would be that MMR-deficient cells would have a normalized sensitivity to ICLs, due to the defective checkpoint and compensation by alternative mechanisms of repair (see model in Figure 7). Ultimately, it will be critical to establish whether a FANCJ/MLH1 complex facilitates ICL repair by promoting homologous recombination or other repair functions. While ablation of BRCA1 binding to FANCJ may not affect the ability of FANCJ to correct the defective response of FA-J cells, the timing or mechanism of repair may be altered. Thus, it is important to consider that the FA-J cells lacking the FANCJ/BRCA1 interaction resist the ICL-induced 4N arrest and sensitivity through an alternative mechanism, perhaps by checkpoint avoidance. Experiments are currently underway to investigate this possibility.
Although MMR proteins are essential to elicit a G2/M arrest, it is presently unclear as to how this arrest is overcome. In cells lacking the active FANCJ helicase and/or a FANCJ/MLH1 interaction, diffusion of the MutLα complex from DNA may lead to an eventual exit from this G2/M arrest. In contrast, this exit may not be achieved in FA-J cells expressing the K52R FANCJ, if the MutLα complex is locked on the DNA by the inactive helicase. It follows that a complete failure to re-enter the cell cycle, as apposed to a slower entry, would be more toxic to cells. Consistent with this, we found that the FA-J K52R expressing cells are shortlived in tissue culture and forced expression of K52R FANCJ in other cell lines is not stable (data not shown).
Given that the FANCJ/MutLα interaction is intact in other FA cells, including FA-A, FA-D2 and FA-D1 (Supplementary Figure 1), loss of this complex is not a general feature of FA cells. However, it remains to be determined whether additional FA–MMR interactions are altered. Moreover, similar to FANCJ deficiency, MLH1 deficiency does not affect the ATR-mediated FANCD2 monoubiquitination (Supplementary Figure 2) (Andreassen et al, 2004; Levitus et al, 2004; Bridge et al, 2005; Litman et al, 2005), suggesting that the FANCJ/MLH1 interaction is not essential for FA-pathway activation. Interestingly, we find that deletions in the MLH1 C-terminus (703–725), which are important for maintaining the stability of PMS2 (Mohd et al, 2006), also disrupted the FANCJ/MLH1 interaction. This finding has implications for MLH1 clinical mutations identified in colon cancer patients, potentially linking MLH1 function not only to PMS2 but also to FANCJ.
In conclusion, these studies have provided the first evidence for a role of the FANCJ/MutLα complex in the ICL-induced response. This work extends the already implicated role of MMR proteins in the ICL response. Further study of the role of FANCJ, BRCA1 and MMR proteins in this process should advance the understanding of how ICL-induced responses are regulated to preserve genomic integrity.
Materials and methods
Cell lines
HeLa, MCF7, 293T, and HeLa S3 cells were grown in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml each). FA-J (EUFA30-F) cells were cultured as previously described (Litman et al, 2005). HCT116 cells were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml each). Hi5 insect cell were grown in Grace's Insect Media supplemented with 10% fetal bovine serum and 1% genetemycin at, 28°C without CO2. FA-J cells were infected with the pOZ retroviral vector (Nakatani and Ogryzko, 2003) containing no insert, WT, K141/142A, or K52R FANCJ inserts, or with the lentiviral vector pLentiV5 (Invitrogen) vector, containing no insert, WT or S990A FANCJ inserts. Stable FA-J pOZ cell lines were generated by sorting pOZ-infected cells with anti-IL-2 magnetic beads (Dyna Beads) and expanding IL-2-positive cells. Stable FA-J pLenti cell lines were generated by blasticidin selection (7 μg/ml).
Purification of a FANCJ complex
A FANCJ complex was purified from nuclear extracts (NE) derived from ∼8 × 109 HeLa cells stably expressing the double tagged FANCJ, by two-step immunoaffinity chromatography, according to the standard method (Nakatani and Ogryzko, 2003). Flag-HA double purified material was electrophoresed in 3–8% Tris-acetate gel (Invitrogen). Individual Silver-stained bands were excised and subsequently analyzed by mass spectrometry (Genomine Inc., South Korea).
