

Abstract
A range of ab initio calculations were carried out on the axial and equatorial anomers of the model carbohydrate 2-ethoxy tetrahydropyran to evaluate the level of theory required to accurately evaluate the glycosyl dihedral angle and the anomeric ratio. Vacuum CCSD(T)/CBS extrapolations at the global minimum yield ΔE = Eequatorial − Eaxial = 1.42 kcal/mol. When corrected for solvent (by the IEFPCM model), zero-point vibrations and entropy, ΔG298 = 0.49 kcal/mol, in excellent agreement with the experimental value of 0.47 ± 0.3 kcal/mol. A new additivity scheme, the layered composite method (LCM), yields ΔE to within 0.1 kcal/mol of the CCSD(T)/CBS result at a fraction of the computer requirements. Anomeric ratios and one-dimensional torsional surfaces generated by LCM and the even more efficient MP2/cc-pVTZ level of theory are in excellent agreement, indicating that the latter is suitable for force-field parameterization of carbohydrates. Hartree-Fock and density functional theory differ from CCSD(T)/CBS for ΔE by ∼1 kcal/mol; they show similar deviations in torsional surfaces evaluated from LCM. A comparison of vacuum and solvent-corrected one- and two-dimensional torsional surfaces indicates the equatorial form of 2-ethoxy tetrahydropyran is more sensitive to solvent than the axial.
INTRODUCTION
The conformational flexibility of polysaccharides is largely determined by the glycosidic linkage. While steric factors approximately locate the global minimum for each linkage type (1), predicting the energies of the other torsional minima and the barriers is substantially more challenging (2,3). There are three reasons. The complex orbital interactions responsible for the anomeric (4–9) and exoanomeric effects (10,11) modulate the glycosyl torsional surface; the number of atoms and degrees of freedom in even a disaccharide is currently beyond the capabilities of high level ab initio methods; and, as follows from the strong interaction of carbohydrates and water, solvent effects must be considered.
This article explores the practical application of ab initio theories to the computation of the anomeric effects, and the mapping of glycosyl torsional surfaces. Calculations are based on the model carbohydrate 2-ethoxy tetrahydropyran (2-Eth-THP, Fig. 1). This compound exhibits both anomeric (the axial configuration is favored over the equatorial despite unfavorable steric interactions) and exoanomeric effects (the gauche form of the torsion angle φ shown in Fig. 1 is preferred). Because hydroxyls are not present, 2-Eth-THP can be described at the complete basis set (CCSD(T)/CBS) level of theory at the global minimum of each configuration (axial and equatorial; see Fig. 1). The ψ-angle is entirely defined by heavy atoms, making 2-Eth-THP a more realistic model of both disaccharides and glycolipids than the more commonly studied 2-methoxy tetrahydropyran (2-Me-THP).
FIGURE 1.

Schematic of axial (left) and equatorial (right) 2-ethoxy tetrahydropyran, including labels showing the glycosyl (φ) and exoglycosyl (ψ) torsion angles.
The essential strategy of this study is to proceed from the CCSD(T)/CBS treatment to progressively more approximate methods that can be applied to wider ranges of points. The first step is to develop a computationally efficient method that yields the energy difference between the axial (ax-2-Eth-THP) and equatorial (eq-2-Eth-THP) configurations with accuracy comparable to CCSD(T)/CBS but at reduced cost. This is denoted the layered composite method (LCM), and incorporates CCSD(T) energies into a layered additivity scheme. This differs from other composite methods that typically employ MP4 energies as a reference and QCISD(T) as the method to account for triples corrections (12–14).
The second step utilizes the LCM one-dimensional (1-D) torsional surfaces of φ and ψ of 2-Eth-THP as targets for a variety of methods because it is presently not feasible to calculate these surfaces at the CCSD(T)/CBS limit. This yields a ranking of methods, and speaks to the practical question of whether a method must yield the correct anomeric ratio when only the relative conformational energies of particular anomers are required. Solvent effects are included at selected levels of theory via implicit models (15–19). This not only affords comparisons to experiment but also lends insight to the differential responses of the axial and equatorial anomers to nonpolar and aqueous environments.
The Methods section outlines the ab initio approaches employed in this study, and describes the layered composite method (LCM) in detail. The Results and Discussion section consists of three subsections. Anomeric Ratios begins with the determination of the optimal LCM combination based on comparison with CCSD(T)/CBS results previously calculated for 2-Me-THP (20) and presently calculated for 2-Eth-THP. Minimum energy (ΔE), zero-point corrected energy (ΔE0), thermally corrected (ΔH298), and free energy (ΔG298) differences are calculated for 2-Eth-THP in vacuum and solvent at various levels of theory, and the deficiencies of some treatments are discussed (20–22). The Glycosyl Torsional Potentials subsection reports two-dimensional (2-D) φ,ψ maps for the 2-Eth-THP glycosyl linkage calculated at the MP2/6-31G(d) level, and proceeds to the 1-D torsional surfaces for the LCM and other relevant levels of theory. Lastly, Effects of Solvation investigates the solvent effects on the 1-D and 2-D surfaces, and the final section presents the Conclusions.
METHODS
Computations were performed using either the Gaussian 03 (23), Q-Chem 3.0 (24), or Molpro 2002 (25) software packages. Full 2-D surfaces were created by rotating around the glycosyl torsion angles φ and ψ in 15° increments and performing optimizations for a total of 576 points on each surface. Using these two surfaces as guides, 1-D glycosyl torsional surfaces of the φ and ψ angles in ax-2-Eth-THP and eq-2-Eth-THP were generated.
In the full 2-D surfaces the selected dihedrals were constrained and the remainder of the molecule optimized at the MP2/6-31G(d) level of theory. Solvation corrections to the φ,ψ surfaces were performed as single point energy corrections and employed the IPCM implicit solvation model (19). RIMP2 and Local MP2 calculations were performed with Q-Chem 3.0. The calculation of ΔG298 was performed after optimizing the global minima of the respective 2-D surfaces at the MP2/cc-pVTZ level of theory and computing the normal modes of vibration at 298.15 K.
One-dimensional surfaces were refined with constrained optimizations at the MP2/cc-pVTZ level of theory. Solvation corrections made to the 1-D φ and ψ surfaces employed the more robust and efficient IEFPCM solvation model (15–18) and were again performed at the MP2/cc-pVTZ level of theory. The corrections performed on the 1-D surfaces employed a dielectric constant of 78.4 to model the effects of water as a solvent. The solvation correction to the anomeric ratio was performed using a dielectric of 10.0 to approximate experimental conditions (26). More explicitly, since the experimental free energy difference was determined in neat liquid 2-Eth-THP we approximated the dielectric constant by using the default radius (2.560 Å) and ε∞ (1.971) of tetrahydrofuran while increasing the dielectric constant (ε) from 7.58 (ε of tetrahydrofuran) to 10.0, thereby effectively increasing the polarity.
