

Abstract
In the narrow segment of an ion conducting pathway, it is likely that a permeating ion influences the positions of the nearby atoms that carry partial or full electronic charges. Here we introduce a method of incorporating the motion of charged atoms lining the pore into Brownian dynamics simulations of ion conduction. The movements of the carbonyl groups in the selectivity filter of the KcsA channel are calculated explicitly, allowing their bond lengths, bond angles, and dihedral angels to change in response to the forces acting upon them. By systematically changing the coefficients of bond stretching and of angle bending, the carbon and oxygen atoms can be made to fluctuate from their fixed positions by varying mean distances. We show that incorporating carbonyl motion in this way does not alter the mechanism of ion conduction and only has a small influence on the computed current. The slope conductance of the channel increases by ∼25% when the root mean-square fluctuations of the carbonyl groups are increased from 0.01 to 0.61 Å. The energy profiles and the number of resident ions in the channel remain unchanged. The method we utilized here can be extended to allow the movement of glutamate or aspartate side chains lining the selectivity filters of other ionic channels.
INTRODUCTION
Three computational approaches have been commonly used to model biological ion channels: Poisson-Nernst-Planck (PNP) theory (1,2), Brownian dynamics (3,4), and molecular dynamics (5,6). Each of these has its strengths and limitations, and involves a degree of approximation. The main defects of the PNP theory are errors stemming from the mean-field assumption. In particular, it ignores the effects of induced surface charges created as a charged particle in electrolyte solutions approaches the water-protein interface. The magnitude of the errors introduced by the mean-field approximation become large when the theory is applied to narrow ionic channels (7,8). By incorporating a term in the PNP equations to account for the barrier created by induced surface charges, the errors can be reduced somewhat (9) but, in doing so, much of the simplicity of the PNP theory, one of its main advantages over the other approaches, is compromised. One of the limitations of molecular dynamics is the computational power required that limits the possible simulation times. Although the calculation of free energy profiles and other properties provide useful information on ion permeation, it is generally not possible to directly calculate channel currents from these simulations. Indeed, only a couple of studies exist in which conductance has been calculated directly using molecular dynamics at large applied potentials in simplified or wide channels (10,11), which highlights the difficulty involved in using such techniques to directly compare simulation results with experimental data. To simulate long enough to see the permeation of ions across a channel and to determine its conductance, we utilize Brownian dynamics, where water molecules that form the bulk of the system are integrated out and only the ions themselves are simulated explicitly. The net effects of incessant collisions between ions and water molecules are lumped together and treated as the frictional and random forces. This treatment of water molecules can be viewed as an approximation of the central limit theorem by using stochastic averaging of water molecules. Also, it has been assumed that atoms forming the transmembrane conduit are rigid and do not move. This rigidity assumption can be intuitively justified via stochastic averaging theory (12–14).
All three approaches are useful in elucidating the mechanisms underlying selectivity and permeation of ions across biological nanotubes. For ion channels with large pore radii, the PNP theory can be fruitfully utilized. To study the mechanisms underlying the selectivity sequences of monovalent ions or to determine the precise conformational changes of the protein when a channel undergoes the transformation from the closed to the open state, one has to rely on molecular dynamics simulations. Thus far, a combined use of molecular and Brownian dynamics has proved to be well suited for studying structure-function relationships in ion channels. Brownian dynamics enables the calculation of conductance properties whereas molecular dynamics can provide input and justification for the parameters of the stochastic simulations (15–17) as well as explain finer details such as size-based selectivity (18).
One main caveat to the application of Brownian dynamics to biological ion channels is the simplifying assumption that the channel is rigid in that all protein atoms remain in their fixed positions. In reality, proteins are malleable and atoms undergo rapid thermal fluctuations (19–21). Moreover, some of the charged or polar residues lining the pore interact with permeant ions Coulombically, and it is possible that the motions of these atoms or side chains, especially those lining the narrow segment of the pore, may influence the dynamics of ion permeation across the channel. Recently, Allen et al. (20) reported that the root mean-square (rms) variations in carbonyl oxygen atoms in the gramicidin channel range from 0.42 to 0.65 Å. The carbonyl oxygens in the selectivity filter of the KscA potassium channels are likely to exhibit the fluctuations of a similar magnitude. It is not known, however, if the rapid motions of the carbonyl groups are in part induced by or correlated with the motions of the permeant ion in the pore. It is also unknown what effects, if any, such fluctuations of polar or charged residues will have on the dynamics of ion permeation. In the selectivity filter of the L-type calcium channel, for example, the charged side chains of four glutamate residues play a crucial role in valence selectivity (22,23). It has been postulated that processing of ions at the EEEE locus relies on motions of these residues (24).
Here, we extended the Brownian dynamics algorithm to explicitly simulate the movement of the carbonyl oxygens in the selectivity filter of the KcsA potassium channel. To do this, the bond length of the carbon and oxygen atoms r is allowed to stretch, and the bond angle θ and dihedral angles χ are allowed to bend in response to the forces acting on them. By changing the coefficients in the equations of motion, we can vary the magnitude of the fluctuations to a desired value. Here we detail the effects of allowing the oxygen atoms in the selectivity filter to rapidly fluctuate from their mean positions on the permeation dynamics. We find that the movements of the K+ ion in the filter and the movements of any oxygen atom are uncorrelated in our simulations, indicating that the Coulomb interaction between the charged and polar atom is not sufficiently strong to rigidly or elastically couple their motions. When the bond-stretching and angle-bending coefficients are successively reduced, the slope conductance of the channel increases by ∼25%. The shape of the energy profiles obtained with three different values of rms fluctuations are broadly similar. Similarly, the number of resident ions in the channel and the dynamics of ion permeation in the selectivity filter remain unaffected by the motion of the carbonyl oxygen atoms.
