

Abstract
Recent molecular cytogenetic data have shown that the constitution of complex chromosome rearrangements (CCRs) may be more complicated than previously thought. The complicated nature of these rearrangements challenges the accurate delineation of the chromosomal breakpoints and mechanisms involved. Here, we report a molecular cytogenetic analysis of two patients with congenital anomalies and unbalanced de novo CCRs involving chromosome 17p using high-resolution array-based comparative genomic hybridization (array CGH) and fluorescent in situ hybridization (FISH). In the first patient, a 4-month-old boy with developmental delay, hypotonia, growth retardation, coronal synostosis, mild hypertelorism, and bilateral club feet, we found a duplication of the Charcot-Marie–Tooth disease type 1A and Smith-Magenis syndrome (SMS) chromosome regions, inverted insertion of the Miller-Dieker lissencephaly syndrome region into the SMS region, and two microdeletions including a terminal deletion of 17p. The latter, together with a duplication of 21q22.3-qter detected by array CGH, are likely the unbalanced product of a translocation t(17;21)(p13.3;q22.3). In the second patient, an 8-year-old girl with mental retardation, short stature, microcephaly and mild dysmorphic features, we identified four submicroscopic interspersed 17p duplications. All 17 breakpoints were examined in detail by FISH analysis. We found that four of the breakpoints mapped within known low-copy repeats (LCRs), including LCR17pA, middle SMS-REP/LCR17pB block, and LCR17pC. Our findings suggest that the LCR burden in proximal 17p may have stimulated the formation of these CCRs and, thus, that genome architectural features such as LCRs may have been instrumental in the generation of these CCRs.
Introduction
The majority of constitutional chromosomal abnormalities are thought to be simple rearrangements, involving less than three breaks in one or two chromosomes. It is known that genomic architectural features such as low-copy repeat (LCR) structures may play an important role in the formation of these abnormalities (Lupski 1998; Stankiewicz and Lupski 2002). LCRs, also termed segmental duplications or duplicons, encompass apparently normal stretches of genomic DNA, often containing genes. LCRs are present in more than one copy in the genome and are defined by a ≥1 kb size and a >90% sequence identity (Bailey et al. 2001, 2002; Eichler 2001). LCRs can mediate recurrent DNA rearrangements such as deletions, duplications, and inversions through chromosome or chromatid misalignment followed by nonallelic homologous recombination (NAHR). The majority of the currently known chromosome microdeletion/duplication syndromes result from NAHR between large (usually >10 kb), highly identical (>95%) LCRs and are, therefore, also termed genomic disorders (Lupski 1998; Stankiewicz and Lupski 2002; Lupski and Stankiewicz 2006). In addition to recurrent events, breakpoints of nonrecurrent rearrangements have been associated also with genomic architectural features including LCRs and AT-rich palindromic sequences (Stankiewicz et al. 2003; Shaw and Lupski 2004, 2005; Kriek et al. 2006; Lee et al. 2006).
Complex chromosome rearrangements (CCRs) are relatively rare events, and can be classified into groups based on the number of breakpoints and type of rearrangement, being (1) three way exchange with three breaks in three involved chromosomes, (2) two way exchange that coincides two separate reciprocal translocations, and (3) exceptional CCRs with multiple breaks and complicated rearrangements (Bartels et al. 2006). CCRs have been observed in phenotypically normal individuals as well as in individuals with mental retardation and/or congenital abnormalities (Pai et al. 1980; Battisti et al. 2003; Kuechler et al. 2005). As a consequence, the characterization of these rearrangements is essential for reaching a proper clinical diagnosis and for estimation of the recurrence risk. In standard clinical practice, this characterization is usually achieved by conventional cytogenetic approaches only, thus submicroscopic imbalances at the breakpoints will remain undetected because of the low resolution of such approaches. Recently, the use of high-resolution molecular techniques such as array-based comparative genomic hybridization (array CGH) have contributed to a growing awareness of the presence of CCRs and cryptic imbalances in patients with MR and/or congenital anomalies (Vissers et al. 2003; Weise et al. 2003; Lespinasse et al. 2004; Patsalis et al. 2004; Shaw-Smith et al. 2004; Vermeulen et al. 2004; Borg et al. 2005; de Vries et al. 2005; Chen et al. 2006; Gajecka et al. 2006; Karmous-Benailly et al. 2006). Although molecular mechanisms have been studied in recurrent, simple rearrangements, the role of genomic architecture underlying the occurrence of nonrecurrent CCRs, remains as yet poorly understood because until recently detailed identification of the exact breakpoints was lacking.
Here, we investigated the potential involvement of genome architectural features such as LCRs in the occurrence of nonrecurrent 17p CCRs in two patients with congenital anomalies. The proximal chromosome 17p arm is associated with a wide variety of recurrent chromosome aberrations resulting from NAHR between LCRs including four well known genomic disorders: Charcot–Marie–Tooth disease type 1A (CMT1A) [MIM 118220], hereditary neuropathy with liabilities to pressure palsies [MIM 162500], Smith-Magenis syndrome (SMS) [MIM 182290] and the Potocki-Lupski syndrome associated with duplication 17p11.2 (Pentao et al. 1992; Reiter et al. 1996; Chen et al. 1997; Potocki et al. 2000; Inoue et al. 2001; Shaw et al. 2002; Bi et al. 2003; Potocki et al. 2007). By applying high-resolution array CGH to these two novel CCRs we observed an unexpected level of complexity. Subsequent FISH analyses revealed the presence of LCRs at the CCR breakpoints, thus providing a potential mechanistic basis for the occurrence of the CCRs studied. We propose that genomic architectural features such as LCRs may underlie the occurrence of these CCRs.
Patients, materials, and methods
Patients
Patient 1–the male proband was born to a 42-year-old woman and 43-year-old man at 39 week’s gestation by Cesarean section. Because of an abnormal ultrasound showing prominent nuchal fold, growth retardation, club feet, and hyperechoic bowel, amniocentesis was performed at 20 weeks of gestation and an abnormal karyotype with 17pter monosomy and 17p13.1p13.3 trisomy was identified. The birth weight was 2,840 g (10th centile), length 51 cm (60th centile), and head circumference 36 cm (50th centile). Apgar scores were 6 and 8 at 1 and 5 min, respectively. The bilateral club feet were corrected by surgery. Echocardiogram revealed bicuspid aortic valve. Physical exam at 11 months revealed weight and height at the 3rd centile while his head circumference had increased to the 75th centile. There was a premature closure of the metopic suture, with anterior fontanel still open (2 × 2 cm) and mild hypertelorism. At the age of 16 months, he was an amiable, interpersonally interactive little boy who had developmental delay with babbling but no clear words. In addition, he suffered from truncal hypotonia but he was able to pull to stand. His skin was dry with marked eczema and the right testis was still undescended.
Patient 2—this 8-year-old, moderately mentally retarded girl was born after an uneventful pregnancy at 40 week’s gestation, with a normal birth weight of 3,400 g. Apgar scores were 3, 5, and 6 at 1 min, 5 min, and 10 min, respectively. Feeding problems were present from birth. Her development was moderately delayed with an IQ score of 50 points at the age of 4 years and 9 months. At the age of 6 years and 10 months, she had a short stature of 110 cm (<3rd centile) and a head circumference of 49 cm (10th centile) with distinctive facial features such as synophrys, upward slanting palpebral fissures, flattened upper-ear helices with large ear lobules, an upturned nasal tip, and a broad mouth with a long philtrum and full lower lip. She had a cardiac murmur; short broad hands with clinodactyly of the fifth fingers, and absence of distal interphalangeal joint creases; short broad feet with short toes and a slight skin syndactyly between the second and third toes bilaterally. She had an outgoing personality and easily approaches strangers. At the age of 8 years, she started to complain about painful feet and developed a bilateral pes cavus deformity. Neurological examination showed areflexia and electromyography studies were consistent with a demyelinating neuropathy. In addition, treatment for precocious puberty was started.