We devised and performed layered composite method calculations, LCM(X,Y) where X = 2,3 and Y = 3,4,5. All LCM calculations employed the Dunning correlation consistent basis sets as denoted by cc-pVXZ (X = D,T,Q,5) (27). The LCM(X,Y) procedure can be considered a combination of G3 (12) and ONIOM (28) theories where X refers to the number of layers (i.e., levels of theory) and Y refers to the largest cardinal number of the Dunning basis sets.
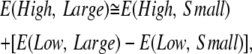 |
(1) |
where High = CCSD(T), Large = cc-pVTZ, Low = MP2, and Small = cc-pVDZ. This leads to the following definition of LCM(2,3):
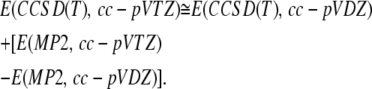 |
(2) |
The LCM(2,3) has a similar form to the G3(MP2)-RAD procedure of Radom and co-workers (29,30), although it uses Dunning basis sets and does not include a high-level correction. In addition to a two-layered composite energy, a three-layered composite energy was formulated using the definition
 |
(3) |
where High = CCSD(T), Intermediate = MP2, Low = HF, Large = cc-pVQZ, Medium = cc-pVTZ, and Small = cc-pVDZ. This leads to LCM(3,4) being defined as
 |
(4) |
All structures were optimized at the MP2/cc-pVTZ level of theory.
To validate the LCM(X,Y) procedure, we performed complete basis set (CBS) extrapolations (31,32). The CBS limit of the SCF total energy was determined by fitting the cc-pVXZ (X = T,Q,5) calculations to the three-parameter function:
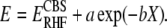 |
(5) |
The CBS limit of the MP2 correlation energy was obtained using a simplified version of the two-point Helgaker extrapolation (33),
 |
(6) |
where A = 5 (the cardinal number of cc-pV5Z) and B = 4 (the cardinal number of cc-pVQZ). To compute the CCSD(T)/CBS limit, Eq. 6 was again employed. The CCSD(T)(D,T) and MP2(D,T) results yield
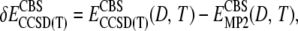 |
(7) |
which leads to the final CBS energy of
 |
(8) |
This extrapolated energy was only computed for the grid-constrained global minimum of the ax-2-Eth-THP and eq-2-Eth-THP anomers.
RESULTS AND DISCUSSION
Anomeric ratios
Many groups have sought to accurately compute anomeric ratios of carbohydrates with most employing Hartree-Fock (HF) or density functional theory (DFT) (34–37). The reasons for the choices in level of theory are twofold: one, until recently efficient computational hardware and software did not exist to perform high level calculations on such large systems; and, two, it has been noted by several groups, but initially by Salzner and Schleyer (21) and then by Tvaroska and Carver (22,35), that Möller Plesset series methods (i.e., MP2 and MP4) significantly overestimate anomeric ratios of model carbohydrates. For example, Tvaroska and Carver reported a 2.23 kcal/mol anomeric energy difference at the MP2/6-31G* level of theory compared to a 1.47 kcal/mol result at the HF/6-31G* level. A very recent article by Weldon et al. (20) investigated this problem by performing CCSD(T) with complete basis set (CBS) extrapolations and concluded that HF and DFT overestimated the equatorial stabilization by 0.7 and 0.4 kcal/mol, respectively (20). This is somewhat at odds with previous studies (21,22,35) that have concluded that MP2 was erroneous. However, these studies were limited to small basis sets and lower levels of theory.
As a first step toward understanding the influence of applied level of theory on the anomeric ratio and other conformational energies, the proposed extrapolations, LCM(X,Y) X = 2,3 and Y = 3,4,5, were applied to 2-Me-THP and compared to the CCSD(T)/CBS anomeric energy difference reported by Weldon et al. (20) (1.27 kcal/mol). The two-layered result (LCM(2,3)), which is obviously deficient in the n-particle direction (i.e., basis set), overestimates the energy difference by 0.16 kcal/mol (1.43 kcal/mol). In contrast, the three-layered result (LCM(3,4)), agrees almost exactly with the much more expensive and tedious CBS extrapolation procedure; i.e., ΔE = 1.26 kcal/mol, which is only 0.01 kcal/mol different from the result of Weldon et al.
To further demonstrate the accuracy of the layering procedure, Table 1 compares the results of full CBS extrapolations on the global minima of ax-2-Eth-THP and eq-2-Eth-THP with LCM (2,3), (2,4), (2,5), (3,4) and (3,5). The LCM results are within 0.1 kcal/mol of the much more costly CBS extrapolations in all cases. For example, LCM(3,4) is 100 times faster than the CCSD(T)/CBS procedure, and requires one-20th of the system resources. This is because LCM(3,4) employs only a CCSD(T)/cc-pVDZ energy, while the CCSD(T)/CBS requires a CCSD(T)/cc-pVTZ energy. Consequently, energies along the 1-D torsion were generated at the LCM(3,4) level, and served as a standard for more approximate methods.
TABLE 1.