METHODS
The motion of ions in and around the channel under the influence of electric and random forces is traced using the Langevin equation, a stochastic version of Newton's equation of motion,
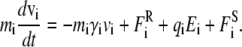 |
(1) |
Here, mi, vi, miγi, and qi are the mass, velocity, friction coefficient, and charge on an ion with index i, whereas 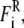 Ei, and
Ei, and 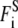 are the random stochastic force, systematic electric field, and short range forces experienced by the ion, respectively. Ions are initially given random positions with a specified mean concentration and Boltzmann distributed velocities within reservoirs attached to either end of the channel that mimic the intracellular and extracellular environment. We calculate the total force acting on each and every ion in the assembly and then calculate new positions for the ions a short time later using an algorithm derived by van Gunsteren and Berendsen (25). A multiple time-step algorithm is used, where a time step of Δt = 100 fs is employed in the reservoirs and 2 fs in the channel where the forces change more rapidly.
are the random stochastic force, systematic electric field, and short range forces experienced by the ion, respectively. Ions are initially given random positions with a specified mean concentration and Boltzmann distributed velocities within reservoirs attached to either end of the channel that mimic the intracellular and extracellular environment. We calculate the total force acting on each and every ion in the assembly and then calculate new positions for the ions a short time later using an algorithm derived by van Gunsteren and Berendsen (25). A multiple time-step algorithm is used, where a time step of Δt = 100 fs is employed in the reservoirs and 2 fs in the channel where the forces change more rapidly.
To enable simulations to be run for long enough to calculate the current passing through the channel, water molecules are not treated explicitly. However, the influence of the individual water molecules on the motion of ions is mimicked through the use of the frictional term (mγv) and random forces (FR). The electrostatic forces acting upon the ions are calculated by solving Poisson's equation using a boundary element method (26). To do this we assign a dielectric constant of εp = 2 to the protein and membrane and εw = 60 to the water inside the channel as in previous Brownian dynamics studies of the KcsA channel (4). Because calculating the electric forces at every step in the simulation is very time consuming, we store precalculated electric fields and potentials due to one and two ion configurations in a system of lookup tables and interpolate values for the given ion positions from these during the simulation (27). Further details of the Brownian dynamics approach are given elsewhere (4,28,22).
At short ranges, the Coulomb interaction between two ions is modified by adding a potential USR(r) that replicates effects of the overlap of electron clouds and hydration. As noted previously (22), molecular dynamics simulations show that the hydration forces between two ions add further structure to the 1/r9 repulsive potential due to the overlap of electron clouds in the form of damped oscillations (29,30). These two effects can be approximately represented by
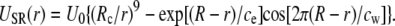 |
(2) |
Here the oscillation length cw = 2.76 Å is given by the water diameter and the other parameters are determined by fitting Eq. 2 to the calculated potentials of mean force (29–32). The potentials of mean force for K-Cl pairs can be replicated in Brownian dynamics simulations using an overall strength of the potential of U0 = 5.4, 2.5, and 1.4 kT for K-Cl, K-K, and Cl-Cl pairs (22). As in previous simulations we use Rc = r1 + r2 for anion-cation pairs and Rc = r1 + r2 + 1.6 Å for like ions (where r1 and r2 are the Pauling radii of each atom) and R = Rc + 0.2 Å for like ions and R = Rc − 0.2 Å otherwise. An exponential drop parameter of ce = 1 Å is used for all ion pairs. The short range force in Eq. 1 is determined from the derivatives of the potential in Eq. 2 as well as from a 1/r9 potential between ions and the channel boundary.
The channel structure we use for this study is the same as that used in previous Brownian dynamics simulations of this channel (4). The model channel is based on the crystal structure of Doyle et al. (33) with the pore expanded at the internal end of the channel using a cylindrical repulsive potential in molecular dynamics to have a radius of 5 Å as illustrated in Fig. 1 A. Here the carbonyl groups lining the selectivity filter are shown in purple and blue. As described previously, the selectivity filter is also expanded slightly such that the minimum radius is 1.4 Å (4). The full charges of −e and +e are assigned to the Glu-118 and Arg-117 residues that guard the intracellular gate.
FIGURE 1.

Model potassium channel. (A) Three of the four subunits of the full experimentally determined protein structure are illustrated. The carbonyl groups of the selectivity filter, including those belonging to the top subunit, are shown. The water-protein interface is shown in gold. Potassium ions are indicated in red. (B) The close-up view of the selectivity filter shows the locations of the 12 carbonyl groups that are allowed to move. The four groups at the top are removed to reveal the ion-conducting path.
Unlike in previous Brownian dynamics simulations in which the protein was taken to be a rigid dielectric environment containing fixed partial charges, in this study the motion of a small number of protein atoms is calculated explicitly. As for the ions, the motion of these atoms is described by the Langevin equation with 2 fs time steps. However, additional forces are applied to these atoms to replicate the covalent bonds binding them together and to the remainder of the protein. These forces take the form of bond stretching, bond angle bending, and dihedral angle bending forces as found in molecular mechanics force fields:
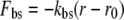 |
(3) |
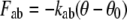 |
(4) |
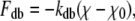 |
(5) |
where r, θ, and χ are the bond lengths, angles, and dihedral angles; r0, θ0, and χ0 are their equilibrium values; and kbs, kab, and kdb are the associated force constants.