We obtained samples from the patients and their family members after acquiring informed consent approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine, Houston, and Radboud University Nijmegen Medical Centre, Nijmegen.
Array-based comparative genomic hybridization
Genomic DNA was isolated from peripheral blood lymphocytes or from lymphoblastoid cell lines by routine procedures. Two pools of reference DNA were used—one containing equal amounts of genomic DNA from ten healthy male blood donors and one containing equal amounts of genomic DNA from ten healthy female blood donors. Isolation of genomic DNA, DNA labeling, hybridization of labeled DNA to a 32,447-BAC array, and spot identification were performed as described elsewhere (de Vries et al. 2005). In brief, 500 ng of genomic DNA from each patient was labeled by random priming with Cy3-dUTP or Cy5-dUTP (Amersham Biosciences) and hybridized to the sex-mismatched reference pool. Test and reference samples were mixed with 120 μg of human Cot-1 DNA (Roche), co-precipitated, and resuspended in 120 μl of a hybridization solution containing 50% formamide, 10% dextran sulfate, 2 × SSC, 4% SDS, and 10 mg/ml of yeast tRNA (Invitrogen). Hybridization and post-hybridization washing procedures were performed using a GeneTac Hybridization Station (Genomic Solutions). An 18-h hybridization at 37°C with active circulation of the probe was performed, followed by five post-hybridization wash cycles in 50% formamide and 2 × SSC at 45°C, and five wash cycles in phosphate buffer at 20°C. Slides were dried by centrifugation and scanned using a GenePix Autoloader 4200AL laser scanner (Axon Instruments). Spot identification and two-color fluorescence intensity measurements were obtained using the GenePix 5.1 software, and all data were entered into a database for subsequent analysis. Following normalization, the log2 transformed test-over-reference ratios were analyzed for loss and gain of genomic regions by a standard Hidden Markov Model (Rabiner 1989; de Vries et al. 2005).
Fluorescent in situ hybridization
PAC and BAC probes specific for human chromosome 17p were selected based on their physical location within the affected 17p region (Inoue et al. 2001; Bi et al. 2002; http://www.genome.ucsc.edu/). DNA was isolated from liquid cultures using Perfectprep Plasmid Mini (Eppendorf, Hamburg, Germany). The relative alignments of the selected BACs were determined by BLAST searches against the high-throughput genome sequence database (http://www.ncbi.nlm.nih.gov/blast) and assembled using the Sequencher software (Gene Codes Corp., Ann Arbor, MI, USA). FISH was essentially performed as described (Stankiewicz et al. 2001a). In brief, 100–200 ng of isolated BAC or PAC DNA was labeled with biotin or digoxigenin by nick-translation (BioNick Labeling System, Invitrogen; DIG-Nick Translation Mix, Roche) and visualized with FITC avidin (Vector) or rhodamine-labeled antibodies (Sigma). Patient’s chromosomes derived from lymphoblast cell lines were counterstained with DAPI (Sigma). The signals from the normal chromosomes 17 were used as internal control. Cells were analyzed using a Zeiss Axioskop fluorescence microscope equipped with an appropriate filter combination and a CCD camera. Monochromatic images were captured and pseudocolored using MacProbe 4.2.2 (Perceptive Scientific Instruments, League City, TX, USA) on a Power Macintosh G4 system.
Computational analyses
For the identification of novel LCRs associated with the CCRs, ∼100 kb fragments of genomic sequence flanking breakpoints for which no LCRs are currently known, were analyzed for sequence homology (http:// www.genome.ucsc.edu; May 2004 freeze). Interspersed repeat sequences within the downloaded DNA sequence were eliminated by RepeatMasker (http:// www.genome.ucsc.edu) and the repeat masked genomic sequences were analyzed using NCBI BLAST2 (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html).
Results
Cytogenetic and molecular analyses of patient 1
Conventional cytogenetic analysis at 550-band resolution in patient 1 (Fig. 1a–c) revealed an abnormal male karyotype with an apparent complex rearrangement involving the short arm of chromosome 17. Parental chromosomes at 600-band resolution were normal. FISH analysis confirmed this complex rearrangement and, in addition, revealed a deletion of 17pter, a duplication of the genomic region containing the SMS and the CMT1A regions, and an inversion involving the Miller-Dieker lissencephaly syndrome (MDLS) region. The presence of these abnormalities in the parental chromosomes was excluded by FISH. The patient’s karyotype was designated as:
46,XY,der(17).ish del(17)(p13.3)dup(17)(p11.2p12)inv(17)(p11.2p13.3) dn.
Subsequently, array CGH was used to refine the breakpoints and genomic sizes of the complex 17p rearrangement. The deletion involving the telomeric region of 17p was determined to be ∼600 kb in size, whereas the duplication containing the SMS and CMT1A region was ∼6.1 Mb in size. Interestingly, array CGH revealed two additional submicroscopic alterations that had remained undetected using previous approaches. These included an ∼2.2 Mb interstitial deletion in 17p12 and an ∼4.4 Mb duplication in 21q22.3 (Fig. 2a, b).
Fig. 1.
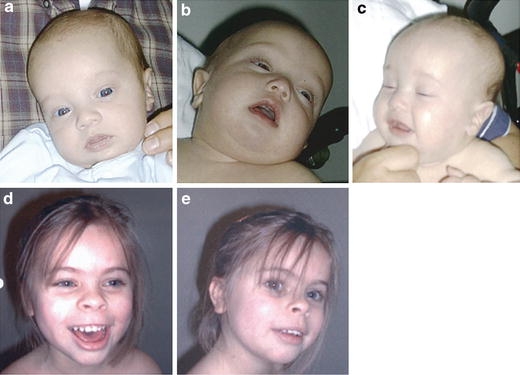
Patient 1 (a–c) at the age of 4 months. Patient 2 (d–e) at the age of 6 years 10 months
Fig. 2.