Anomeric energy differences (ΔE) defined as Eequatorial − Eaxial
| Level of theory | ΔE (ΔE0, ΔH298, ΔG298) |
|---|---|
| HF/cc-pVDZ//HF/cc-pVDZ | 1.28 (0.98, 1.03, 0.92) |
| HF/cc-pVTZ//HF/cc-pVTZ | 0.64 (0.35, 0.44, 0.27) |
| HF/cc-pVTZ/IEFPCM//HF/cc-pVTZ | −0.11 [−0.40, −0.31, −0.48] |
| HF/cc-pVTZ/IEFPCM//HF/cc-pVTZ/IEFPCM | −0.19 [−0.48, −0.39, −0.56] |
| HF/cc-pVQZ//HF/cc-pVQZ | 0.55 (0.28, 0.32, 0.26) |
| MP2/6-31G(d)//MP2/6-31G(d) | 2.30 (2.02, 2.06, 2.01) |
| MP2/cc-pVDZ//MP2/cc-pVDZ | 2.02 (1.73, 1.78, 1.68) |
| MP2/cc-pVTZ//MP2/cc-pVDZ | 2.10 (1.80, 1.86, 1.69) |
| MP2/cc-pVTZ//MP2/cc-pVTZ | 1.60 (1.36, 1.39, 1.41) |
| MP2/cc-pVQZ//MP2/cc-pVTZ | 1.54 [1.31, 1.33, 1.35] |
| MP2/cc-pV5Z//MP2/cc-pVTZ | 1.54 [1.31, 1.33, 1.35] |
| MP2/cc-pVTZ/IEFPCM//MP2/cc-pVTZ | 0.96 [0.72, 0.75, 0.77] |
| MP2/cc-pVTZ/IEFPCM//MP2/cc-pVTZ/IEFPCM | 0.75 [0.51, 0.54, 0.56] |
| LMP2(TRIM)/cc-pVTZ//MP2/cc-pVTZ | 1.63 [1.39, 1.42, 1.44] |
| LMP2(DIM)/cc-pVTZ//MP2/cc-pVTZ | 1.30 [1.06, 1.09, 1.11] |
| RIMP2/cc-pVTZ//RIMP2/cc-pVTZ | 1.51 [1.27, 1.30, 1.32] |
| CCSD(T)/cc-pVDZ//MP2/cc-pVTZ | 2.00 [1.76, 1.78, 1.58] |
| CCSD(T)/cc-pVTZ//MP2/cc-pVTZ | 1.47 [1.23, 1.26, 1.28] |
| CCSD(T)/CBS//MP2/cc-pVTZ | 1.42 [1.19, 1.21, 1.23] |
| CCSD(T)/CBS/IEFPCM//MP2/cc-pVTZa | 0.68 [0.44, 0.47, 0.49] |
| B3LYP/6-311+G(d,p)//B3LYP/6-311+G(d,p) | 0.44 (0.55, 0.43, 0.96) |
| B3LYP/cc-pVTZ//B3LYP/cc-pVTZ | 0.63 (0.45, 0.45, 0.58) |
| B3LYP/cc-pVTZ/IEFPCM//B3LYP/cc-pVTZ | −0.07 [−0.25, −0.25,−0.12] |
| B3LYP/cc-pVTZ/IEFPCM//B3LYP/cc-pVTZ/IEFPCM | −0.13 [−0.31, −0.31,−0.18] |
| LCM(2,3) | 1.49 [1.25, 1.28, 1.30] |
| LCM(2,4) | 1.41 [1.17, 1.20, 1.22] |
| LCM(2,5) | 1.41 [1.17, 1.20, 1.22] |
| LCM(3,4) | 1.35 [1.11, 1.14, 1.16] |
| LCM(3,5) | 1.39 [1.15, 1.18, 1.20] |
| LCM(3,5)/IEFPCM* | 0.64 [0.41, 0.43, 0.45] |
| Experimental (26) | 0.47 ± 0.3 |
A positive value indicates the axial anomer is more stable than the equatorial form. Values in the parentheses are zero-point corrected (ΔE0), enthalpy (ΔH298), and free energy (ΔG298) differences, respectively. Values in brackets were not computed but instead were projected using the MP2/cc-pVTZ vibrational information.
Applied a solvation correction based on the neat liquid dielectric (0.75 kcal/mol), computed at the MP2/cc-pVTZ/IEFPCM//MP2/cc-pVTZ/IEFPCM level of theory. Experimental value is the free energy difference (ΔGexp) in the neat fluid.
While the above results indicate that the proposed composite methods can yield very good agreement with CBS results, additional calculations were undertaken to determine if even more computationally accessible methods could also yield satisfactory agreement. The results confirm that HF overstabilizes equatorial conformations of THP derivatives. The ΔE at the MP2/cc-pVTZ level shows that ax-2-Eth-THP is preferred by 1.60 kcal/mol, which predicts 93.7% axial population. On the other hand, the experimental free energy difference (ΔGexp) shows a 68% (ΔGexp = 0.47 ± 0.3 kcal/mol) axial population for neat 2-Eth-THP (26). However, both HF/cc-pVQZ (ΔE = 0.55 kcal/mol) and HF/cc-pVTZ (ΔE = 0.64 kcal/mol) are easily within the error of the original experiment and would lead to equatorial populations of 71.7% and 74.7%, respectively. These results are in accord with those of Weldon et al. (20), who show that ΔE is not directly comparable to ΔH298 or ΔG298 for 2-hydroxy- and 2-methoxy-THP. To explore this point, the enthalpic and entropic contributions, computed at 298.18 K, at the highest level of theory reasonable (MP2/cc-pVTZ) were investigated to determine their effect on the relative energies. After adding entropic corrections to the energy difference (ΔE → ΔG298) at the MP2/cc-pVTZ//MP2/cc-pVTZ level, the free energy difference decreased to 1.41 kcal/mol (91.5%) in favor of the axial conformer. This again is a substantial overestimation of the axial population in solution. Applying this correction to the LCM(3,4) extrapolation results in a ΔG298 of 1.16 kcal/mol (87.6%), which is still overly favorable to the axial conformer.
Free energy calculations, even in the gas phase, show an increased entropy for the equatorial conformer, lowering the ΔG298. These results indicate that the anomeric effect is a balance of both enthalpic and entropic effects, and when attempting to use correlated levels of theory to accurately compute anomeric ratios of carbohydrates it is essential to include the thermal corrections. The anomeric energy difference computed at the CCSD(T)/cc-pVTZ//MP2/cc-pVTZ level only corrected the MP2 energy gap by 0.13 kcal/mol. This parallels the results of Weldon et al. (20), who demonstrated that increasing level of theory from MP2 to CCSD(T) is relatively basis-set-independent and accounts for only ∼0.1–0.2 kcal/mol. This is one of the properties that makes additivity-based extrapolations possible.
Based on the present results, the seemingly correct results of HF may be attributed to three major factors: overstabilization of the equatorial anomer; cancellation of errors between a lack of correlation treatment and a lack of solvation effects; and neglect of thermal (enthalpic and/or entropic) effects. These observations allow us to conclude that the use of HF to obtain anomeric energy differences is not appropriate.
At this stage, it was deemed appropriate to determine the contribution of solvation effects to the axial and equatorial relative energy. To account for these effects, the anomeric ratio was computed at both the MP2/cc-pVTZ and MP2/cc-pVTZ/IEFPCM levels of theory. Performing these calculations yielded solvation corrections (ΔEgas − ΔEsolvent) of 0.64 (grid-constrained) and 0.75 kcal/mol (fully relaxed). Fully relaxed solvation corrections were also computed at the HF/cc-pVTZ and B3LYP/cc-pVTZ levels of theory to examine the methodological dependence of this effect. There is good agreement between the MP2 correction (0.75 kcal/mol) and HF (0.86 kcal/mol) and B3LYP (0.74 kcal/mol). In addition, the validity of a static solvation correction was examined by performing HF/cc-pVTZ/IEFPCM//HF/cc-pVTZ and B3LYP/cc-pVTZ/IEFPCM//B3LYP/cc-pVTZ calculations. This resulted in solvation corrections of 0.79 and 0.68 kcal/mol for the HF and B3LYP levels, respectively. This confirmed relatively small minima shifts from the fully relaxed IEFPCM optimizations and validates our use of reaction field corrections at the gas-phase optimized structures. From this point forward, the MP2 fully relaxed solvation correction (0.75 kcal/mol) will be employed. Applying the MP2 solvation correction to LCM(3,5) and CCSD(T)/CBS (Table 1) resulted in ΔG298 values of 0.45 and 0.49 kcal/mol, respectively. The results are in excellent agreement with the experimental value of 0.47 ± 0.3 (26). From this analysis it is clear that, even very high-level, vacuum-based ab initio methods (i.e., CCSD(T) extrapolations, LCM(X,Y), etc.) cannot reproduce the ΔGexp of this model carbohydrate. However, even a simple solvation correction can yield good agreement with experiment.