Immunoprecipitations, immunoblotting and Abs
Cells were harvested and lysed in 150 mM NETN lysis buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin) for 30 min on ice. Cell extracts were clarified by centrifugation. The cell lysates were boiled in SDS loading buffer. For immunoprecipitation assays, cells lysates were incubated with protein-A beads, and either FANCJ (E67 or E47), MLH1 (BD Bioscience) or Myc (9e10) Abs at 4°C for 2 h. Beads were subsequently washed and boiled in SDS loading buffer. Proteins were separated using SDS–PAGE and electrotransferred to nitrocellulose membranes. Membranes were blocked in 5% milk phosphate-buffered saline (PBS)/Tween and incubated with primary Ab for 1 h. Abs for Western blot analysis included anti-MLH1 (BD Bioscience, 1:500), anti-PMS2 (BD Biosciences, 1:200), anti-BRCA1(ms110, hybridoma cell, 1:3), anti-FANCJ (Monoclonal pool 2G7, 2C10,1B4), anti-FANCD2 (Fanconi Anemia Research Foundation) and Myc (9E10, hybridoma cell, 1:3). Membranes were washed, and incubated with horseradish peroxidase-linked secondary Abs (Amersham, 1:5000), and detected by chemiluminescence (Amersham).
ICL-induced 4N accumulation and sensitivity assays
FA-J cells reconstituted with either vector, WT FANCJ, K141/142A FANCJ, K52R FANCJ, or S990A FANCJ were either treated with 0.5 μg/ml of melphalan (Sigma) or left untreated, and incubated for 65 h. Cells were fixed with 90% methanol in PBS and were then incubated 10 min with PBS containing 30 μ/ml DNase-free RNase A and 50 μg/ml propidium iodide. A total of 1 × 104 cells was analyzed using a FACs Calibur instrument (Becton-Dickinson, San Jose, CA). Aggregates were gated out and the percentage of cells with 4N DNA content was calculated using Modfit software.
The FA-J cells reconstituted with either vector, WT FANCJ, K141/142A FANCJ, K52R FANCJ, or S990A FANCJ were seeded on 24-well plates 1000 cells per well and incubated overnight. The cells were either left untreated or treated with increasing doses of MMC for 1 h and incubated for 8 days. Finally, the cells were collected by trypsinization and counted using a hemacytometer. The percentage of live cells at each concentration was calculated using the untreated controls as the baseline growth.
MCF7 cells transfected with either empty vector or the 128–158 FANCJ eGFP construct were seeded at 500 cells per well in 96-well plates, and incubated overnight. The cells were either left untreated or treated with increasing concentrations of MMC for 1 h and incubated 4–5 days. The percentage of live cells was calculated by ATP content, as previously described (Litman et al, 2005).
Plasmid construction and in vitro translation
The WT and S990A FANCJ pLentiviral vectors were a gift from J Chen (Yu et al, 2003). The pCDNA3-myc.his vector (Invitrogen) was digested by Not1/Apa1, and different FANCJ fragments generated by PCR and digested Not1 and Apa1 were inserted. Primers are available upon request. The WT FANCJ pOZ-FH vector was generated by PCR cloning. Specifically, 5′ Xho1 and a 3′ Not1 restriction sites were added by using primers: 5′–3′ CGCTCGAGGCCACCATGTCTTCAATGTGGTCTGAATATACAATT and 5′–3′ CAGCGGCCGCCTTAAAACCAGGAAACATGCCTTTATT. The PCR product was digested XhoI and NotI and subcloned into the pOZ-FH vector. The K52R, S990A and K141/142A pOZ vectors were generated with the QuickChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) by using the FANCJ-pOZ as a template and the following primers: (K52R) 5′–3′ CCCACAGGAAGTGGAAGGAGCTTAGCCTTAGCC and 5′–3′ GGCTAAGGCTAAGCTCCTTCC-ACTTCCTGTGGG; (S990A)5′-3′ TCCAGATCCACAGCCCCAACTTTCAAC and 5′–3′ GTTGAAAGTTGGGGCTGTGGATCTGGA; (K141/142A) 5′–3′ GCAAAGTTATCTGCT GCGGCACAGGCATCCATATAC and 5′-3′ GTATATGGATGCCTGTGCCGCAGCAGATAACTTTGC. The same set of primers was used to generate the K141/142 A-pCDNA3 and K141/142 A-pVL132, by using the WT FANCJ pCDNA3myc.his- (Cantor et al, 2001) and pVL132Flag-tagged (Cantor et al, 2004) constructs, respectively. The Q143E and S145A pCDNA3 were generated with the QuickChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) by using the following primers: (Q143E) 5′–3′ GCTGCAAAGTTATCTGCTAAGAAAGAGGCATCCATATACAG and 5′–3′ CTGTATATGGATGCCTCTTTCTTAGCAGATAACTTTGCAGC; or (S145A) 5′–3′ TCTGCTAAGAAACAGGCAGCCATATACAGAGAT GAA and 5′–3′ TTCATCTCTGTATATGGCTGCCTGTTTCTTAGCAGA. All DNA constructs were confirmed by DNA sequencing. MLH1 protein was synthesized in vitro by coupled transcription and translation using the T7 Quick-coupled TnT kit (Pormega) and MLH1 pCDNA3 vector as a template (Plotz et al, 2003) gift from Guido Plotz (Homburg/Saar, Germany). The WT-MLH1 pcDNA3 vector (Invitrogen) was digested NotI/Apa1 and different MLH1 fragments were generated by PCR and digested Not1 and Apa1 products were inserted. Primers are available upon request.