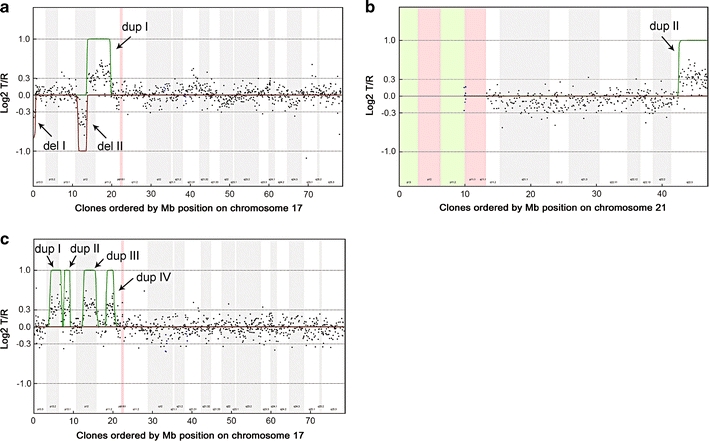
Chromosome 17 (patient 1 and 2) and chromosome 21 (patient 1) array CGH profiles. On the x-axis, clones are ordered by Mb position on chromosome 17 and 21, respectively, and on the y-axis log2T/R ratios are shown. Hidden Markov Model was used to identify the duplications (green lines) and deletions (red lines) in patient 1 and 2. a In patient 1, chromosome 17 shows two interspersed deletions of ∼600 kb (17p13.3–Del I) and ∼2.2 Mb (17p12–Del II), respectively, and a duplication of ∼6.1 Mb (Dup I). b Additionally, a 4.4 Mb duplication of 21q22.3 was observed (Dup II). c Patient 2 showed four interspersed duplications on 17p11–p13 (Dup I–Dup IV), in total comprising ∼8.8 Mb of genomic sequence. All alterations were shown to be de novo
Dual color FISH experiments were performed to independently map each of the chromosome breakpoints (Fig. 3; Table 1). FISH analyses confirmed the deletion of the 17p telomeric region (Fig. 3a) as well as the dup(17)(p11.2p12) (Fig. 3b). The FISH results of the duplication were consistent with a tandem duplication (Fig. 3b). Interestingly, the MDLS region was found to be inserted into the middle SMS-REP/LCR17pB block in the genomic region involved in SMS (Fig. 3c). Unexpectedly, the duplicated fragment 21q22.3-qter was localized on der(17), likely representing the product of an unbalanced translocation t(17;21)(p13.3;q22.3) (Fig. 3d). FISH analyses on the parental chromosomes excluded the presence of a balanced t(17;21)(p13.3;q22.3). Based on these results, the patient’s karyotype was redefined as:
46,XY,der(17).ish del(17)(p13.3)del(17)(p12p12)dup(17)(p11.2p12)ins(17)(p11.2p13.3p13.2)t(17;21)(p13.3;q22.3).arr cgh 17p13.3(CTD-2348K1→RP11-143G11)x1,17p12x(RP11-165H21→RP11-590H8)x1,17p12p11.2(RP11-687M21→CTD-2019P4)x3,21q22.3(RP11-282I20→RP11-1000I21)x3 dn.
Fig. 3.
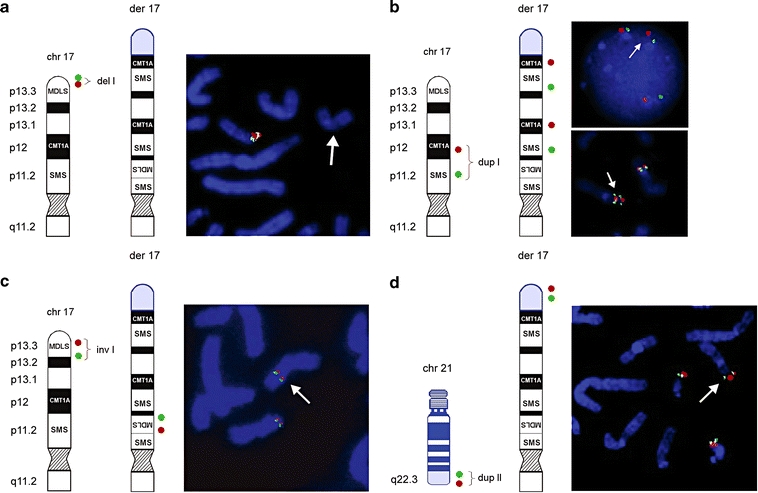
Ideograms and FISH results of patient 1. Schematic representation of a normal 17p and der(17) (black and white) with translocated chromosome 21 material (blue). The location of the FISH probes are shown on the left side of each figure panel; der(17) is indicated on FISH pictures by a white arrow. a Terminal deletion of 17pter was validated using BAC clones RP11-1260E13 (red) and CTD-2326F1 (green) (del I). b FISH with PMP22-specific PAC RP1-150M12 (red) RA11-specific and BAC RP11-525O11 (green) revealed direct duplication of the CMT1A and SMS regions in 17p12p11.2 (dup I). c FISH with PAC RP1-95H6 (red; adapted from Chong et al. 1997) and BAC GS-202L17 (green; adapted from Knight et al. 2000) showed inverted insertion of the MDLS region into the SMS region. d Array CGH also identified a duplication of 21q22.3 (dup II). Additional FISH analysis using BAC clones RP11-40L10 (green) and RP11-16B19 (red) revealed that the duplicated material of 21q22.3 was translocated onto der(17). Summary of FISH results is provided in Table 1
Table 1.
Summary of the array CGH and FISH results patient 1
| Chromosome band | Clonea | Distance from pter (Mb)e | Der(17) | Technique(s) | ||
|---|---|---|---|---|---|---|
| 17p13.3 | CTD-2348K1 | 0.1 | – | Array CGH | ||
| 17p13.3 | CTD-2326F1 | (Green) | 0.1 | – | Array CGH/FISH | |
| 17p13.3 | CTD-2573J8 | 0.2 | – | Array CGH | Del I | |
| 17p13.3 | RP11-1260E13 | (Red) | 0.3 | – | FISH | |
| 17p13.3 | RP11-143G11 | 0.6 | – | Array CGH | ||
| 17p13.3 | RP11-488O17 | 0.6 | Normal | Array CGH | ||
| 17p13.3 | RP11-411G7 | 0.6 | Normal | FISH | ||
| 17p13.3 | RP1-95H6b | (Red) | 2.7 | Inv ins | FISH | |
| 17p13.2 | RP11-148L19 | 4.1 | Inv ins | FISH | ||
| 17p13.2 | GS-202L17c | (Green) | 4.9 | Inv ins | FISH | Middle SMS-REP/LCR17pB |
| 17p13.2 | RP11-373N8 | 5.5 | Inv ins | FISH | ||
| 17p12 | RP11-466J1 | 11.1 | Normal | Array CGH | ||
| 17p12 | RP11-22L4 | 11.2 | Normal | FISH | ||
| 17p12 | RP11-271N1 | 11.4 | Normal | Array CGH | ||
| 17p12 | RP11-165H21 | 11.5 | – | Array CGH | ||
| 17p12 | RP11-802E21 | 11.6 | – | Array CGH | ||
| 17p12 | RP11-64L11 | 13.1 | – | FISH | Del II | |
| 17p12 | RP11-352N24 | 13.2 | – | Array CGH | ||
| 17p12 | RP11-590H8 | 13.4 | – | Array CGH | ||
| 17p12 | RP11-112H7 | 13.6 | Normal | Array CGH | ||
| 17p12 | CTD-2120F7 | 13.7 | Normal | Array CGH | ||
| 17p12 | RP11-687M21 | 13.9 | ++ | Array CGH | ||
| 17p12 | RP11-78J16 | 14.1 | ++ | Array CGH | ||
| 17p12 | RP1-150M12d | (Red) | 15.4 | ++ | FISH | |
| 17p11.2 | RP11-209J20 | 16.3 | ++ | FISH | ||
| 17p11.2 | RP11-525O11 | (Green) | 17.6 | ++ | FISH | Dup I |
| 17p11.2 | RP1-178F10 | 18.4 | ++ | FISH | ||
| 17p11.2 | RP11-28B23 | 18.7 | ++ | FISH | ||
| 17p11.2 | RP11-1113L8 | 19.3 | ++ | FISH | ||
| 17p11.2 | RP11-277B5 | 19.4 | ++ | FISH | ||
| 17p11.2 | CTD-2019P4 | 19.6 | ++ | Array CGH/FISH | ||
| 17p11.2 | CTD-2020J20 | 19.7 | Normal | Array CGH | ||
| 17p11.2 | RP11-78O07 | 19.7 | Normal | FISH | ||
| Chromosome band | Clone | Distance from pter (Mb)e | Der(21) | Technique(s) | ||
| 21q22.3 | RP11-690D9 | 42.5 | Normal | Array CGH | ||
| 21q22.3 | RP11-282I20 | 42.6 | ++ | Array CGH | ||
| 21q22.3 | RP11-40L10 | (Green) | 44.2 | ++ | FISH | Dup II |
| 21q22.3 | RP11-16B19 | (Red) | 45.1 | ++ | FISH | Located on der(17) |
| 21q22.3 | RP11-1000I21 | 46.8 | ++ | Array CGH |
‘–’ Deleted, ‘++’ duplicated, ‘inv ins’ inverted inversion. Extent of deletion and duplication is indicated at the right. Breakpoint-associated LCRs are indicated in bold
aClones indicated by red or green are shown in Fig. 3a–d in the respective colors
bLIS1-specific PAC clone (adapted from Chong et al. 1997)
cAdapted from Knight et al. 2000
dPMP22-specific PAC clone
eDistances from pter to the clone midpoints
Cytogenetic and molecular analyses of patient 2
Conventional cytogenetic analysis at 550-band resolution in patient 2 (Fig. 1d–e) showed a normal 46, XX karyotype. Multiplex ligation-dependent probe amplification revealed normal disomic copy numbers of the subtelomeric regions. Subsequent array CGH revealed four interspersed microduplications involving the short arm of chromosome 17, in total comprising 8.8 Mb of genomic sequence (Table 2). De novo occurrence was established by a similar array CGH analysis with DNA obtained from the accompanying parents (Fig. 2c; de Vries et al. 2005). Duplication I in 17p13.2 was determined as ∼2.9 Mb in size; duplication II in 17p13.1 was assessed as ∼1.4 Mb in size; duplication III in 17p12 was estimated as ∼2.9 Mb in size and included the PMP22 gene known to cause CMT1A disease with dosage-specific overexpression of PMP22 (Patel et al. 1992). Duplication IV in 17p11.2 was determined as being ∼1.5 Mb in size.