Given the popularity of density functional theory, we also computed the anomeric ratio employing the most widely used functional (B3LYP) and a common Pople basis set (6-311+G(d,p)). Optimizing both the axial and equatorial conformers at this level of theory led to a 0.44 kcal/mol anomeric energy difference. As previously noted, B3LYP, in addition to HF, also suffers from overstabilization of the equatorial anomer. As reported by Weldon et al. (20), this effect, using DFT, accounts for an ∼0.4 kcal/mol lowering of ΔE. However, using the 6-311+G(d,p) basis set, the difference is nearly 1 kcal/mol. After confirming that these were minima on their relative potential energy surfaces, we added the entropic corrections and arrived at a 0.96 kcal/mol ΔG298, which is in better agreement with the ∼0.4 kcal/mol value observed for 2-methoxy and 2-hydroxy THP.
To further investigate the equatorial overstabilization of HF and DFT we examined the trends in going from ΔE → ΔG298 and vacuum → solvated and compared this to MP2 results. When examining the ΔE alone, HF and DFT appear to be doing very well; however, when entropic effects are included, free energy differences are underestimated. When solvation corrections are incorporated, which bring MP2 results to within the experimental error, HF and DFT invert the relative stability as compared to experiment. HF predicts the equatorial conformer to be 0.19 kcal/mol more stable than the axial, and B3LYP predicts 0.13 kcal/mol equatorial favorability. This result arises because both entropic effects and solvation effects stabilize the equatorial form. A comparison of the −0.19 and −0.13 free energy differences to the experimental value of 0.47 yields equatorial overstabilization of 0.66 and 0.60 kcal/mol, respectively, for HF and B3LYP. These are slightly larger than the 0.4 kcal/mol predicted by Weldon and co-workers.
Glycosyl torsional potentials
2-D φ,ψ maps
We performed constrained (φ, ψ) 2-D surface optimizations at the MP2/6-31G(d) level for the glycosyl linkage of both ax-2-Eth-THP and eq-2-Eth-THP. These surfaces (Fig. 2), which were created to identify all possible minima and maxima, show the respective conformers to have multiple broad minima located near the φ equals 60° and 285° regions. An additional local minimum appears on the eq-2-Eth-THP surface (Fig. 2, right) at the 60°,195° (φ, ψ) point. This broad low energy region, which is purely an exocyclic effect, comes about because of stabilization of the exocyclic oxygen lone-pair by the remaining portion of the exocyclic substituent (i.e., ethyl moiety). At the 60°,195° minimum, the exocyclic lone-pair orients to maximize hyperconjugation with the adjacent C-H anti-bonding orbitals. This effect also contributes to the stabilization of the global minimum of 300°,195°.
FIGURE 2.

Relaxed φ,ψ surfaces for the axial (left) and equatorial (right) 2-Eth-THP glycosyl linkage computed at the MP2/6-31G(d) level of theory, with vacuum (top) and solvent (IPCM) (bottom).
1-D φ surfaces
To our knowledge, no high-level ab initio study has been published examining the glycosyl torsional potentials of ax-2-Eth-THP and eq-2-Eth-THP. Most researchers have used either HF or DFT to map these potentials because of problems associated with computing the anomeric effect and the cost of obtaining full 1-D surfaces at higher levels of theory. In the past, 2-Me-THP has typically been the largest model employed to study anomeric and exoanomeric effects and to parameterize modern carbohydrate force fields (37–40). Mapping the φ- and ψ-torsional potentials of ax-2-Eth-THP and eq-2-Eth-THP will allow for a comparison with 2-Me-THP.
Booth et al. (41) determined experimentally the three conformations that are most highly populated on the φ-surfaces of axial and equatorial 2-Me-THP. These correspond to the φ = 60° region on the ax-2-Eth-THP surface and the φ = 60° and 300° regions on the eq-2-Eth-THP surface (Fig. 3). Our results confirm this assertion.
FIGURE 3.

1-D Axial (top) and equatorial (bottom) φ-surfaces.
Examining the φ-surface of ax-2-Eth-THP (Table 2, Fig. 3, top) in detail reveals a deep, well-defined minimum at 60° at all levels of theory. HF and B3LYP flatten this portion of the surface making 60° and 75° nearly isoenergetic. This result should have little effect on the populations, as there are no other well-defined minima that would contribute significantly while sampling this surface.
TABLE 2.
Relative energies of the φ-torsional potentials for axial and equatorial 2-Eth-THP
| Axial φ-torsional profile
|
Equatorial φ-torsional profile
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Level of theory | 60° | 135° | 150° | 240° | 0° | 60° | 135° | 210° | 300° |
| HF/cc-pVDZ//HF/cc-pVDZ | 0.00 | 3.43 | 3.57 | 10.62 | 4.57 | 2.87 | 8.24 | 4.40 | 0.00 |
| HF/cc-pVTZ//HF/cc-pVTZ | 0.06 | 3.05 | 3.17 | 10.14 | 4.85 | 2.93 | 7.78 | 3.91 | 0.00 |
| HF/cc-pVTZ//HF/cc-pVTZ (ΔG) | 0.22 | 3.05 | 2.48 | 10.21 | 5.08 | 3.39 | 8.02 | 3.17 | 0.31 |
| HF/cc-pVQZ//HF/cc-pVQZ | 0.07 | 2.94 | 3.06 | 9.95 | 4.93 | 2.94 | 7.63 | 3.78 | 0.01 |
| MP2/cc-pVDZ//MP2/cc-pVTZ | 0.00 | 4.28 | 4.19 | 9.94 | 5.34 | 2.50 | 7.71 | 4.35 | 0.00 |
| MP2/cc-pVTZ//MP2/cc-pVTZ | 0.00 | 3.90 | 3.74 | 9.09 | 5.39 | 2.42 | 7.14 | 4.01 | 0.00 |
| CCSD(T)/cc-pVDZ//MP2/cc-pVTZ | 0.00 | 4.16 | 4.08 | 9.61 | 5.20 | 2.45 | 7.57 | 4.33 | 0.00 |
| LCM(2,3) | 0.00 | 3.79 | 3.63 | 8.76 | 5.25 | 2.37 | 7.00 | 3.99 | 0.00 |
| LCM(3,4) | 0.00 | 3.68 | 3.53 | 8.57 | 5.32 | 2.37 | 6.83 | 3.84 | 0.00 |
| B3LYP/6-311+G(d,p)//B3LYP/6-311+G(d,p) | 0.00 | 3.33 | 3.39 | 8.82 | 5.20 | 2.69 | 7.36 | 4.10 | 0.00 |
| B3LYP/6-311+G(d,p)//B3LYP/6-311+G(d,p) (ΔG) | 0.04 | 3.83 | 2.90 | 9.43 | 5.69 | 2.63 | 7.92 | 3.33 | 0.14 |
| MP2/6-31G(d)//MP2/6-31G(d) | 0.00 | 4.51 | 3.99 | 10.39 | 5.28 | 2.73 | 7.96 | 4.56 | 0.00 |
| MP2/cc-pVTZ/IEFPCM//MP2/cc-pVTZ | 0.00 | 3.44 | 2.98 | 7.34 | 6.28 | 2.40 | 5.72 | 2.64 | 0.21 |
All values in kcal/mol. Entries with nonzero values at φ = 60° or 300° have energy minima slightly shifted from these values.