In this study only 16 carbonyl groups from residues Thr-54, Val-55, Gly-56, and Tyr-57 lining the selectivity filter in each of the four subunits were treated explicitly. The 12 of 16 carbonyl groups that are rendered mobile are highlighted in Fig. 1 B. The top four carbonyl groups are removed for clarity. In reality, the carbon atom of a carbonyl group is bonded to three atoms: the carbonyl oxygen, the backbone α-carbon, and the nitrogen from the next residue. To avoid having two additional bonds on the carbon atom, we modeled the carbonyl bonds as F-C=O, where F is a fixed point, located midway between the α-carbon and nitrogen. Thus, the bond length between the fixed position F and the carbon atom is allowed to increase or decrease and the bond angle between the fixed point F and the carbon atom is allowed to change. Similarly, the bond length and bond angle between the carbon and oxygen atoms in the carbonyl group are allowed to change from their equilibrium position. Thus, there are four force constants: two bond-stretching constants and two angle-bending constants.
It is not clear exactly what force constants should be used so that fluctuations of the carbonyl groups would mimic those occurring in reality. As we use partial charges from the CHARMM22 parameter set, an obvious starting point is to use the force constants determined for carbonyl bonds from this parameter set as well. We note here that the default values of the bond-stretching constants between C=O and F-C (taken as the average of the values for the bonds with the Cα and nitrogen), used in CHARMM22, are 4.32 × 102 and 1.83 × 102 J/m2 (620 and 260 kcal/mol/Å2), respectively. The corresponding values used in CHARMM22 for the angle-bending constants are 1.77 × 10−8 and 1.14 × 10−8 J/rad2 (80 and 50 kcal/mol/Å2). However, as we are simulating the motion of only a few protein atoms, and in particular because the α-carbon and the nitrogen atoms to which the carbonyl carbon atoms are attached are assumed to be stationary, it is likely that the motion of the oxygen atoms will be underestimated using these values. To this end, we use a range of differing force constants so that we can examine the rms fluctuations of the oxygen and carbon atoms in the carbonyl groups from their starting positions. These fluctuations can be compared to values determined in molecular dynamics simulations to gain a better appreciation of the most realistic force constants to use in our simulation. As we have included only the O and C atoms in each carbonyl we have not considered the dihedral angle in these calculations.
Short range K+-oxygen forces are applied in a similar manner to the short range ion-ion interactions applied previously in Brownian dynamics simulations. For this purpose the parameters Rc = 2.7 Å, R = 2.5 Å, U0 = 1.0, and cw = 1.0 are employed. The electrostatic forces acting upon the oxygen atoms are calculated using Poisson's equation and are stored in lookup tables as for the ions.
RESULTS
The carbonyl atoms fluctuate rapidly from their original positions, with the magnitude of fluctuations increasing steadily as the force constants are reduced. Three traces illustrated in Fig. 2 A highlight the fluctuations obtained with the bond-stretching and angle-bending constants set at high (red), intermediate (green), and low (blue) values. The rms fluctuations calculated from the three traces are, respectively, 0.09, 0.15, and 0.61 Å. The rms fluctuations of the atoms lining the selectivity filter, inferred from the crystallographic B-factor and molecular dynamics simulations of the KcsA channel, are on the order of 0.75–1.0 Å (34). This value is close to that observed from molecular dynamics simulations using the CHARMM22 default values for the carbonyl bond. When these values are used in the Brownian dynamics code, the rms fluctuations of the carbonyl oxygen atoms in the selectivity filter is much lower at only 0.15 Å, as illustrated in green in Fig. 2 A. This discrepancy probably arises from the fact that in our BD simulation the carbonyl carbon is attached to a fixed point rather than to the remainder of the protein that would be mobile. It is only when the force constants are reduced by a factor of 6 that the rms fluctuation observed in BD simulations equals that deduced from the B-factor of the KcsA channel.
FIGURE 2.

Fluctuations of carbonyl oxygen atoms in the selectivity filter. (A) The motion of one carbonyl oxygen atom from its equilibrium position is plotted against time. The C=O bond-stretching force constants used to obtain the traces exhibited are: red, 25.92 × 102 J/m2; green, 4.32 × 102 J/m2; blue, 0.81 × 102 J/m2. Values of the angle bending and F-C constants are given in Table 1. (B) Rms fluctuations of the carbonyl oxygen atoms from their equilibrium positions (solid circles) and their original positions (open circles) are plotted against the log of the C=O bond-stretching constants. The other force constants used to obtain each measured point are given in Table 1.
The measured fluctuations of the carbonyl oxygens at various values of the force constants are tabulated in Table 1. We measured the rms fluctuations in two different ways: fluctuations from the mean position of the atoms (rmsd) and fluctuations from the initial positions of the atoms (rmsd0). Both values are tabulated in Table 1, but we quote the second values hereafter. Fig. 2 B shows the rms fluctuations of the oxygens from their original positions as a function of the force constants. The rms fluctuations increase from 0.09 to 0.74 Å as the bond-stretching and angle-bending constants are reduced from the largest to smallest values used. Here, the rmsd is plotted against the logarithm of the bond-stretching constant. The steepest increases occur when the bond-stretching coefficient is further reduced below 1.6 × 102 J/m2.