Table 2.
Summary of the array CGH and FISH results patient 2
| Chromosome band | Clonea | Distance from pter (Mb)b | Der(17) | Technique(s) | ||
|---|---|---|---|---|---|---|
| 17p13.2 | RP11-810M2 | (Green) | 4.0 | Normal | Array CGH/FISH | |
| 17p13.2 | RP11-597I9 | (Red) | 4.1 | ++ | Array CGH/FISH | |
| 17p13.2 | RP11-106A7 | 4.3 | ++ | Array CGH/FISH | ||
| 17p13.2 | RP11-217O7 | 4.3 | ++ | Array CGH | ||
| 17p13.1 | RP11-816H10 | 6.7 | ++ | Array CGH | Dup I | |
| 17p13.1 | RP11-417F20 | 6.9 | ++ | array CGH/FISH | ||
| 17p13.1 | RP11-558E15 | 7.1 | ++ | Array CGH | ||
| 17p13.1 | RP11-222J21 | (Green) | 7.2 | ++ | Array CGH/FISH | |
| 17p13.1 | RP11-98D15 | (Red) | 7.3 | Normal | Array CGH/ FISH | |
| 17p13.1 | RP11-205D17 | 7.3 | Normal | Array CGH | ||
| 17p13.1 | RP13-626G5 | 7.4 | Normal | Array CGH | ||
| 17p13.1 | RP11-63C7 | 7.7 | ++ | Array CGH | ||
| 17p13.1 | RP11-441N13 | 7.8 | ++ | Array CGH/FISH | ||
| 17p13.1 | RP11-452D1 | 8.1 | ++ | Array CGH/FISH | ||
| 17p13.1 | RP11-111I3 | 9.0 | ++ | Array CGH/FISH | Dup II | |
| 17p13.1 | RP11-85B7 | 9.1 | ++ | Array CGH | ||
| 17p13.1 | RP11-342E3 | 9.1 | ++ | Array CGH/FISH | ||
| 17p13.1 | RP11-324C22 | 9.2 | ++ | Array CGH | ||
| 17p13.1 | RP11-482E13 | 9.2 | Normal | Array CGH/FISH | ||
| 17p13.1 | RP11-589N11 | 9.2 | Normal | Array CGH | ||
| 17p12 | RP11-560N10 | 12.3 | Normal | Array CGH/FISH | ||
| 17p12 | RP11-674I3 | 12.5 | Normal | Array CGH | ||
| 17p12 | RP11-746E8 | 12.5 | ++ | FISH | ||
| 17p12 | RP11-333I5 | 12.6 | ++ | Array CGH/FISH | ||
| 17p12 | RP11-601N13 | (Green) | 13.9 | ++ | FISH | |
| 17p12 | RP11-686G16 | 15.1 | ++ | Array CGH | Dup III | |
| 17p12 | RP11-726O12 | (Red) ) | 15.2 | ++ | Array CGH/FISH | |
| 17p12 | RP11-385D13 | 15.4 | ++ | FISH | ||
| 17p12 | RP11-640N23 | 15.5 | ++ | Array CGH | ||
| 17p12 | RP11-640I15 | 15.6 | ++ | FISH | LCR17pA | |
| 17p12 | CTD-3157E16 | 15.7 | Normal | FISH | ||
| 17p12 | RP11-59N13 | 15.7 | Normal | Array CGH | ||
| 17p11.2 | RP11-484D23 | 18.3 | Normal | Array CGH | ||
| 17p11.2 | RP11-667E24 | 18.4 | Normal | Array CGH | ||
| 17p11.2 | RP1-178F10 | 18.4 | Normal | FISH | ||
| 17p11.2 | RP11-448D22 | (Green) | 18.6 | ++ | FISH | Middle SMS-REP/LCR17pB |
| 17p11.2 | RP11-304M17 | 18.6 | ++ | Array CGH | ||
| 17p11.2 | CTD-2145A24 | (Red) | 18.7 | ++ | FISH | Dup IV |
| 17p11.2 | RP11-137E6 | 20.0 | ++ | Array CGH/ FISH | ||
| 17p11.2 | CTD-2022C7 | 20.0 | ++ | Array CGH | ||
| 17p11.2 | RP11-121A13 | 20.1 | ++ | FISH | LCR17pC | |
| 17p11.2 | CTD-2313N10 | 20.3 | Normal | Array CGH | ||
| 17p11.2 | RP11-185K8 | 20.4 | Normal | Array CGH |
‘++’ duplicated. Extent of duplication is indicated at the right. Breakpoint-associated LCRs are indicated in bold
aClones indicated by red or green are shown in Fig. 4a–d in the respective colors
bDistances from pter to the clone midpoints
Dual color FISH analysis was used to independently confirm the array CGH results (Fig. 4; Table 2). In addition, the parental chromosomes were evaluated for predisposing rearrangements within chromosome 17p potentially explaining the complexity of the CCR, however, no such rearrangements were observed. The patients karyotype was finally designated as:
46,XX.ish dup(17)(p13.1p13.2)dup(17)(p13.1p13.1)dup(17)(p12p12)dup(17)(p11.2p11.2).arr cgh 17p13.1p13.2(RP11-597I9→RP11-222J21)x3,17p13.1(RP11-63C7→RP11-324C22)x3, 17p12(RP11-333I5→RP11-640N23)x3,17p11.2(RP11-304M17→CTD-2022C7)x3 dn.
