

Abstract
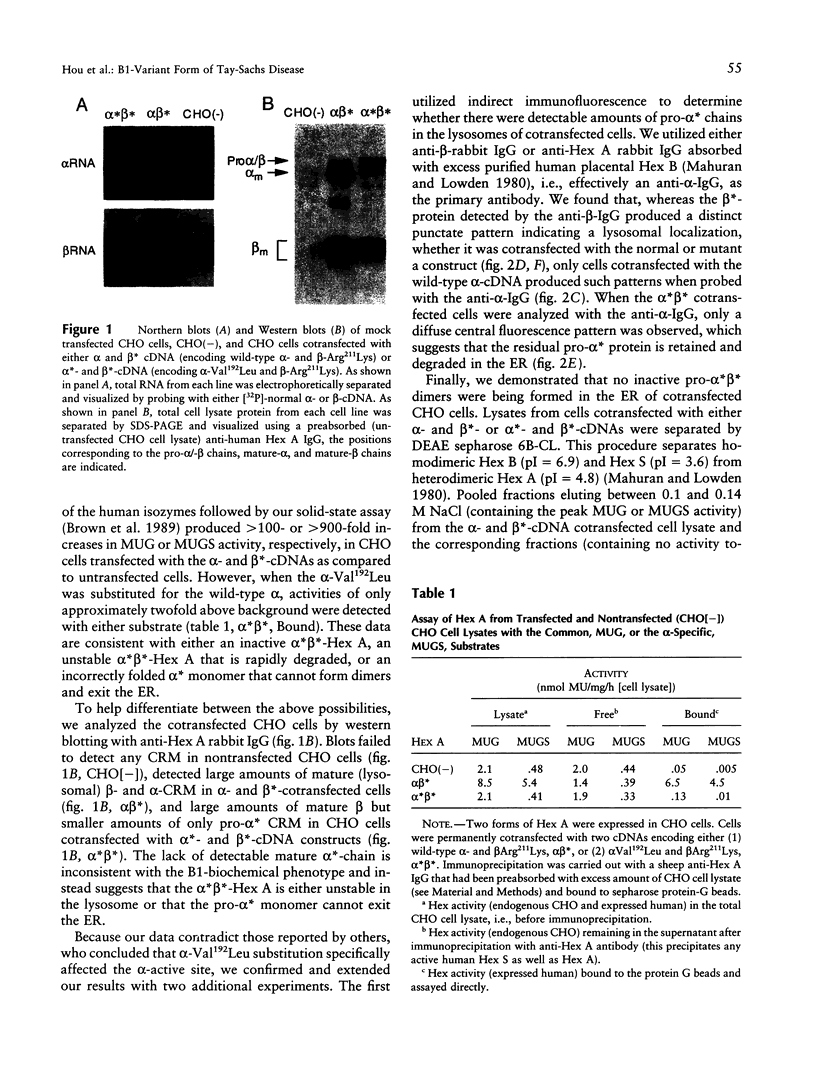
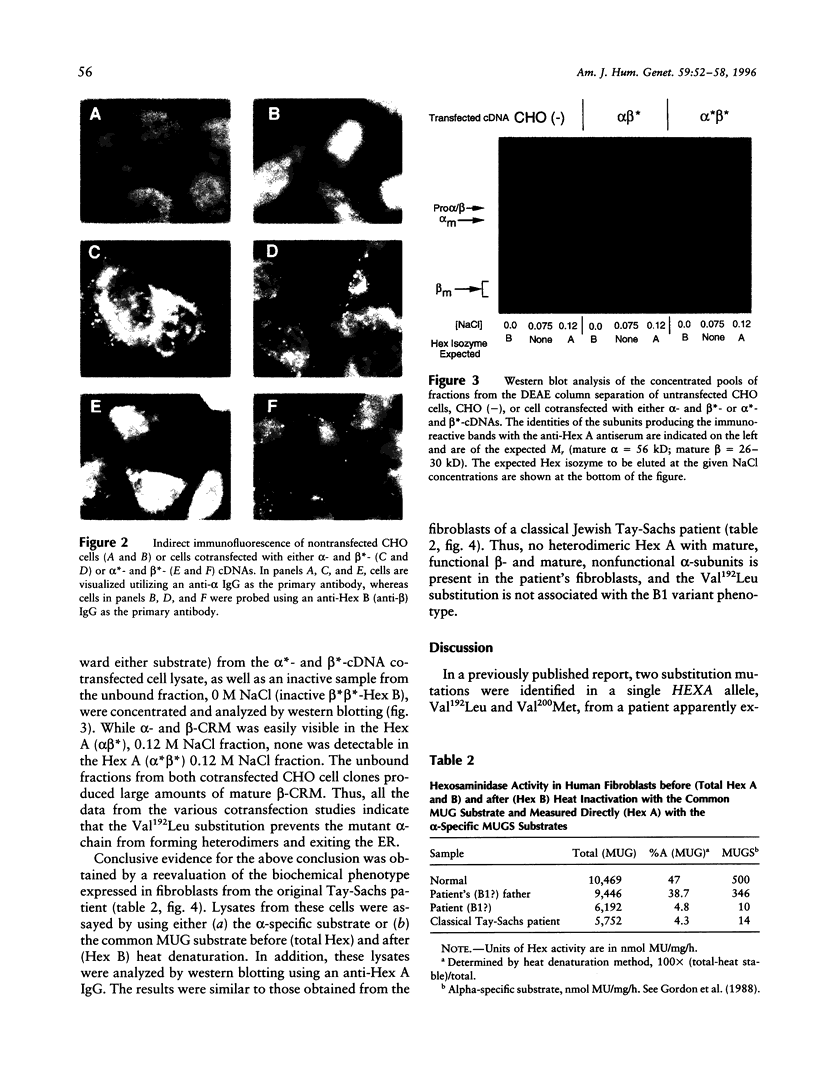
Substitution mutations adversely affecting the alpha-subunit of beta-hexosaminidase A (alphabeta) (EC 3.2.1.52) result in Tay-Sachs disease. The majority affect the initial folding of the pro-alpha chain in the endoplasmic reticulum, resulting in its retention and degradation. A much less common occurrence is a mutation that specifically affects an "active-site" residue necessary for substrate binding and/or catalysis. In this case, hexosaminidase A is present in the lysosome, but it lacks all alpha-specific activity. This biochemical phenotype is referred to as the "B1-variant form" of Tay-Sachs disease. Kinetic analysis of suspected B1-variant mutations is complex because hexosaminidase A is heterodimeric and both subunits possess similar active sites. In this report, we examine a previously identified B1-variant mutation, alpha-Val192Leu. Chinese hamster ovary cells were permanently cotransfected with an alpha-cDNA-construct encoding the substitution and a mutant beta-cDNA (beta-Arg211Lys), encoding a beta-subunit that is inactive but normal in all other respects. We were surprised to find that the Val192Leu substitution, produced a pro-alpha chain that did not form alpha-beta dimers and was not transported to the lysosome. Finally, we reexamined the hexosaminidase activity and protein levels in the fibroblasts from the original patient. These data were also not consistent with the biochemical phenotype of the B1 variant of Tay-Sachs disease previously reported to be present. Thus, we conclude that the Val192Leu substitution does not specifically affect the alpha-active site.