TABLE 1.
Root mean-square fluctuations of carbonyl oxygens
| C=O
|
F-C
|
Fluctuations
|
|||
|---|---|---|---|---|---|
| Bond stretch (× 102 J/m2) | Angle bend (× 10−8 J/rad2) | Bond stretch (× 102 J/m2) | Angle bend (× 10−8 J/rad2) | rmsd (× 10−11 m) | rmsd0 (× 10−11 m) |
| 25.92 | 10.62 | 10.98 | 6.84 | 0.60 | 0.87 |
| 12.96 | 5.31 | 5.49 | 3.42 | 0.72 | 0.98 |
| 4.32 | 1.77 | 1.83 | 1.14 | 1.03 | 1.51 |
| 2.16 | 0.88 | 0.92 | 0.57 | 1.34 | 1.86 |
| 1.80 | 0.74 | 0.76 | 0.46 | 1.46 | 2.08 |
| 1.62 | 0.66 | 0.69 | 0.42 | 1.53 | 2.19 |
| 1.35 | 0.55 | 0.57 | 0.37 | 1.79 | 2.40 |
| 0.99 | 0.41 | 0.42 | 0.26 | 3.17 | 4.30 |
| 0.86 | 0.35 | 0.36 | 0.22 | 3.86 | 5.23 |
| 0.81 | 0.33 | 0.35 | 0.21 | 4.20 | 6.07 |
| 0.72 | 0.29 | 0.31 | 0.19 | 5.17 | 7.35 |
Analysis of crystal structures of KcsA (35), molecular (36,37), and previous Brownian dynamics (4) models suggest that the conduction of K+ through the selectivity filter involves concerted motion of multiple ions. In our simulations, as in these previous studies, the selectivity filter is usually occupied by at least two ions and that the entry of a third ion is required to knock one of the resident ions out of the filter. The two ions bound in the selectivity filter spend on average 16 ns at the applied potential of −100 mV, executing random motions from their equilibrium positions before a successful conduction event takes place. At the same time, the nearby oxygen and carbon atoms of the carbonyl groups are also undergoing thermal fluctuations from their equilibrium positions. Characteristic oscillation periods of atomic motions, such as the bond stretching and angle bending, are between 10 and 90 fs (19). Thus, the timescale of a conduction event is ∼5–6 orders of magnitude slower than the timescale of atomic thermal fluctuations. Because the rings of oxygen atoms surround the resident ion in the binding site, it is possible that the thermal motions of the ion are tightly coupled to those of the oxygen atoms. Here we show that no such coupling exists in our simulations.
The degree of correlated motion between permeant ions and the mobile carbonyl oxygen atoms, is shown in Fig. 3. Here we plot the average correlation between each ring of oxygen atoms against the axial position of the ion in the pore. A value of 1.0 represents perfectly correlated motion between the atoms, 0 no correlation, and −1.0 anticorrelated behavior. To obtain this data, we monitor the velocities of one of the two ions in the selectivity filter. In the course of a simulation period lasting 0.2 μs, this test ion wanders between z = 7 to z = 20 Å. The relative probability of the ion being at a given axial position is shown in Fig. 3 A. The velocity of the ion, as well as that of the carbonyl oxygen atoms are recorded at each time step. The degree of correlated motion between the ion and each oxygen for that time step is then calculated from the dot product of the velocities of the ion (vi) and the oxygen atom (vj):
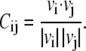 |
(6) |
FIGURE 3.

Correlation between the motion of a resident K+ ion and each of the four rings of oxygen atoms. The relative probability of an ion being at various axial positions in the selectivity filter during a simulation period of 0.2 μs is shown in panel A. The correlations between the motion of the ion when it resides at various axial positions in the filter and that of the four oxygen atoms of the Thr-54 (red) and Val-55 (blue) residues are shown in panel B, and with the Gly-56 (green) and Tyr-57 (red) residues are plotted in panel C. The locations of the carbonyl oxygens are shown panel D.
Finally, the correlation is averaged over 0.2 μs of simulation and averaged within each ring of oxygen atoms. The correlation coefficients between the test ion at a given axial position and the first (Thr-54), second (Val-55), third (Gly-56), and fourth (Tyr-57) rings of carbonyl oxygen atoms are shown in Fig. 4, B and C. All values hover around zero, indicating that the movement of the ion in the filter does not influence the movement of the oxygen atoms surrounding it, or vice versa. The locations of the carbonyl rings along the z axis are shown in Fig. 3 D.
FIGURE 4.

The current-voltage curves obtained using three different sets of the force constants. The force constants used for panels A, B, and C correspond to those used to construct Fig. 2 A. Each data point represents the average of 24 to 32 sets of simulations, each lasting 2 × 106 time steps (or 0.2 μs). Error bars in this and following figures have a length of mean ± 1 SE and are not shown when they are smaller than the data points.