All levels of theory agree on the shape of the φ-surface global minimum of eq-2-Eth-THP (Fig. 3, bottom). The minimum, which is nearly isoenergetic at φ-values of 285° and 300°, is fairly broad. Another minimum, which corresponds to Booth's prediction of a second participating conformation, appears at φ = 60°. The LCM(2,3) and LCM(3,4) results are ∼2.3 kcal/mol above the global minimum. This is nearly 1 kBT lower in energy than the HF results (2.9 kcal/mol) and ∼0.5 kBT lower than the B3LYP and MP2/6-31G(d) results (2.7 kcal/mol). These differences cannot be ignored as they would likely lead to significant population differences when sampling on their respective surfaces. At the barrier of this transition, HF again does not agree with LCM, whereas B3LYP and MP2/6-31G(d) behave more favorably. HF/cc-pVDZ underestimates the barrier by 0.7 kcal/mol, while the larger basis sets (HF/cc-pVXZ X = T,Q) more closely match the LCM(3,4) results (Table 2). These results clearly show that using a deficient double-ζ quality basis set will lead to significant differences not only in populations but also in predicted rates of conversion between relevant states. In contrast, the MP2/cc-pVTZ surfaces are in excellent agreement with LCM(3,4) φ-surfaces with the largest deviations being observed in the vicinity of the inaccessible high-energy region (φ = 285°) on the axial surface (Fig. 3, top).
To examine the effects of entropy on the 1-D φ surfaces, frequencies (and thus ΔG298) were computed at the HF/cc-pVTZ and B3LYP/6-311+G(d,p) levels employing the extreme points listed in Table 2. At both levels of theory, the ax-2-Eth-THP free energy surface shows a clearly defined minimum at the 150°. The ΔE between 60° and 150° is 3.11 and 3.39 kcal/mol for HF and B3LYP, respectively. However, when entropy is accounted for, these free energy differences (ΔG298) drop to 2.49 and 2.86 kcal/mol. In addition, the barrier that separates these two points along the surface increases from 3.33 to 3.83 kcal/mol at the B3LYP level while the HF barrier remains virtually unchanged. Although the MP2 frequencies are currently too expensive to perform the same analysis it is expected that similar trends would be observed thus deepening the 150° minima and increasing the barrier that separates it from the global minimum at 60°.
An analogous effect is observed when examining ΔE → ΔG298 on the eq-2-Eth-THP φ-surface. For example, the energy surface shows a shoulder region located at 210° that appears to be a very shallow minimum at the LCM(3,4) level of theory; however, this does not exist with HF and B3LYP. When transitioning from energy to free energy, the ΔG298 between the global minimum (300°) and 210° drops from 3.91 to 3.17 kcal/mol at HF/cc-pVTZ and 4.10 to 3.33 kcal/mol at B3LYP/6-311+G(d,p). More importantly, an increase occurs at 225° that creates a true barrier on the B3LYP (3.90 → 4.27 kcal/mol) free energy surface; however, the barrier actually decreases on the HF surface, but does so less than the decrease at the 210° minimum.
1-D ψ-surfaces
Next, ψ-surfaces (Fig. 4, Table 3) of ax-2-Eth-THP and eq-2-Eth-THP were computed keeping their respective φ-values (60° for ax-2-Eth-THP and 300° for eq-2-Eth-THP) fixed at their lowest energy values as determined from the full MP2/6-31G(d) 2-D surface maps. In contrast to the φ-surfaces, which are governed by fairly deep minima, the ψ-potentials have one well-defined minimum and two broad shallow minima on either side of the global minimum.
FIGURE 4.

1-D Axial (top) and equatorial (bottom) ψ-surfaces.
TABLE 3.
Relative energies of the ψ-torsional potentials for axial and equatorial 2-Eth-THP
| Axial ψ-torsional profile
|
Equatorial ψ-torsional profile
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Level of theory | 75° | 120° | 180° | 240° | 270° | 105° | 135° | 180° | 240° | 270° |
| HF/cc-pVDZ//HF/cc-pVDZ | 1.64 | 1.34 | 0.00 | 2.33 | 1.75 | 1.09 | 1.20 | 0.00 | 1.56 | 1.54 |
| HF/cc-pVTZ//HF/cc-pVTZ | 1.74 | 1.52 | 0.00 | 2.37 | 2.00 | 1.50 | 1.43 | 0.00 | 1.77 | 1.68 |
| HF/cc-pVTZ//HF/cc-pVTZ (ΔG) | 2.43 | 1.95 | 0.00 | 2.94 | 2.01 | 1.10 | 2.01 | 0.00 | 2.29 | 1.51 |
| HF/cc-pVQZ//HF/cc-pVQZ | 1.76 | 1.56 | 0.00 | 2.38 | 2.07 | 1.60 | 1.50 | 0.00 | 1.80 | 1.69 |
| MP2/cc-pVDZ//MP2/cc-pVTZ | 1.11 | 1.38 | 0.00 | 1.92 | 1.10 | 0.44 | 1.05 | 0.00 | 1.49 | 0.96 |
| MP2/cc-pVTZ//MP2/cc-pVTZ | 1.13 | 1.60 | 0.00 | 1.87 | 1.40 | 0.83 | 1.24 | 0.01 | 1.69 | 1.16 |
| CCSD(T)/cc-pVDZ//MP2/cc-pVTZ | 1.06 | 1.29 | 0.00 | 1.79 | 1.01 | 0.43 | 1.04 | 0.02 | 1.42 | 0.91 |
| LCM(2,3) | 1.08 | 1.51 | 0.00 | 1.74 | 1.31 | 0.82 | 1.23 | 0.03 | 1.62 | 1.11 |
| LCM(3,4) | 1.10 | 1.55 | 0.00 | 1.74 | 1.39 | 0.92 | 1.31 | 0.04 | 1.66 | 1.12 |
| B3LYP/6-311+G(d,p)//B3LYP/6-311+G(d,p) | 1.28 | 1.48 | 0.00 | 1.96 | 1.59 | 1.31 | 1.38 | 0.00 | 1.71 | 1.40 |
| B3LYP/6-311+G(d,p)//B3LYP/6-311+G(d,p) (ΔG) | 1.16 | 2.27 | 0.00 | 2.87 | 1.86 | 1.22 | 2.20 | 0.00 | 2.44 | 1.34 |
| MP2/6-31G(d)//MP2/6-31G(d) | 1.16 | 1.39 | 0.00 | 1.92 | 1.32 | 0.54 | 1.14 | 0.05 | 1.55 | 1.13 |
| MP2/cc-pVTZ/IEFPCM//MP2/cc-pVTZ | 1.04 | 1.51 | 0.02 | 1.77 | 1.29 | 1.27 | 1.46 | 0.08 | 1.60 | 0.70 |
All values in kcal/mol. Entries with nonzero values at ψ = 120° or 180° have energy minima slightly shifted from these values.