The FANCJ fragment (amino acids (aa) 128–158) was generated by PCR. An eGFP expression vector was generated by subcloning PCR fragments of eGFP into pcDNA3.1 (Invitrogen, Carlsbad, California) (gift from Dr Andrew Kung) to create new unique restrictions sites within the active loop of eGFP. The annealed FANCJ fragment and the empty eGFP pCDNA3 vector were digested with BamH1 and EcoR1 restriction enzymes for 1 h at 37°. The FANCJ fragment and eGFP pCDNA3 vector were ligated using Quick ligase (NEB) for 1 h at room temperature. Primers covered FANCJ sequence from 128–158 aa.
ELISA studies
Purified recombinant MLH1 protein was diluted to a concentration of 1 ng/μl in Carbonate buffer (0.016 M Na2CO3, 0.034 M NaHCO3, pH 9.6) and added to appropriate wells of a 96-well microtiter plate (50 μl/well), which was incubated at 4°C. 3% bovine serum albumin (BSA) was used in the coating step for control reactions. The samples were aspirated, and the wells were blocked for 2 h at 30°C with blocking buffer (PBS, 0.5% Tween 20 and 3% BSA). The procedure was repeated. Purified recombinant FANCJ protein was diluted in blocking buffer, and the indicated concentrations were added to the appropriate wells of the ELISA plate (50 μl/well), which was incubated for 1 h at 30°C. For EtBr or DNaseI treatment, 50 μg/ml EtBr or DNaseI (2 μg/ml) was included in the incubation with FANCJ during the binding step in the corresponding wells. The samples were aspirated, and the wells were washed five times before addition of rabbit polyclonal anti-FANCJ Ab (Sigma, B-1310) that was diluted 1:5000 in Blocking buffer. Wells were then incubated at 30°C for 1 h. Following three washings, horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:5000) was added to the wells, and the samples were incubated for 30 min at 30°C. After washing five times, any FANCJ bound to the immobilized MLH1 was detected using OPD substrate (Sigma). The reaction was terminated after 3 min with 3 N H2SO4, and absorbance readings were taken at 490 nm.
Recombinant protein and helicase assays
Hi5 cells were infected with pVL132 K141/142A FANCJ and incubated for 72 h. Cells were collected, lysed in Insect Lysis Buffer (Roche) containing protease inhibitors (Roche) for 30 min at 4°C, and subsequently cleared by centrifugation. The WT, K52R, and K141/142A FANCJ proteins were purified as previously described (Cantor et al, 2004). Briefly, K141/142A FANCJ-Flag was IPed with 50 μl of FlagM2-conjugated beads for 2 h at 4°C. Beads were washed three times in 500 mM NETN (500 mM NaCl, 0.5% NP-40, 1 mM EDTA and 20 mM Tris–HCl (pH8.0)), followed by a final wash with 150 mM NETN, and K141/142A FANCJ protein was eluted twice using 3 × Flag peptide. Elutions were pooled and dialyzed overnight in storage buffer. Helicase assay reaction mixtures (20 μl) contained 40 mM Tris–HCl (pH 7.6), 25 mM KCl, 5 mM MgCl2, 2 mM dithiothreitol, 2% glycerol, 100 ng/μl BSA, 2 mM ATP, 10 fmol of the specified duplex DNA substrate (0.5 nM DNA substrate concentration), and the indicated concentrations of FANCJ helicase. Helicase reactions were initiated by the addition of FANCJ, and then incubated at 30°C for 15 min. Reactions were quenched in the presence of a 10-fold excess of unlabeled oligonucleotide with the same sequence as the labeled strand, to prevent reannealing, and products were resolved on non-denaturing 12% (19:1 acrylamide:bisacrylamide) polyacrylamide gels, and quantitated as previously described (Gupta et al, 2005).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We are grateful to Hans Joenje (UMC, Netherlands) for the FA-J fibroblasts (EUFA30-F), Guido Plotz (Homburg/Saar, Germany) for the MLH1 pCDNA3 vector and Junjie Chen (Mayo Clinic, MN) for the WT and S990A FANCJ pLenti vectors. We thank Teresa M Wilson (Univ. MD Baltimore) for purified MutLα. This work was funded by a grant from the Mary Kay Ash Charitable Foundation, and is supported by grant IRG 93–033 from the American Cancer Society. We are also grateful to Martha Berman and Robert Lipp of the Bari Lipp Foundation for their support, and to the Fanconi Anemia Research Foundation for FA cells and Abs.