It is worth comparing the amount of correlated motion in these simulations with that seen in molecular dynamics simulations of the KcsA channel. In such molecular simulations we find that there is a degree of correlated motion between an ion in the selectivity filter and the oxygen atoms in contact with it, although there is no correlation to the more distant atoms. The correlation coefficient for the eight immediate neighboring oxygen atoms of the ion in the molecular simulations of the KcsA channel calculated as described above are ∼0.22. One possible reason for not observing such a correlation in our simulation is that the random force that acts independently upon the ion in the pore as well as the carbonyl atoms, which is an integral part of the Brownian dynamics technique, is overwhelming any correlated motion. To test this hypothesis we examined the effect of altering the random force in three different ways. First, we removed the random force on the ions and oxygen atoms in the selectivity filter of the pore in our Brownian dynamics simulations. This manipulation (which is the equivalent of removing the coupling of these atoms to the temperature bath) resulted in correlated motion between the ion and the neighboring oxygen atoms with a correlation coefficient of ∼0.19. Second, we removed the random force from just ions, while leaving it on the carbonyl atoms. Third, we set the random force acting on the oxygen atoms to be equal to that on the ion when they are within 3.5 Å of each other. As expected, assigning the identical random force at time step to the carbonyl atoms and resident ions yielded a correlation coefficient of ∼0.55. These procedures, however, do not accurately reflect the physical reality. Conduction does not take place when the random force that imparts the kinetic energy to the resident ions is removed from the total forces acting on the atoms in the assembly. Under these conditions, the ions and atoms stay relatively frozen in the local energy minima as they are not surrounded by explicitly simulated atoms that can impart kinetic energy nor are they receiving this from random forces. Removing the random force from just the ions does not overcome this problem since some of the atoms adjacent to the ions, such as surrounding water and other protein atoms, are still not being simulated explicitly, and the ion still gains a reduced kinetic energy. Similarly, coupling the motion of the ion to that of the nearby oxygen atoms by imparting the identical random forces to them cause the ion to stay fixed in the location of the energy minima, thereby preventing it to climb out of the well. It is not clear if one can devise a satisfactory method of inducing correlated motion in the Brownian dynamics framework.
Even if the thermal motions of the permeant ions and carbonyl atoms are not correlated, it is still possible that the presence of the ions has a significant effect on the positions of the carbonyl groups by altering their mean position. To examine this possibility, we compared the results of two simulations carried out with weak force constants: one in which ions were prevented from entering the channel, and one in which they were not. Surprisingly, the mean radial position of all the carbonyl oxygens in the two simulations were within 0.02 Å. The presence of ions affects some of the oxygen positions more than others, but the difference in the mean radial position with and without ions was <0.1 Å in all cases. The ions have a greater effect on the thermal fluctuations of the oxygen atoms, however, increasing their magnitude by ∼17%.
Fig. 4 shows the current-voltage relationships obtained with three different sets of force constants with a symmetric solution of 300 mM KCl in both reservoirs. The control curve (Fig. 4 A, solid circles) is obtained with the force constants that rendered the rms fluctuations of the oxygen atoms to be 0.09 Å. Use of the default force constants from CHARMM22 (rms = 0.15 Å) has only a small influence on the current as shown in Fig. 4 B. Superimposed on this curve is the fitted curve across the control data shown in Fig. 4 A (dashed line). By reducing the force constants to 18% of the values used in CHARMM, we are able to increase the rms fluctuations of the carbonyl oxygen atoms from their original positions to 0.61 Å. This value roughly corresponds to the rms fluctuations of the gramicidin carbonyl groups measured during molecular dynamics simulations (20). The current-voltage curve obtained with these weak force constants is illustrated in Fig. 4 C. The outward currents are slightly larger than for the control when the intermediate force constants are used, while the inward currents are larger than the control when the weak force constants are used.
To check if the conductance changes in any systematic way as the carbonyl oxygen atoms are made more mobile, we change the force constants and measure the current across the channel. The inward current measured for a range of force constants with an applied potential of −100 mV are plotted in the form of histogram in Fig. 5 A. In these simulations, the C=O and F-C bond-stretching and angle-bending constants are altered together as indicated in Table 1. The conductance fluctuates up and down from the control value of 114.3 pS, ranging from 62% to 126% of this value. The outward and inward currents obtained with applied potentials of + 60 mV (solid circles) and −100 mV are plotted against rms fluctuations in Fig. 5 B and C. The measurements shown open circles are reproduced from Fig. 5 A, this time the currents being plotted against rms fluctuations. The outward currents increase consistently when the carbonyl oxygens fluctuate between 0.1 and 0.2 Å. Also, the inward currents decrease when the carbonyl oxygens fluctuate between 0.3 and 0.5 Å. Thus, there is a nonmonotonous relationship between the amplitude of fluctuations of carbonyl groups around their mean positions and the current. Most likely, small shifts in the locations of the binding sites of the residence ions in the selectivity filter cause the observed reduction or enhancement of the current by up to ±25%. We have not carried out detailed analyses for the causes of these changes in the currents with the magnitude of carbonyl fluctuations.
FIGURE 5.

Currents obtained at various force constants under the applied potential of −100 and +60 mV. (A) The currents measured at an applied potential of −100 mV obtained by using various sets of force constants are shown. The number accompanying each bar represents the C=O bond-stretching constant. The other three force constants used are tabulated in Table 1. (B and C) The inward and outward currents measured at the applied potentials of +60 (solid circles) and −100 mV (open circles) are plotted against rms fluctuations. The data plotted in solid circles are reproduced from panel A.
It is notable that the four rings of carbonyl oxygens do not all have the same magnitude of thermal fluctuation, which may explain the slight asymmetric effect of introducing carbonyl mobility on the channel current. The outermost ring appears the most mobile, possibly since it has less atoms packed around it. With the weak force constants used in Figs. 4 and 5, the rms fluctuations of the rings are 0.50, 0.61, 0.63, and 0.94 Å, respectively. The additional freedom of the outer ring may assist ion entry into the selectivity filter and account for the slightly larger inward currents seen in Fig. 4 C.