The axial ψ-surface global minimum is very broad and is centered between 150° and 195°. The energetic variation of this region is maximized at the 150° position and is only ∼0.5 kcal/mol higher in energy than the global minimum, which is positioned at 180°. On each side of the global minimum there is a shallow minima that lies ∼1 kcal/mol higher in energy. The local minimum at ψ = 75°–90° is extremely shallow, and is not even found at HF levels of theory. All correlated levels confirm this minimum, but predict a depth of only 0.2–0.4 kcal/mol.
At ψ = 270° another shallow minimum occurs. The LCM(3,4) extrapolation predicts this to be 1.39 kcal/mol above the global minimum with a barrier between them of only 1.74 kcal/mol leaving a well-depth of 0.35 kcal/mol. At the HF levels of theory we again observe sporadic behavior with HF/cc-pVXZ (X = D,T,Q; Table 3). These results are nearly a full kBT higher in energy than the extrapolated results and would lead to significant population differences if using these surfaces for simulations. On the other hand, the rather cost-effective MP2/cc-pVTZ level of theory reproduces the higher level curves with an average error of <0.05 kcal/mol.
Similar to the axial surface the equatorial ψ-surface has its broad global minimum spanning the 180°–195° region. Results from the LCM(3,4) calculations show a local minimum (ψ = 105°) lying 0.92 kcal/mol above the global minimum with a barrier between them of only 0.39 kcal/mol. HF results, which show no minimum at ψ = 105°, lie significantly higher in energy than the LCM results. Although B3LYP agrees with LCM results slightly better than HF, the barrier between this point and the global minimum is only 0.07 kcal/mol and is questionable at this level of theory.
An additional minimum exists (ψ = 270°) on the equatorial surface. This minimum is not predicted by HF, and is only weakly predicted by B3LYP with only a 0.31 kcal/mol barrier and total energy difference of 1.40 kcal/mol. However, LCM(3,4) predicts ΔE = 1.12 kcal/mol with a barrier going to the global minimum of 0.54 kcal/mol.
Analysis of the ψ-surfaces shows that both the axial and equatorial potentials are very soft and this degree of freedom will be quite floppy. Therefore, it is essential that an adequate level of theory be chosen to properly characterize it. The axial and equatorial φ-surfaces are not nearly as soft and thus it has been possible to use lower levels of theory. However, even on well-defined surfaces such as equatorial φ (Fig. 3, bottom), significant differences can be observed by using too low a level of theory (i.e., HF). For example, there is nearly a full kBT energy difference between our LCM results and the best HF level when comparing ΔE of φ = 300° and 60° on the eq-2-Eth-THP surface. In contrast to this behavior, the MP2/cc-pVTZ surfaces closely mirror the LCM(3,4) results with an average error, at the extrema, of <0.06 kcal/mol.
Examining the ΔG298 values on the 1-D ax-2-Eth-THP ψ surface (at the HF/cc-pVTZ and B3LYP/6-311+G(d,p)) reveals that entropy tends to destabilize the extreme points that we examined. The only exception to this was the 75° point on the B3LYP surface, which was stabilized slightly (0.12 kcal/mol). Although entropy destabilizes the extreme points, the minima (75° and 240°) actually become better defined. This is because the transition points along the path are destabilized more than the minima. For example, on the B3LYP surface, the 240° barrier increases by 0.91 kcal/mol. whereas the 270° minimum only increases by 0.27 kcal/mol. The exception to this is the 75° region on the HF surface where no minimum was previously predicted.
In contrast to the behavior of the ax-2-Eth-THP ψ-surface, the eq-2-Eth-THP ψ-surface does encounter slight entropic stabilization at the minima. However, like the ax-2-Eth-THP surfaces, the barriers become larger when going from energy to free energy, thus creating more well-defined minima. Again, this analysis was not feasible at the MP2 level of theory, but it is expected that observed trends will remain consistent.
Effects of solvation
The impact of aqueous solvation on the conformational properties of the 2-Eth-THP glycosyl linkage was investigated by IEFPCM (1-D surfaces) and IPCM (2-D surfaces) reaction field models. Energies for selected points of the 1-D surfaces are listed in Tables 2 and 3 for the φ- and ψ-torsions, respectively. The axial φ-surface global minimum retains the same shape as vacuum ab initio methods predict. However, a well-defined minimum appears at φ = 165° with a well depth of 0.7 kcal/mol. Our solvation-corrected curve (at the MP2/cc-pVTZ/IEFPCM level) effectively stabilizes the vacuum surface and shows a maximum correction of ∼1.8 kcal/mol at the high energy φ = 240° structure. Qualitatively, solvation has little effect on the surface, but quantitatively the shallow minimum that vacuum levels predicted is stabilized by 1.0 kcal/mol and the barrier connecting this to the global minimum is lowered by 0.3 kcal/mol. The axial ψ-surface shows very little qualitative or quantitative effects from solvation.
The equatorial φ-surface shows larger solvent shifts than does the axial. The first and most important change on this surface is the dramatic stabilization at φ = 135°. Vacuum calculations show this to be the global maximum whereas solvation predicts the global maximum to be at φ = 0°/360°. This amounts to lowering the high energy barrier between global/local minima by 1.5 kcal/mol. In conjunction, solvation raises the barrier at φ = 0°/360° from 4.9 to 6.3 kcal/mol (1.4 kcal/mol). The positions of the global and local minima also shift slightly from 300° to 285° and 60° to 75°, respectively. In addition, the shoulder region of this surface, located at ∼210°, becomes a shallow minimum.
The equatorial ψ-surface also undergoes changes with respect to the vacuum. The global minimum, which was broad and spanned ψ = 180°/190° region, is now even broader, encompassing ψ = 150° as well. The minimum located at 105° is significantly destabilized by solvent. The vacuum minimum, which was 0.8 kcal/mol above the global minimum, is raised 0.5 kcal/mol in energy. In contrast, the vacuum minimum located at 270° is stabilized by solvent and the ΔE is lowered from 1.2 kcal/mol to 0.7 kcal/mol with the barrier height remaining nearly unchanged.
Observing the role solvent corrections had in modifying the 1-D glycosyl torsional potentials, a solvation correction was added to the full 2-D (φ,ψ) vacuum surfaces of ax-2-Eth-THP and eq-2-Eth-THP (Fig. 2, bottom). As observed for the 1-D torsional potentials, solvent has the largest effect on high energy regions of the φ,ψ surfaces. In particular, the φ ∼ 180° region on the 2-D surfaces of both ax-2-Eth-THP and eq-2-Eth-THP show significant (2–3 kcal/mol) stabilization when solvent is introduced. This corresponds to the highest energy transition state on the respective 1-D potentials. In addition to the changes seen at the φ maximum region, the equatorial map shows more variation at ψ ∼ 285°, which encompasses the global minimum region (Fig. 2, right).