References
- Akkari YM, Bateman RL, Reifsteck CA, D'Andrea AD, Olson SB, Grompe M (2001) The 4N cell cycle delay in Fanconi anemia reflects growth arrest in late S phase. Mol Genet Metab 74: 403–412 [DOI] [PubMed] [Google Scholar]
- Andreassen PR, D'Andrea AD, Taniguchi T (2004) ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 18: 1958–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilina G, Ceccotti S, Martinelli S, Hampson R, Bignami M (1998) N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosourea sensitivity in mismatch repair-defective human cells. Cancer Res 58: 135–141 [PubMed] [Google Scholar]
- Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K (2005) The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37: 953–957 [DOI] [PubMed] [Google Scholar]
- Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R (2003) The mismatch repair system is required for S-phase checkpoint activation. Nat Genet 33: 80–84 [DOI] [PubMed] [Google Scholar]
- Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM (2004) The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA 101: 2357–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM (2001) BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105: 149–160 [DOI] [PubMed] [Google Scholar]
- Cejka P, Stojic L, Mojas N, Russell AM, Heinimann K, Cannavo E, di Pietro M, Marra G, Jiricny J (2003) Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J 22: 2245–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Levran O, Jurickova I, Maas C, Kapur R, Schindler D, Henry R, Milton K, Batish SD, Cancelas JA, Hanenberg H, Auerbach AD, Williams DA (2005) A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol Ther 5: 978–984 [DOI] [PubMed] [Google Scholar]
- Dao V, Modrich P (1998) Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J Biol Chem 273: 9202–9207 [DOI] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Berns A, Radman M, te Riele H (1995) Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 82: 321–330 [DOI] [PubMed] [Google Scholar]
- Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P (1996) Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci USA 93: 6443–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrillaux B, Aurias A, Dutrillaux AM, Buriot D, Prieur M (1982) The cell cycle of lymphocytes in Fanconi anemia. Hum Genet 62: 327–332 [DOI] [PubMed] [Google Scholar]
- Fiumicino S, Martinelli S, Colussi C, Aquilina G, Leonetti C, Crescenzi M, Bignami M (2000) Sensitivity to DNA cross-linking chemotherapeutic agents in mismatch repair-defective cells in vitro and in xenografts. Int J Cancer 85: 590–596 [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM (2006) Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 20: 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Sharma S, Sommers JA, Jin Z, Cantor SB, Brosh RM Jr (2005) Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem 280: 25450–25460 [DOI] [PubMed] [Google Scholar]
- Hall MC, Jordan JR, Matson SW (1998) Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J 17: 1535–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich MC, Hoatlin ME, Zigler AJ, Silvey KV, Bakke AC, Keeble WW, Zhi Y, Reifsteck CA, Grompe M, Brown MG, Magenis RE, Olson SB, Bagby GC (1998) DNA cross-linker-induced G2/M arrest in group C Fanconi anemia lymphoblasts reflects normal checkpoint function. Blood 91: 275–287 [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297: 606–609 [DOI] [PubMed] [Google Scholar]
- Jasin M (2002) Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene 21: 8981–8993 [DOI] [PubMed] [Google Scholar]
- Kaiser TN, Lojewski A, Dougherty C, Juergens L, Sahar E, Latt SA (1982) Flow cytometric characterization of the response of Fanconi's anemia cells to mitomycin C treatment. Cytometry 2: 291–297 [DOI] [PubMed] [Google Scholar]
- Kupfer GM, D'Andrea AD (1996) The effect of the Fanconi anemia polypeptide, FAC, upon p53 induction and G2 checkpoint regulation. Blood 88: 1019–1025 [PubMed] [Google Scholar]
- Kupfer GM, Yamashita T, Naf D, Suliman A, Asano S, D'Andrea AD (1997) The Fanconi anemia polypeptide, FAC, binds to the cyclin-dependent kinase, cdc2. Blood 90: 1047–1054 [PubMed] [Google Scholar]