We have also examined how some of the other factors that determine the mechanisms of ion conduction that can be derived from Brownian dynamics change when the carbonyl groups are allowed to move. The number of ions in the selectivity filter, when the carbonyl groups are held with strong, intermediate, and weak force constants, as used to construct the current-voltage curves remain unchanged. The average numbers of resident ions in the extracellular half of the channel, excluding the region just outside of the selectivity filter ranges from 2.36 to 2.64 and remained more or less constant regardless of the force constants placed on the carbonyl groups. Fig. 6 shows where ions reside in the channel during these simulations. When the carbonyl groups are rendered virtually immobile (Fig. 6 A) or allowed to fluctuate widely by assigning weak force constants (Fig. 6 C), the histograms reveal three prominent binding sites, two in the selectivity filter (z = 11 and z = 19 Å) and another one near the inner chamber (z = 9 Å). The preferred sites where the resident ions dwell become altered when the intermediate force constants are used (Fig. 6 B). The resident ion located near the intracellular side of the selectivity filter shifts its position inward and outward by ∼3 Å, giving the appearance that there are four distinct binding sites in the filter, in addition to the one in the central chamber.
FIGURE 6.

Average number of ions in the pore with no applied potential using three different sets of the force constants. The force constants used for panels A, B, and C correspond to those used to construct Fig. 2 A. The channel is divided into 100 sections, and the average number of ions in each section is calculated over a simulation period of 0.2 μs.
Finally, the single-ion energy profile along the channel constructed while the carbonyl oxygens undergo rms fluctuations of 0.61 Å is broadly similar in shape to that constructed while the carbonyl atoms are held less flexible (rmsd0 = 0.15 Å) or virtually rigid (rmsd0 = 0.09 Å). The three profiles are illustrated in Fig. 7. There is a small but consistent broadening of the profile near the extracellular entrance of the channel with an increase in the rms fluctuations of the carbonyl groups. To create these energy profiles, we fix the test ion on the axis of the channel at the specified z-position and calculate its average potential energy during a 0.2-μs simulation while the carbonyl atoms undergo thermal fluctuations.
FIGURE 7.

The potential energy profiles seen by a single ion traversing the pore. The three curves are obtained using three different sets of the force constants as in Fig. 2 A. A schematic outline of the channel is shown in panel A.
DISCUSSION
The potential of mean force constructed from the gramicidin pore is exquisitely sensitive to the positions of the atoms forming the pore. The rms fluctuations of the carbonyl oxygen atoms during molecular dynamics simulations vary from ∼0.42 to 0.63 Å depending on their positions (20). The free energy profile calculated from molecular dynamics simulations using the flexible structure shows two binding sites of ∼8 kT in depth near the entrances of the pore that are separated by a central barrier whose height with respect to the binding well is ∼20 kT (38). This potential of mean force is related to the force experienced by an ion as it traverses across the pore, averaged over a large number of instantaneous structural configurations (20). The central barrier increases from ∼20 kT to ∼29 kT and the well depth changes from −7 kT to +2 kT when the backbone atoms or all the peptide atoms are rendered rigid (39). Although these profiles, whether they are computed from a rigid structure or from a flexible structure, do not appear to be able to reproduce observed currents (40), they highlight the need for relaxing one of the assumptions made in semimicroscopic computational methods, such as PNP theory and Brownian dynamics: that the protein remains rigid. Extrapolating from the differences observed between the profiles obtained from the flexible and rigid gramicidin structures, one would expect that semimicroscopic approaches could make errors of up to several orders of magnitude if they assume that the atoms lining the pore are rigid. It is desirable thus to extend the Brownian dynamics algorithm such that some of the atoms forming the channel conduit can be explicitly simulated.
With this aim in mind, we have introduced a method of allowing some of the atoms in the protein to undergo fluctuations in Brownian dynamics simulations. In the simulations we report, we only allowed the carbonyl oxygen and carbon atoms along the selectivity filter to move, but the method could be extended to include all the atoms lining the pore, including backbone atoms and side chains of the aspartic and glutamic residues. Doing this allows the effects of thermal fluctuations of the atoms on the dynamics of ion permeation to be uncovered. If thermal fluctuations are found to have little effect on permeation dynamics, then this may support the use of rigid structures, provided that appropriate average structures are available. If not, future semimicroscopic simulations should aim to incorporate the important degrees of protein motion.
Without detailed investigation, it is not obvious whether the motion of the charged or polar residues forming the ion-conducting pathway will have discernible effects on the channel conductance. Many dynamical systems occurring in nature are composed of two parts: one that is moving very quickly relative to other parts of the systems that are moving slowly. The behavior of any such system can be rigorously approximated with averaging theory (12–14). These results show that the parts of the system moving on the fast timescale perceive the slowly moving portions as being constant, and the slowly moving portions only see a time-averaged effect from the fast timescale parts. Thus, in accord with the ergodicity theorem, the time average in a large system will converge to, and can be replaced with, the configuration average weighted by the Boltzmann factor (41).