Fig. 4.
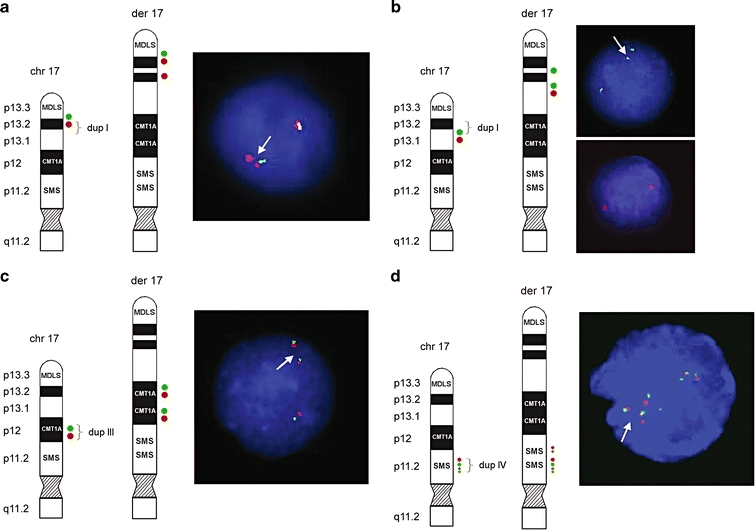
Ideograms and FISH results of patient 2. Schematic representation of the normal 17p and der(17). The locations of the FISH probes are shown on the left side of each figure panel. a The distal breakpoint of duplication I showed a relatively simple fluorescence signal pattern with probes RP11-810M2 (green; normal) and RP11-597I9 (red; duplicated). b The proximal breakpoint of duplication I showed a duplicated signal for RP11-222J21 (green) and a normal signal for RP11-98D15 (red). c Direct orientation of duplication III was shown using BAC clones RP11-601N13 (green) and RP11-726O12 (red). d For the distal breakpoint of duplication IV, BAC clones RP11-448D22 (green) and CTD-2145A24 (red) showed duplicated signals on der(17), indicating that both middle SMS-REP and LCR17pB are duplicated as a block. Four red signals on der(17) representing two normal and two duplicated copies of LCR17pA/B (dup III) and LCR17pB (dup IV) and four green signals depicting three normal copies of SMS-REPs and the duplicated middle SMS-REP. Summary of FISH results is provided in Table 2
Overview of the 17p breakpoints in both patients
In total, 17 breakpoints were identified and molecularly characterized at one BAC clone resolution in these two patients, with nine breaks in patient 1 and eight breaks in patient 2. In patient 1, one of the nine breakpoints (inverted insertion of the MDLS region) was mapped within the middle SMS-REP/LCR17pB block using FISH with these LCR flanking clones RP1-178F10 and RP11-28B23. In patient 2, three of the eight breakpoints were associated with LCRs in proximal 17p. The proximal breakpoint of duplication III was located in LCR17pA, between subunits LCR17pA/B and LCR17pA/D using FISH with LCR17pA/B- specific BAC clone RP11-640I15 and LCR17pA/D- and LCR17pA/C specific BAC clone CTD-3157E16 that are known to cross-hybridize to their homologous copies LCR17pB, LCR17pD and LCRq7pC, respectively (Stankiewicz et al. 2004). BAC clone RP11-640I15 showed four fluorescent signals on der(17) (representing two normal and two duplicated copies of LCR17pA/B (dup III) and LCR17pB (dup IV) and CTD-3157E16 three signals on der(17) in analyzed interphase nuclei (depicting normal, not duplicated copies of LCR17pA/D LCR17pC, and LCR17pD). In duplication IV the distal breakpoints was mapped in the middle of SMS-REP, between PAC clone RP1-178F10 (flanks the middle SMS-REP on the telomeric side) and BAC clone RP11-448D22 (middle SMS-REP-specific) (Fig. 4d). The proximal breakpoint of dup IV was mapped in LCR17pC using FISH with its specific BAC clone RP11-121A13. DNA sequence analysis of the genomic regions surrounding the remaining 13 chromosome breakpoints revealed no significant homology or evidence of low-copy repeats. Figure 5 shows an overview of the proximal 17p breakpoints and its association with genomic architectural features observed in both patients. In addition, a selected set of breakpoints previously reported in other patients with 17p aberrations that are associated with LCRs is displayed.
Fig. 5.
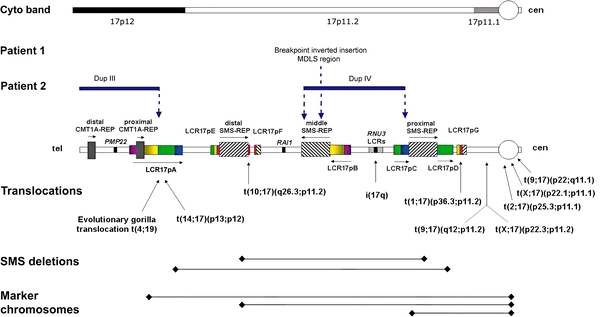
Schematic diagram of breakpoints for DNA rearrangements in 17p. Top: Cytogenetic chromosome bands 17p12, and sub-bands 17p11.2 and 17p11.1 are shown. Breakpoints of the chromosome rearrangements in patients 1 and 2 associated with architectural features in proximal 17p are shown by vertical dotted blue arrows. In patient 1, one breakpoint is located within the middle SMS-REP/LCR17pB block, whereas in patient 2 breakpoints are located within LCR17pA (proximal Dup III), middle SMS-REP (distal Dup IV) and LCR17pC (proximal Dup IV). Duplication III and IV in patient 2 are indicated by horizontal blue lines. Bottom: Previously identified rearrangements associated with LCRs in 17p. Breakpoints of translocations and isochromosome 17q are indicated by vertical black arrows whereas common ∼4 Mb and uncommon ∼5 Mb SMS deletions and marker chromosomes are indicated by black horizontal lines. The LCR17p structures are depicted in colors to better represent their positional orientation with respect to each other; the shaded rectangles and horizontal black arrows represent the orientation of the LCRs
Discussion
The availability of molecular genome profiling techniques such as array CGH have markedly enhanced the resolution of chromosome studies and enabled high-resolution genome analysis, thus proving a more accurate method for the identification and delineation of chromosomal rearrangements (Vissers et al. 2003; Shaw-Smith et al. 2004; Cheung et al. 2005; de Vries et al. 2005; Johnston et al. 2007). As a result, precise definitions of CCRs and their true complexity can now be better established (Astbury et al. 2004; Thienpont et al. 2006). An apparent underestimation of the full complexity of CCRs is well demonstrated in patient 1, in whom a complex karyotype was identified, including an inverted insertion of the MDLS region into the middle SMS-REP/LCR17pB block, two microdeletions (terminal and interstitial in 17p12) and a microduplication involving both SMS and CMT1A chromosome regions. Furthermore, a duplication of 21q22.3qter translocated onto 17p13.3 was identified. The interstitial deletion on 17p12, as well as the duplication of 21q22.3, were not identified prior to array CGH analysis. Thus, by unraveling the complexity of CCRs using array CGH, the added value of this technique to conventional karyotyping was demonstrated. It is, however, noteworthy that current array CGH techniques are incapable of detecting balanced alterations such as inversions, and only provide information regarding genomic gains and losses. FISH can augment the study of CCRs by providing genomic positional and orientational information of imbalances. This is well demonstrated by the apparently balanced inverted insertion of the MDLS region into the middle SMS-REP/LCR17pB block in patient 1, which would have escaped attention if array CGH would have been the only technique employed.