A general conclusion drawn from the full 2-D solvation maps is that equatorial conformations typically undergo more dramatic stabilization as a consequence of adding solvent. Specifically, the higher energy (and lower energy to a lesser extent) regions associated with equatorial conformations incur greater solvent stabilization as compared to their axial counterparts. This result is expected due to the extended conformations that equatorial anomers can adopt; however, the present maps allow us to observe the exact regions in which solvent has the largest effects.
CONCLUSIONS
This is the first high-level, ab initio study on the model carbohydrate 2-ethoxy tetrahydropyran. This compound is a mimic for the glycosyl linkage in disaccharides and glycolipids. We performed constrained (φ,ψ) vacuum optimizations at the MP2/6-31G(d) level of theory (15° intervals) and determined 2-D potential energy surfaces for both the axial and equatorial anomers of 2-Eth-THP. These surfaces provide the approximate locations of all minima and transition states connecting them. Three minima exist on the axial surface with the global minimum at 60°,185°. The equatorial surface displays four minima, with the 300°,195° point being the lowest in energy.
We computed the anomeric ratios (ΔE and ΔG298) for ax-2-Eth-THP and eq-2-Eth-THP at various levels of theory. We developed and applied an energy additivity scheme, LCM(X,Y); X = 2,3 and Y = 3,4,5, to predict high level results for the anomeric ratios and compared these results to previous theoretical and experimental work. In agreement with recent work published by Weldon et al. (20), it is shown that HF and DFT benefit from cancellation of errors and tend to overstabilize equatorial carbohydrate conformations. We also determined that, for THP derivatives, performing extrapolations with high level (e.g., CCSD(T)) estimates in vacuum and correcting for thermal effects is not enough to achieve results that are in agreement with solution-based experiments. However, including solvation effects, even at the implicit level, corrects gas phase results, and is essential for comparing computed anomeric ratios with experimental free energy differences (ΔG298).
Glycosyl torsional potentials were examined by mapping constrained 1-D surfaces at numerous levels of theory. Using these surfaces as foundations we performed LCM(2,3) and LCM(3,4) extrapolations of all four possible φ- and ψ-surfaces of ax-2-Eth-THP and eq-2-Eth-THP. From this it was determined that variations in the 1-D surfaces, predicted with lower levels of theory such as HF, DFT, and MP2 in combination with small basis sets, can be significant and that care must be taken if accurate potentials are desired. In contrast, the MP2/cc-pVTZ level of theory yields very good agreement with the fully extrapolated φ- and ψ-constrained torsional potentials; the surfaces are typically within 0.15 kcal/mol of the full three-layered, LCM(3,4), results. However, if possible, we suggest using the two-layered approach, especially at high energy regions of the surfaces. The improved results are due to the coupled cluster and basis set corrections that are employed in the layered schemes.
In addition, entropic effects were examined on the 1-D torsional potentials. It was determined that, in general, entropy helps to stabilize minima, although not necessarily directly. For example, in many cases minima were destabilized by entropy, but their associated barriers to the global minimum were destabilized to a larger extent. This effectively increased their stability and resulted in more well-defined minima.
Also examined were solvation effects on the 1-D and 2-D φ-and ψ-surfaces. These results suggest that solvation effects are more important in stabilizing equatorial carbohydrates (e.g., eq-2-Eth-THP, β-Glucose) than their axial anomeric forms. A particularly clear example is the equatorial φ glycosyl torsional potential, which changes qualitatively when using an implicit solvent model.
This study not only examines some fundamental aspects of carbohydrate structural and stereoelectronic properties, but also lays the groundwork for the parameterization of a CHARMM (42,43) compatible carbohydrate force field. Vacuum and solvent maps, like the ones produced in the current work, will be invaluable not only to groups that are interested in parameterizing new force fields, but also to those interested in correcting current force fields via the empirical correction procedures.
Acknowledgments
We thank the following for helpful insight and fruitful discussions: Daron Freedberg, Joseph Larkin, John Brady, and Richard Venable. Use of the LoBoS (www.lobos.nih.gov) and Biowulf (biowulf.nih.gov) supercomputers is also acknowledged and appreciated.
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute, and by National Institutes of Health grant GM No. 70855 to A.D.M. Jr. D.M. thanks the University of Sydney for a Sesqui Postdoctoral Fellowship.
Editor: Steven D. Schwartz.
References
- 1.Rao, V. S. R., P. K. Quasba, P. V. Balaji, and R. Chandrasekaran. 1998. Conformation of Carbohydrates. Harwood, Amsterdam, The Netherlands.
- 2.Brady, J. W., and R. K. Schmidt. 1993. The role of hydrogen-bonding in carbohydrates—molecular dynamics simulations of maltose in aqueous solution. J. Phys. Chem. 97:958–966. [Google Scholar]
- 3.Metzler, D. E. 2001. Biochemistry, 2nd Ed. Harcourt Academic Press, San Diego, CA.
- 4.Edward, J. T. 1955. Stability of glycosides to acid hydrolysis. Chem. Ind. (London). 36:1102–1104. [Google Scholar]
- 5.Lemieux, R. U., and P. Chu. 1958. Abstracts of Papers. 133rd National Meeting of the American Chemical Society, San Francisco, CA. American Chemical Society, Washington, DC.
- 6.Lemieux, R. U. 1971. Effects of unshared pairs of electrons and their solvation on conformational equilibria. Pure Appl. Chem. 25:527–548. [Google Scholar]
- 7.Eliel, E. 1972. Conformational analysis in heterocyclic systems—recent results and applications. Angew. Chem. Int. Ed. Engl. 11:739–750. [Google Scholar]
- 8.Szarek, W. A., and D. Horton. 1979. Anomeric Effect: Origin and Consequences. ACS Symposium Series, No. 87. American Chemical Society, Washington, DC.
- 9.Kirby, A. J. 1983. The Anomeric Effect and Related Stereoelectric Effects at Oxygen. Springer Verlag, Berlin.