It is not entirely clear, however, if the results of the average theory can be invoked for the dynamics of ion permeation in biological ion channels. Permeant ions are generally trapped in steep energy wells formed by charged residues. Intuitively, estimations from a model in which all the distant atoms are fixed rigidly should correspond with those obtained from molecular dynamics. Thermal fluctuations of distant atoms in the protein are unlikely to have discernible influences on the processes of ion permeation across the pore. A conduction event in an ion channel typically takes place at about five to six orders of magnitude slower timescale compared to the characteristic atomic oscillation periods (19). On the other hand, the forces acting on the ions and those acting on the protein are approximately of the same magnitude, thus indicating that the fluctuations of the atoms in the protein are of the same order of magnitude as those of ions in the pore.
In contrast to the gramicidin pore, the energy profile derived from the KcsA potassium channel with flexible carbonyl oxygens lining the selectivity filter does not differ appreciably from that obtained with the carbonyl atoms fixed rigidly at their initial positions (Fig. 7). The motion of the resident ion does not influence the motion of the carbonyl oxygen atoms lining the pore in any significant way (Fig. 3). Also, the average number of ions in the selectivity filter and the wide chamber remains unchanged (Fig. 6). Two resident ions occupy the selectivity filter in a stable equilibrium. When this equilibrium is disrupted by the entry of a third ion, the outermost ion is expelled instantaneously from the narrow filter (42). The slope conductance estimated when the carbonyl groups are rigidly held is 23% lower than those obtained when the intermediate and weak force constants (Fig. 4). The current-concentration curves, not shown here, obtained using the three different sets of force constants, are also broadly similar. It is possible that the lack of correlation observed in the Brownian dynamics simulations may influence these results, but one might expect that the inclusion of correlated motion would act to keep the ion-oxygen distances more constant, and thus the results would be closer to those observed using a fixed protein structure. Thus, at least for the KcsA channel, the salient conclusions drawn about the mechanisms of ion permeation obtained with a rigid protein in Brownian dynamics remain valid.
In this context, we note that, although the single-ion energy profile shows a smooth well (Fig. 7), its depth is such that the channel will hold multiple ions. The presence of multiple ions constrained in the well and their mutual interactions lead to the preferred positions at which the ions reside, as shown in the dwell histograms illustrated in Fig. 6. The results illustrated here are broadly similar to those seen in previous simulations with a rigid protein (4). It would be of interest to carry out more detailed analysis of the occupancy of ions in the channel and the effective volumes statistically occupied by ions in each well. From such an analysis, it should be possible to calculate a multiion free energy profile and deduce the entropic and enthalpic contributions to it. There is, however, one potential difficulty in carrying out such an analysis and making inferences about the enthalpic interactions of ions with the oxygens. The potential energy we calculate based on macroscopic electrostatics implicitly include entropic contributions from the water surrounding ions and proteins. The extent of the contributions made by the implicit water has to be estimated to arrive at the true figure for the entropy.
The approach developed here should be useful for examining other ion channels in which the motions of side chains or backbone atoms lining the pore are believed to influence ion conduction. By incorporating some important degrees of protein flexibility into mesoscopic simulations, we are able to examine their effect on observable properties such as the channel current. Some degree of caution must be exercised, however, as the user has to identify the protein motions they are interested in before conducting simulations. Also, there is no clear a priori way of determining the force constants that should be used when only a limited number of atoms are treated in this way; thus, these must be altered such that the motion of the mobile protein atoms approximates that seen in more detailed simulations.
Acknowledgments
Matthew Hoyles provided helpful advice and suggestions throughout the course of this study, for which we are grateful.
This work was supported by grants from the National Health & Medical Research Council of Australia.
Editor: Peter Tieleman.
References
- 1.Eisenberg, R. S. 1996. Computing the field in proteins and channels. J. Membr. Biol. 150:1–25. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg, R. S. 1998. Ionic channels in biological membranes: electrostatic analysis of a natural nano-tube. Comtemp. Phys. 39:447–466. [Google Scholar]
- 3.Chung, S. H., M. Hoyles, T. W. Allen, and S. Kuyucak. 1998. Study of ionic currents across a model membrane channel using Brownian dynamics. Biophys. J. 77:793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung, S. H., T. W. Allen, and S. Kuyucak. 2002. Conducting-state properties of the KcsA potassium channel from molecular and Brownian dynamics simulations. Biophys. J. 82:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crouzy, S., T. B. Woolf, and B. Roux. 1994. A molecular dynamics study of gating in dioxolane-linked gramicidin A channels. Biophys. J. 67:1370–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tieleman, D. P., and H. J. C. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 74:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moy, G., B. Corry, S. Kuyucak, and S. H. Chung. 2000. Tests of continuum theories as models of ion channels. I. Poisson-Boltzmann theory versus Brownian dynamics. Biophys. J. 78:2349–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corry, B., S. Kuyucak, and S. H. Chung. 2000. Tests of continuum theories as models of ion channels. II. Poisson-Nernst-Planck theory versus Brownian dynamics. Biophys. J. 78:2364–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corry, B., S. Kuyucak, and S. H. Chung. 2003. Dielectric self-energy in Poisson-Boltzmann and Poisson-Nernst-Planck models of ion channels. Biophys. J. 84:3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crozier, P. S., R. L. Rowley, N. B. Holladay, D. Hendersen, and D. D. Busath. 2001. Molecular dynamics simulation of continuous current flow through a model biological membrane channel. Phys. Rev. Lett. 86:2467–2470. [DOI] [PubMed] [Google Scholar]
- 11.Askimentiev, A., and K. Schulten. 2005. Imaging α-hemolysin with molecular dynamics: ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 88:3745–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kushner, H. 1984. Approximation and Weak Convergence Methods for Random Processes, with Applications to Stochastic Systems Theory. MIT Press, Cambridge, MA.