A total of 106 genes in patient 1 and 133 genes in patient 2 were affected by genomic imbalances (gains and losses) because of the CCRs. For patient 1, the number of genes affected is less than expected based on the average number of 15 genes/Mb on chromosome 17 and 6 genes/Mb on chromosome 21. It thus seems that the CCR in patient 1 affected relatively gene poor regions. For patient 2, the total number of genes affected was in concordance with the expected number of genes affected by the CCR. Given the large number of genes affected in each patient and the fact that only a fraction of these genes may be subject to gene dosage effect causing a phenotype, it is difficult to correlate any specific gene with the observed phenotypes. However, a few genes affected by the CCRs are well-established dosage sensitive genes, including RAI1 in patient 1 and PMP22 in patient 2, which are known to cause disease when duplicated. Recently, duplication of RAI1 has been shown to cause a physical and behavioral phenotype termed Potocki–Lupski syndrome (Potocki et al. 2007). However, due to the young age of patient 1, as well as the imbalances of other genomic regions, assessing phenotype-genotype correlations is not possible. One of the genes affected by the CCR in patient 2 is the dosage-sensitive PMP22 gene. Duplication of this gene causes CMT1A, a common inherited neuropathy characterized by myelin degeneration (Patel et al. 1992). Indeed, the first clinical signs of CMT1A were present in this patient.
Genome profiling techniques provide a readily accessible platform for the delineation of complex rearrangements at the breakpoint level, thus allowing the study of the molecular mechanisms underlying such rearrangements. For recurrent as well as nonrecurrent alterations, it has been shown that genomic architectural features such as LCRs and smaller repetitive elements including Alu sequences are capable of mediating and/or stimulating the occurrence of deletions and duplications (Pentao et al. 1992; Chen et al. 1997; Shaw and Lupski 2005). LCRs in proximal 17p comprise more than 23% of the analyzed genomic sequence, which is approximately fourfold higher than might be expected based on virtual analysis of the entire human genome (Stankiewicz et al. 2003). It might be anticipated that the breakpoints of the uncommon nonrecurrent chromosomal aberrations as observed in this study occurred by chance alone within one of these LCRs. However, of five breakpoints that occurred within the proximal part of 17p, four were located within a LCR structure. The remaining ten breakpoints were located in the LCR-poor distal fragment of 17p. Thus, it appears that the location of the breakpoints is associated with local genome architecture, i.e, LCRs.
Based on the abundance of genomic architectural features in proximal 17p, it remains to be determined whether the two CCRs described in this study are representative for other CCRs. However, a comparable genomic complexity has been observed in other genomic regions. For instance, LCRs in 22q11.2 have been shown to be responsible for recurrent and nonreccurrent chromosome deletions, duplications, and translocations (Shaikh et al. 2001; Spiteri et al. 2003). Thus, it can be expected that additional complex rearrangements with genomic architecture playing a mediating role, will be identified. It is, however, noteworthy that chromosomes known to be rich in LCR structures, including chromosome 17 and 22, are not overrepresented in large series of CCRs reported (Chen et al. 2006). It could therefore be the case that the potential role of LCRs in generating CCRs is restricted to a number of exceptional cases, such as the cases presented here, or to CCRs affecting only one chromosome, as is the case for the CCR in patient 2.
The molecular characterization of the complex rearrangements enabled the identification of nine and eight breakpoints in the CCRs in patient 1 and 2, respectively. Of all these 17 breakpoints, four involved known LCRs being the middle SMS-REP/LCR17pB in patient 1 and the middle SMS-REP, LCR17pA and LCR17pC in patient 2. The remaining breakpoints did not reveal any significant similarity and/or any evidence for the presence of LCRs. The proximal breakpoint of duplication III in patient 2 was located within LCR17pA. This genomic region has previously been found to be involved in several other constitutional and evolutionary rearrangements, including t(4;19) in Gorilla gorilla (Stankiewicz et al. 2001a, b, 2003, 2004; Shaw et al. 2004; Lupski and Stankiewicz 2005; Yatsenko et al. 2005; Ou et al. 2006). The distal breakpoint of this duplication, however, was not located within a genomic segment sharing sequence homology with LCR17pA. Breakpoints of duplication IV were mapped within non-homologous LCR copies, middle SMS-REP and LCR17pC. The middle SMS-REP/LCR17pB was also insertion target of the MDLS region in patient 1. These observations suggest that other recombination mechanisms, such as nonhomologous end-joining (NHEJ), may have facilitated these CCRs. NHEJ may play a prominent role in nonrecurrent rearrangements as has been shown for PLP1 deletions and duplications (Inoue et al. 2002; Lee et al. 2006). It remains to be determined to what extent NHEJ is a mechanism for genomic rearrangements, since other potential replication-based mechanisms may occur with nonrecurrent rearrangements (Lee et al. 2006).
The detailed molecular information gathered on the breakpoints does, however, allow for speculation on the mechanism underlying CCR formation in our patients. We propose models based upon the principle of parsimony and the minimal amount of breaks required for formation of the CCRs. Although predisposing inversion(s) and translocations rendering susceptibility to rearrangements in the offspring similar to what is known for several genomic disorders have been excluded in the parental chromosomes (Axton 2006; Lupski 2006), alternative mechanisms of CCR formation cannot be fully excluded. The complex rearrangement observed in patient 1 includes nine breakpoints, with one insertion, two microdeletions, one microduplication, and one translocation. We propose that the breaks in der(17), 17p13.3, 17p12, and 17p11.2 as well as the break in chromosome 21q22.3 arose simultaneously. This resulted in the insertion of the MDLS region into the middle SMS-REP/LCR17pB block, loss of the subtelomeric region 17p13.3, and part of 17p12, and duplication of the CMT1A and SMS regions. Additionally, the duplicated 21q22.3 region was translocated to the 17p subtelomeric region (Fig. 6a), most likely by a telomere healing mechanism. The complex rearrangement in patient 2 includes eight breaks. For the most parsimonious mechanism, a single breakpoint is involved in both, the formation of, as well as the insertion of, the duplicated segment. Thus, for each duplication observed in patient 2, one of the breakpoints is involved in the formation of the duplication and provides at the same time the site for inserting the duplicated segment. We suggest that also in this case all breaks in chromosome 17 occurred concurrently, resulting in four interspersed directly orientated microduplications (Fig. 6b).
Fig. 6.