- Levitus M, Rooimans MA, Steltenpool J, Cool NF, Oostra AB, Mathew CG, Hoatlin ME, Waisfisz Q, Arwert F, de Winter JP, Joenje H (2004) Heterogeneity in Fanconi anemia: evidence for 2 new genetic subtypes. Blood 103: 2498–2503 [DOI] [PubMed] [Google Scholar]
- Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H (2005) The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37: 934–935 [DOI] [PubMed] [Google Scholar]
- Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD (2005) The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet 37: 931–933 [DOI] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8: 255–265 [DOI] [PubMed] [Google Scholar]
- Miglierina R, Le Coniat M, Berger R (1991) A simple diagnostic test for Fanconi anemia by flow cytometry. Anal Cell Pathol 3: 111–118 [PubMed] [Google Scholar]
- Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen M, Huo X, Buermeyer AB (2006) Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst) 5: 347–361 [DOI] [PubMed] [Google Scholar]
- Mohindra A, Hays LE, Phillips EN, Preston BD, Helleday T, Meuth M (2002) Defects in homologous recombination repair in mismatch-repair-deficient tumour cell lines. Hum Mol Genet 11: 2189–2200 [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Ogryzko V (2003) Immunoaffinity purification of mammalian protein complexes. Methods Enzymol 370: 430–444 [DOI] [PubMed] [Google Scholar]
- O'Brien V, Brown R (2006) Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis 27: 682–692 [DOI] [PubMed] [Google Scholar]
- Plotz G, Raedle J, Brieger A, Trojan J, Zeuzem S (2003) N-terminus of hMLH1 confers interaction of hMutLalpha and hMutLbeta with hMutSalpha. Nucleic Acids Res 31: 3217–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala-Trepat M, Rouillard D, Escarceller M, Laquerbe A, Moustacchi E, Papadopoulo D (2000) Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp Cell Res 260: 208–215 [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Hsieh P (2003) DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol 57: 579–608 [DOI] [PubMed] [Google Scholar]
- Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N (2006) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38: 1239–1241 [DOI] [PubMed] [Google Scholar]
- Spampinato C, Modrich P (2000) The MutL ATPase is required for mismatch repair. J Biol Chem 275: 9863–9869 [DOI] [PubMed] [Google Scholar]
- Thompson LH (2005) Unraveling the Fanconi anemia-DNA repair connection. Nat Genet 37: 921–922 [DOI] [PubMed] [Google Scholar]
- Thompson LH, Hinz JM, Yamada NA, Jones NJ (2005) How Fanconi anemia proteins promote the four Rs: replication, recombination, repair, and recovery. Environ Mol Mutagen 45: 128–142 [DOI] [PubMed] [Google Scholar]
- Trojan J, Zeuzem S, Randolph A, Hemmerle C, Brieger A, Raedle J, Plotz G, Jiricny J, Marra G (2002) Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology 122: 211–219 [DOI] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10: 373–385 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927–939 [PMC free article] [PubMed] [Google Scholar]
- Yamada M, O'Regan E, Brown R, Karran P (1997) Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res 25: 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Dao V, Modrich P (1998) MutS and MutL activate DNA helicase II in a mismatch-dependent manner. J Biol Chem 273: 9197–9201 [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Yoshioka Y, Hsieh P (2006) ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell 22: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J (2003) The BRCT domain is a phospho-protein binding domain. Science 302: 639–642 [DOI] [PubMed] [Google Scholar]
- Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ (2002) hMutSbeta is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol 22: 2388–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Wang X, Legerski RJ, Glazer PM, Li L (2006) Repair of DNA interstrand cross-links: interactions between homology-dependent and homology-independent pathways. DNA Repair (Amst) 5: 566–574 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3