- 13.Sanders, J. A., and F. Verhulst. 1985. Averaging Methods in Nonlinear Dynamics Systems. Springer Verlag, New York.
- 14.Kushner, H. 1997. Stochastic Approximation Algorithms and Applications. Springer-Verlag, New York.
- 15.Allen, T. W., S. Kuyucak, and S. H. Chung. 1999. The effect of hydrophobic and hydrophilic channel walls on the structure and diffusion of water and ions. J. Chem. Phys. 111:7985–7999. [Google Scholar]
- 16.Im, W., and B. Roux. 2002. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 322:851–869. [DOI] [PubMed] [Google Scholar]
- 17.Noskov, S. Yu., W. Im, and B. Roux. 2004. Ion permeation through the α-homolysin channel: theoretical studies based on Brownian dynamics and Poisson-Nernst-Planck electrodiffusion theory. Biophys. J. 87:2299–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen, T. W., A. Bliznyuk, A. P. Rendell, S. Kuyucak, and S. H. Chung. 2000. The potassium channel: structure, selectivity and diffusion. J. Chem. Phys. 112:8191–8204. [Google Scholar]
- 19.Feenstra, K. A., B. Hess, and J. C. Berendsen. 1999. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comp. Chem. 20:786–798. [DOI] [PubMed] [Google Scholar]
- 20.Allen, T. W., O. S. Andersen, and B. Roux. 2004. On the importance of atomic fluctuations, protein flexibility, and solvent in ion permeation. J. Gen. Physiol. 124:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noskov, S. Yu., S. Bernéche, and B. Roux. 2004. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 431:830–834. [DOI] [PubMed] [Google Scholar]
- 22.Corry, B., T. W. Allen, S. Kuyucak, and S. H. Chung. 2001. Mechanisms of permeation and selectivity in calcium channels. Biophys. J. 80:195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corry, B., and S. H. Chung. 2006. Mechanisms of valence selectivity in biological ion channels. Cell. Mol. Life Sci. 63:301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellinor, P. T., J. Yang, W. A. Sather, J.-F. Zhang, and R. W. Tsien. 1995. Ca2+ channel selectivity at a single locus for high-affinity Ca2+ interactions. Neuron. 15:1121–1132. [DOI] [PubMed] [Google Scholar]
- 25.van Gunsteren, W. F., and H. J. C. Berendsen. 1982. Algorithms for Brownian dynamics. Mol. Phys. 45:637–647. [Google Scholar]
- 26.Hoyles, M., S. Kuyucak, and S. H. Chung. 1996. Energy barrier presented to ions by the vestibule of the biological membrane channel. Biophys. J. 70:1628–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoyles, M., S. Kuyucak, and S. H. Chung. 1998. Computer simulation of ion conductance in membrane channels. Phys. Rev. E. 58:3654–3661. [Google Scholar]
- 28.Li, S. C., M. Hoyles, S. Kuyucak, and S. H. Chung. 1998. Brownian dynamics study of ion transport in the vestibule of membrane channels. Biophys. J. 74:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guàrdia, E., R. Rey, and J. A. Padró. 1991. Potential of mean force by constrained molecular dynamics: a sodium chloride ion-pair in water. Chem. Phys. 155:187–195. [Google Scholar]
- 30.Guàrdia, E., R. Rey, and J. A. Padró. 1991. Na+-Na+ and Cl−-Cl− ion pairs in water: mean force potentials by constrained molecular dynamics. J. Chem. Phys. 95:2823–2831. [Google Scholar]
- 31.Guàrdia, E., A. Robinson, and J. A. Padró. 1993. Mean force potential for the calcium-chloride ion pair in water. J. Chem. Phys. 99:4229–4230. [Google Scholar]
- 32.Guàrdia, E., and J. A. Padró. 1996. On the influence of ionic charge on the mean force potential of ion pairs in water. J. Chem. Phys. 104:7219–7222. [Google Scholar]
- 33.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 34.Roux, B. 2005. Ion conduction and selectivity in K+ channels. Annu. Rev. Biophys. Biomol. Struct. 34:153–171. [DOI] [PubMed] [Google Scholar]
- 35.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 36.Bernéche, S., and B. Roux. 2003. A microscopic view of ion conduction through the K+ channel. Proc. Natl. Acad. Sci. USA. 100:8644–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khalili-Araghi, F., E. Tajkhorshid, and K. Schulten. 2006. Dynamics of K+ ion conduction through Kv1.2. Biophys. J. 91:L72–L74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen, T. W., T. Bastug, S. Kuyucak, and S. H. Chung. 2003. Gramicidin A as a test ground for molecular dynamics force fields. Biophys. J. 84:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bastug, T., A. Gray-Weale, S. M. Patra, and S. Kuyucak. 2006. Role of protein flexibility in ion permeation: a case study in gramicidin A. Biophys. J. 90:2285–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corry, B., and S. H. Chung. 2005. Influence of protein flexibility on the electrostatic energy landscape in gramicidin A. Eur. Biophys. J. 34:208–216. [DOI] [PubMed] [Google Scholar]
- 41.Liu, J. S. 2001. Monte Carlo Strategies in Scientific Computing. Springer, New York.
- 42.Allen, T. W., S. Kuyucak, and S. H. Chung. 1999. Molecular dynamics study of the KcsA potassium channel. Biophys. J. 77:2502–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]