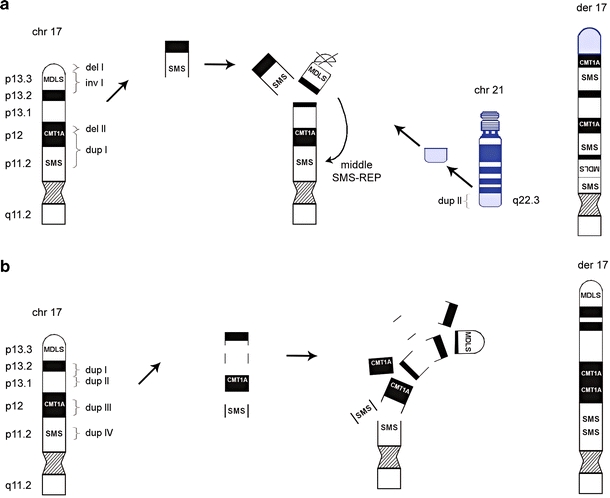
Schematic representation of the proposed mechanism for CCR formation in patient 1 and patient 2. a The complex rearrangement observed in patient 1 includes nine breakpoints, with one insertion, two microdeletions and two microduplications. We propose that the breaks in chromosome 17, p13.3, p12 and p11.2 as well as the break in chromosome 21q22.3 arose simultaneously. This resulted in the insertion of the MDLS region into the middle SMS-REP/LCR17pB block, loss of telomeric 17p and part of 17p12 and duplication of the CMT1A region. Additionally, the duplicated 21q22.3 fragment was translocated to the 17p subtelomeric region. b The complex rearrangement in patient 2 included eight breaks. We suggest that breaks in chromosome 17, p13.2, p13.1, p12 and p11.2 occurred at the same time, resulting in four interspersed directly orientated microduplications. Arrows do not represent a chronological order of events
In conclusion, the human genome contains many genomic architectural features such as LCRs, of which several have been identified at the breakpoints of (recurrent) chromosome rearrangements. As such, these chromosome rearrangements do not appear to represent random events but, instead, result from underlying genomic architectural features. Here, we present two patients with unique de novo CCRs, of which several breakpoints are located within LCRs. These results provide evidence for the first time that in addition to previously reported chromosome deletions, duplications, inversions, translocations, and marker chromosomes, genomic architectural features such as LCRs are mechanistically important for the origin of some CCRs.
Acknowledgments
We thank the patients and their parents for participation in this research, and Margie Withers, Irene Janssen, Saskia van der Velde-Visser, and Christel Beumer for excellent technical assistance. This work was supported by the Netherlands Genomics Initiative (050-72-407) (LELMV), the Netherlands Organisation for Health Research and Development (907-00-058; BBAdV and 912-04-047; JAV), the National Institute of Child Health and Development (PO1 HD39420) (JRL) and the Baylor College of Medicine Mental Retardation Research Center (HD24064).
References
- Astbury C, Christ LA, Aughton DJ, Cassidy SB, Fujimoto A, Pletcher BA, Schafer IA, Schwartz S (2004) Delineation of complex chromosomal rearrangements: evidence for increased complexity. Hum Genet 114:448–457 [DOI] [PubMed]
- Axton M (2006) Structural variants deconstruct the genome. Nat Genet 38:959 [DOI] [PubMed]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 11:1005–1017 [DOI] [PMC free article] [PubMed]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE (2002) Recent segmental duplications in the human genome. Science 297:1003–1007 [DOI] [PubMed]
- Bartels I, Starke H, Argyriou L, Sauter SM, Zoll B, Liehr T (2006) An exceptional complex chromosomal rearrangement (CCR) with eight breakpoints involving four chromosomes (1;3;9;14) in an azoospermic male with normal phenotype. Eur J Med Genet doi:10.1016/j.ejmg.2006.10.007 [DOI] [PubMed]
- Battisti C, Bonaglia MC, Giglio S, Anichini C, Pucci L, Dotti MT, Zuffardi O, Federico A (2003) De novo double translocation 3;13 and 4;8;18 in a patient with mental retardation and skeletal abnormalities. Am J Med Genet A 117:207–211 [DOI] [PubMed]
- Bi W, Yan J, Stankiewicz P, Park SS, Walz K, Boerkoel CF, Potocki L, Shaffer LG, Devriendt K, Nowaczyk MJ, Inoue K, Lupski JR (2002) Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res 12:713–728 [DOI] [PMC free article] [PubMed]
- Bi W, Park SS, Shaw CJ, Withers MA, Patel PI, Lupski JR (2003) Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am J Hum Genet 73:1302–1315 [DOI] [PMC free article] [PubMed]
- Borg K, Stankiewicz P, Bocian E, Kruczek A, Obersztyn E, Lupski JR, Mazurczak T (2005) Molecular analysis of a constitutional complex genome rearrangement with 11 breakpoints involving chromosomes 3, 11, 12, and 21 and a approximately 0.5-Mb submicroscopic deletion in a patient with mild mental retardation. Hum Genet 118:267–275 [DOI] [PubMed]
- Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, Lupski JR (1997) Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 17:154–163 [DOI] [PubMed]
- Chen CP, Chern SR, Lee CC, Lin CC, Li YC, Hsieh LJ, Chen WL, Wang W (2006) Prenatal diagnosis of de novo t(2;18;14)(q33.1;q12.2;q31.2), dup(5)(q34q34), del(7)(p21.1p21.1), and del(10)(q25.3q25.3) and a review of the prenatally ascertained de novo apparently balanced complex and multiple chromosomal rearrangements. Prenat Diagn 26:138–146 [DOI] [PubMed]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL (2005) Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med 7:422–432 [DOI] [PubMed]
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith AC, Dobyns WB, Ledbetter DH (1997) A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 6:147–155 [DOI] [PubMed]
- de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, Smeets D, Sistermans EA, Feuth T, van Ravenswaaij-Arts CM, Geurts van Kessel A, Schoenmakers EF, Brunner HG, Veltman JA (2005) Diagnostic genome profiling in mental retardation. Am J Hum Genet 77:606–616 [DOI] [PMC free article] [PubMed]
- Eichler EE (2001) Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet 17:661–669 [DOI] [PubMed]
- Gajecka M, Glotzbach CD, Shaffer LG (2006) Characterization of a complex rearrangement with interstitial deletions and inversion on human chromosome 1. Chromosome Res 14:277–282 [DOI] [PubMed]
- Inoue K, Dewar K, Katsanis N, Reiter LT, Lander ES, Devon KL, Wyman DW, Lupski JR, Birren B (2001) The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res 11:1018–1033 [DOI] [PMC free article] [PubMed]
- Inoue K, Osaka H, Thurston VC, Clarke JT, Yoneyama A, Rosenbarker L, Bird TD, Hodes ME, Shaffer LG, Lupski JR (2002) Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet 71:838–853 [DOI] [PMC free article] [PubMed]
- Johnston JJ, Walker RL, Davis S, Facio F, Turner JT, Bick DP, Daentl DL, Ellison JW, Meltzer PS, Biesecker LG (2007) Zoom-in comparative genomic hybridisation arrays for the characterisation of variable breakpoint contiguous gene syndromes. J Med Genet 44:e59 [DOI] [PMC free article] [PubMed]
- Karmous-Benailly H, Giuliano F, Massol C, Bloch C, De Ricaud D, Lambert JC, Perelman S (2006) Unbalanced inherited complex chromosome rearrangement involving chromosome 8, 10, 11 and 16 in a patient with congenital malformations and delayed development. Eur J Med Genet 49:431–438 [DOI] [PubMed]
- Knight SJ, Lese CM, Precht KS, Kuc J, Ning Y, Lucas S, Regan R, Brenan M, Nicod A, Lawrie NM, Cardy DL, Nguyen H, Hudson TJ, Riethman HC, Ledbetter DH, Flint J (2000) An optimized set of human telomere clones for studying telomere integrity and architecture. Am J Hum Genet 67:320–332 [DOI] [PMC free article] [PubMed]
- Kriek M, White SJ, Szuhai K, Knijnenburg J, van Ommen GJ, den Dunnen JT, Breuning MH (2006) Copy number variation in regions flanked (or unflanked) by duplicons among patients with developmental delay and/or congenital malformations; detection of reciprocal and partial Williams-Beuren duplications. Eur J Hum Genet 14:180–189 [DOI] [PubMed]