- 10.Lemieux, R. U., A. A. Pavia, J. C. Martin, and K. A. Watanabe. 1969. Solvation effects on conformational equilibria. Studies related to conformational properties of 2-methoxytetrahydropyran and related methyl glycopyranosides. Can. J. Chem. 47:4427–4439. [Google Scholar]
- 11.Praly, J. P., and R. U. Lemieux. 1987. Influence of solvent on the magnitude of the anomeric effect. Can. J. Chem. 65:213–223. [Google Scholar]
- 12.Curtiss, L. A., K. Raghavachari, P. C. Redfern, V. Rassolov, and J. A. Pople. 1998. Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys. 109:7764–7776. [Google Scholar]
- 13.DeYonker, N. J., T. R. Cundari, and A. K. Wilson. 2006. The correlation consistent composite approach (CCCA): an alternative to the Gaussian-n methods. J. Chem. Phys. 124:114104–114122. [DOI] [PubMed] [Google Scholar]
- 14.Klauda, J. B., S. L. Garrison, J. Jiang, G. Arora, and S. I. Sandler. 2004. HM-IE: quantum chemical hybrid methods for calculating interaction energies. J. Phys. Chem. A. 108:107–112. [Google Scholar]
- 15.Cances, M. T., B. Mennucci, and J. Tomasi. 1997. A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107:3032–3041. [Google Scholar]
- 16.Mennucci, B., and J. Tomasi. 1997. Continuum solvation models: a new approach to the problem of solute's charge distribution and cavity boundaries. J. Chem. Phys. 106:5151–5158. [Google Scholar]
- 17.Mennucci, B., J. Cances, and M. T. Tomasi. 1997. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B. 101:10506–10517. [Google Scholar]
- 18.Tomasi, J., B. Mennucci, and M. T. Cances. 1999. The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. Theochem. 464:211–226. [Google Scholar]
- 19.Foresman, J. B., T. A. Keith, K. B. Wiberg, J. Snoonian, and M. J. Frisch. 1996. Solvent effects. 5. Influence of cavity shape, truncation of electrostatics, and electron correlation ab initio reaction field calculations. J. Phys. Chem. 100:16098–16104. [Google Scholar]
- 20.Weldon, A. J., T. L. Vickrey, and G. S. Tschumper. 2005. Intrinsic conformational preferences of substituted cyclohexanes and tetrahydropyrans evaluated at the CCSD(T) complete basis set limit: implications for the anomeric effect. J. Phys. Chem. A. 109:11073–11079. [DOI] [PubMed] [Google Scholar]
- 21.Salzner, U., and P. v. R. Schleyer. 1994. Ab initio examination of anomeric effects in tetrahydropyrans, 1, 3-dioxanes, and glucose. J. Org. Chem. 59:2318–2155. [Google Scholar]
- 22.Tvaroska, I., and J. P. Carver. 1994. Ab-initio molecular-orbital calculation of carbohydrate model compounds. 2. Conformational-analysis of axial and equatorial 2-methoxytetrahydropyrans. J. Phys. Chem. 98:9477–9485. [Google Scholar]
- 23.Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, et al. 2004. Gaussian 03, Rev. c.02. Gaussian, Wallingford, CT.
- 24.Shao, Y., L. Fusti-Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. Distasio, R. C. Lochan, et al. 2006. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 8:3172–3191. [DOI] [PubMed] [Google Scholar]
- 25.Werner, H.-J., P. J. Knowles, R. Lindh, M. Schütz, P. Celani, T. Korona, F. R. Manby, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, and D. L. Cooper, et al. 2005. Molpro, Ver. 2002.10, a package of ab initio programs. See http://www.molpro.net.
- 26.Pierson, G. O., and O. A. Runquist. 1968. A conformational analysis of some 2-alkoxytetrahydropyrans. J. Org. Chem. 33:2572–2574. [Google Scholar]
- 27.Dunning, T. H. 1989. Gaussian-basis sets for use in correlated molecular calculations. 1. The atoms boron through neon and hydrogen. J. Chem. Phys. 90:1007–1023. [Google Scholar]
- 28.Maseras, M., and K. Morokuma. 1995. IMOMM: a new integrated ab-initio plus molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J. Comput. Chem. 16:1170–1179. [Google Scholar]
- 29.Henry, D. J., C. J. Parkinson, and L. J. Radom. 2002. An assessment of the performance of high-level theoretical procedures in the computation of the heats of formation of small open-shell molecules. J. Phys. Chem. A. 106:7927–7936. [Google Scholar]
- 30.Henry, D. J., M. B. Sullivan, and L. J. Radom. 2003. G3-rad and g3x-rad: modified Gaussian-3 (g3) and Gaussian-3x (g3x) procedures for radical thermochemistry. J. Chem. Phys. 118:4849–4860. [Google Scholar]
- 31.Feller, D. 1992. Application of systematic sequences of wave-functions to the water dimer. J. Chem. Phys. 96:6104–6114. [Google Scholar]
- 32.Feller, D. 1993. The use of systematic sequences of wave-functions for estimating the complete basis set, full configuration-interaction limit in water. J. Chem. Phys. 98:7059–7071. [Google Scholar]
- 33.Helgaker, T., W. Klopper, H. Koch, and J. Noga. 1996. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 106:9639–9646. [Google Scholar]
- 34.Cramer, C. J., D. G. Truhlar, and A. D. French. 1997. Exo-anomeric effects on energies and geometries of different conformations of glucose and related systems in the gas phase and aqueous solution. Carbohydr. Res. 298:1–14. [Google Scholar]
- 35.Tvaroska, I., and J. P. Carver. 1998. The anomeric and exo-anomeric effects of a hydroxyl group and the stereochemistry of the hemiacetal linkage. Carbohydr. Res. 309:1–9. [Google Scholar]
- 36.Momany, F. A., and J. L. Willett. 2000. Computational studies on carbohydrates. I. Density functional ab initio geometry optimization on maltose conformations. J. Comput. Chem. 21:1204–1219. [Google Scholar]
- 37.Senderowitz, H., C. Parish, and W. C. Still. 1996. Carbohydrates: united atom AMBER* parameterization of pyranoses and simulations yielding anomeric free energies. J. Am. Chem. Soc. 118:2078–2086. [Google Scholar]
- 38.Woods, R. J., R. A. Dwek, C. J. Edge, and B. Fraser-Reid. 1995. Molecular mechanical and molecular dynamical simulations of glycoproteins and oligosaccharides. 1. Glycam-93 parameter development. J. Phys. Chem. 99:3832–3846. [Google Scholar]
- 39.Stortz, C. A. 2004. Comparative performance of MM3(92) and two tinker (TM) MM3 versions for the modeling of carbohydrates. J. Comput. Chem. 26:471–483. [DOI] [PubMed] [Google Scholar]
- 40.Perez, S., A. Imberty, S. B. Engelsen, J. Gruza, K. Mazeau, J. Jimenez-Barbero, A. Poveda, J. F. Espinosa, B. P. van Eyck, G. Johnson, A. D. French, and M. Louise, et al. 1998. A comparison and chemometric analysis of several molecular mechanics force fields and parameter sets applied to carbohydrates. Carbohydr. Res. 314:141–155. [Google Scholar]
- 41.Booth, H., M. Dixon, and S. A. Readshaw. 1992. Experimental studies of the anomeric effect. 5. The influence of some solvents on the conformational equilibria in 2-methoxy-tetrahydropyran and 2-(2′, 2′, 2′-trifluoroethoxy)-tetrahydropyran. Tetrahedron. 48:6151–6160. [Google Scholar]
- 42.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM—a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 43.MacKerell, A. D., D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, and L. Kuchnir, et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]