- Kuechler A, Ziegler M, Blank C, Rommel B, Bullerdiek J, Ahrens J, Claussen U, Liehr T (2005) A highly complex chromosomal rearrangement between five chromosomes in a healthy female diagnosed in preparation for intracytoplasmatic sperm injection. J Histochem Cytochem 53:355–357 [DOI] [PubMed]
- Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR (2006) Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet 15:2250–2265 [DOI] [PubMed]
- Lespinasse J, Bugge M, Rethore MO, North MO, Lundsteen C, Kirchhoff M (2004) De novo complex chromosomal rearrangements (CCR) involving chromosome 1, 5, and 6 resulting in microdeletion for 6q14 in a female carrier with psychotic disorder. Am J Med Genet A 128:199–203 [DOI] [PubMed]
- Lupski JR (1998) Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 14:417–422 [DOI] [PubMed]
- Lupski JR (2006) Genome structural variation and sporadic disease traits. Nat Genet 38:974–976 [DOI] [PubMed]
- Lupski JR, Stankiewicz P (2005) Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 1:e49 [DOI] [PMC free article] [PubMed]
- Lupski JR, Stankiewicz P (2006) (eds) Genomic disorders: the genomic basis of disease. Humana Press, Totowa [DOI] [PubMed]
- Ou Z, Jarmuz M, Sparagana SP, Michaud J, Decarie JC, Yatsenko SA, Nowakowska B, Furman P, Shaw CA, Shaffer LG, Lupski JR, Chinault AC, Cheung SW, Stankiewicz P (2006) Evidence for involvement of TRE-2 (USP6) oncogene, low-copy repeat and acrocentric heterochromatin in two families with chromosomal translocations. Hum Genet 120:227–237 [DOI] [PubMed]
- Pai GS, Thomas GH, Mahoney W, Migeon BR (1980) Complex chromosome rearrangements. Report of a new case and literature review. Clin Genet 18:436–444 [DOI] [PubMed]
- Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U (1992) The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot–Marie–Tooth disease type 1A. Nat Genet 1:159–165 [DOI] [PubMed]
- Patsalis PC, Evangelidou P, Charalambous S, Sismani C (2004) Fluorescence in situ hybridization characterization of apparently balanced translocation reveals cryptic complex chromosomal rearrangements with unexpected level of complexity. Eur J Hum Genet 12:647–653 [DOI] [PubMed]
- Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR (1992) Charcot–Marie–Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat Genet 2:292–300 [DOI] [PubMed]
- Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR (2000) Molecular mechanism for duplication 17p11.2—the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet 24:84–87 [DOI] [PubMed]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CMB, Eifert A, Friedman E, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR (2007) Characterization of the Potocki-Lupski syndrome [dup(17)(p11.2p11.2)] and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet 80:633–649 [DOI] [PMC free article] [PubMed]
- Rabiner LR (1989) A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE Inst Electr Electron Eng 77:257–286
- Reiter LT, Murakami T, Koeuth T, Pentao L, Muzny DM, Gibbs RA, Lupski JR (1996) A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet 12:288–297 [DOI] [PubMed]
- Shaikh TH, Kurahashi H, Emanuel BS (2001) Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet Med 3:6–13 [DOI] [PubMed]
- Shaw CJ, Lupski JR (2004) Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 13 Spec No 1:R57–R64 [DOI] [PubMed]
- Shaw CJ, Lupski JR (2005) Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet 116:1–7 [DOI] [PubMed]
- Shaw CJ, Bi W, Lupski JR (2002) Genetic proof of unequal meiotic crossovers in reciprocal deletion and duplication of 17p11.2. Am J Hum Genet 71:1072–1281 [DOI] [PMC free article] [PubMed]
- Shaw CJ, Withers MA, Lupski JR (2004) Uncommon Smith-Magenis syndrome deletions can be recurrent by utilizing alternate LCRs as homologous recombination substrates. Am J Hum Genet 75:75–81 [DOI] [PMC free article] [PubMed]
- Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, Firth H, Sanlaville D, Winter R, Colleaux L, Bobrow M, Carter NP (2004) Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet 41:241–248 [DOI] [PMC free article] [PubMed]
- Spiteri E, Babcock M, Kashork CD, Wakui K, Gogineni S, Lewis DA, Williams KM, Minoshima S, Sasaki T, Shimizu N, Potocki L, Pulijaal V, Shanske A, Shaffer LG, Morrow BE (2003) Frequent translocations occur between low copy repeats on chromosome 22q11.2 (LCR22s) and telomeric bands of partner chromosomes. Hum Mol Genet 12:1823–1837 [DOI] [PubMed]
- Stankiewicz P, Lupski JR (2002) Genome architecture, rearrangements and genomic disorders. Trends Genet 18:74–82 [DOI] [PubMed]
- Stankiewicz P, Parka SS, Holder SE, Waters CS, Palmer RW, Berend SA, Shaffer LG, Potocki L, Lupski JR (2001a) Trisomy 17p10–p12 resulting from a supernumerary marker chromosome derived from chromosome 17: molecular analysis and delineation of the phenotype. Clin Genet 60:336–344 [DOI] [PubMed]
- Stankiewicz P, Park S-S, Inoue K, Lupski JR (2001b) The evolutionary chromosome translocation 4;19 in Gorilla gorilla is associated with microduplication of the chromosome fragment syntenic to sequences surrounding the human proximal CMT1A-REP. Genome Res 1:1205–1210 [DOI] [PMC free article] [PubMed]
- Stankiewicz P, Shaw CJ, Dapper JD, Wakui K, ShaVer LG, Withers M, Elizondo L, Park S-S, Lupski JR (2003) Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet 72:1101–1116 [DOI] [PMC free article] [PubMed]
- Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR (2004) Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res 14:2209–2220 [DOI] [PMC free article] [PubMed]
- Thienpont B, Gewillig M, Fryns JP, Devriendt K, Vermeesch J (2006) Molecular cytogenetic characterization of a constitutional complex intrachromosomal 4q rearrangement in a patient with multiple congenital anomalies. Cytogenet Genome Res 114:338–341 [DOI] [PubMed]
- Vermeulen S, Menten B, Van Roy N, Van Limbergen H, De Paepe A, Mortier G, Speleman F (2004) Molecular cytogenetic analysis of complex chromosomal rearrangements in patients with mental retardation and congenital malformations: delineation of 7q21.11 breakpoints. Am J Med Genet A 124:10–18 [DOI] [PubMed]
- Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, Straatman H, van der Vliet W, Huys EH, van Rijk A, Smeets D, van Ravenswaaij-Arts CM, Knoers NV, van der Burgt I, de Jong PJ, Brunner HG, Geurts van Kessel A, Schoenmakers EF, Veltman JA (2003) Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 73:1261–1270 [DOI] [PMC free article] [PubMed]
- Weise A, Rittinger O, Starke H, Ziegler M, Claussen U, Liehr T (2003) De novo 9-break-event in one chromosome 21 combined with a microdeletion in 21q22.11 in a mentally retarded boy with short stature. Cytogenet Genome Res 103:14–16 [DOI] [PubMed]
- Yatsenko SA, Treadwell-Deering D, Krull K, Glaze D, Horz M, Stankiewicz P, Lupski JR, Potocki L (2005) Trisomy 17p10–p12 due to mosaic supernumerary marker chromosome: delineation of molecular breakpoints and clinical phenotype and comparison to other proximal 17p segmental duplications. Am J Med Genet 138A:175–180 [DOI] [PubMed]