Full text
PDF




Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Ainsworth P. J., Coulter-Mackie M. B. A double mutation in exon 6 of the beta-hexosaminidase alpha subunit in a patient with the B1 variant of Tay-Sachs disease. Am J Hum Genet. 1992 Oct;51(4):802–809. [PMC free article] [PubMed] [Google Scholar]
- Akli S., Chelly J., Lacorte J. M., Poenaru L., Kahn A. Seven novel Tay-Sachs mutations detected by chemical mismatch cleavage of PCR-amplified cDNA fragments. Genomics. 1991 Sep;11(1):124–134. doi: 10.1016/0888-7543(91)90109-r. [DOI] [PubMed] [Google Scholar]
- Banerjee P., Boyers M. J., Berry-Kravis E., Dawson G. Preferential beta-hexosaminidase (Hex) A (alpha beta) formation in the absence of beta-Hex B (beta beta) due to heterozygous point mutations present in beta-Hex beta-chain alleles of a motor neuron disease patient. J Biol Chem. 1994 Feb 18;269(7):4819–4826. [PubMed] [Google Scholar]
- Brown C. A., Mahuran D. J. Active arginine residues in beta-hexosaminidase. Identification through studies of the B1 variant of Tay-Sachs disease. J Biol Chem. 1991 Aug 25;266(24):15855–15862. [PubMed] [Google Scholar]
- Brown C. A., Neote K., Leung A., Gravel R. A., Mahuran D. J. Introduction of the alpha subunit mutation associated with the B1 variant of Tay-Sachs disease into the beta subunit produces a beta-hexosaminidase B without catalytic activity. J Biol Chem. 1989 Dec 25;264(36):21705–21710. [PubMed] [Google Scholar]
- Coulter-Mackie M. B. Molecular characterization of both alleles in an unusual Tay-Sachs disease B1 variant. Am J Hum Genet. 1994 Jun;54(6):1126–1127. [PMC free article] [PubMed] [Google Scholar]
- Fernandes M., Kaplan F., Natowicz M., Prence E., Kolodny E., Kaback M., Hechtman P. A new Tay-Sachs disease B1 allele in exon 7 in two compound heterozygotes each with a second novel mutation. Hum Mol Genet. 1992 Dec;1(9):759–761. doi: 10.1093/hmg/1.9.759. [DOI] [PubMed] [Google Scholar]
- Gordon B. A., Gordon K. E., Hinton G. G., Cadera W., Feleki V., Bayleran J., Hechtman P. Tay-Sachs disease: B1 variant. Pediatr Neurol. 1988 Jan-Feb;4(1):54–57. doi: 10.1016/0887-8994(88)90026-4. [DOI] [PubMed] [Google Scholar]
- Hou Y., Tse R., Mahuran D. J. Direct determination of the substrate specificity of the alpha-active site in heterodimeric beta-hexosaminidase A. Biochemistry. 1996 Apr 2;35(13):3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- Kytzia H. J., Sandhoff K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J Biol Chem. 1985 Jun 25;260(12):7568–7572. [PubMed] [Google Scholar]
- Leinekugel P., Michel S., Conzelmann E., Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992 Mar;88(5):513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- Mahuran D. J. The biochemistry of HEXA and HEXB gene mutations causing GM2 gangliosidosis. Biochim Biophys Acta. 1991 Feb 22;1096(2):87–94. doi: 10.1016/0925-4439(91)90044-a. [DOI] [PubMed] [Google Scholar]
- Mahuran D., Lowden J. A. The subunit and polypeptide structure of hexosaminidases from human placenta. Can J Biochem. 1980 Apr;58(4):287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- Mahuran D., Novak A., Lowden J. A. The lysosomal hexosaminidase isozymes. Isozymes Curr Top Biol Med Res. 1985;12:229–288. [PubMed] [Google Scholar]
- McDowell G. A., Mules E. H., Fabacher P., Shapira E., Blitzer M. G. The presence of two different infantile Tay-Sachs disease mutations in a Cajun population. Am J Hum Genet. 1992 Nov;51(5):1071–1077. [PMC free article] [PubMed] [Google Scholar]
- Ohno K., Suzuki K. Mutation in GM2-gangliosidosis B1 variant. J Neurochem. 1988 Jan;50(1):316–318. doi: 10.1111/j.1471-4159.1988.tb13266.x. [DOI] [PubMed] [Google Scholar]
- Sandhoff K., Harzer K., Wässle W., Jatzkewitz H. Enzyme alterations and lipid storage in three variants of Tay-Sachs disease. J Neurochem. 1971 Dec;18(12):2469–2489. doi: 10.1111/j.1471-4159.1971.tb00204.x. [DOI] [PubMed] [Google Scholar]
- Sandhoff K. Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease. FEBS Lett. 1969 Aug;4(4):351–354. doi: 10.1016/0014-5793(69)80274-7. [DOI] [PubMed] [Google Scholar]
- Wu B. M., Tomatsu S., Fukuda S., Sukegawa K., Orii T., Sly W. S. Overexpression rescues the mutant phenotype of L176F mutation causing beta-glucuronidase deficiency mucopolysaccharidosis in two Mennonite siblings. J Biol Chem. 1994 Sep 23;269(38):23681–23688. [PubMed] [Google Scholar]
- Xie B., Wang W., Mahuran D. J. A Cys138-to-Arg substitution in the GM2 activator protein is associated with the AB variant form of GM2 gangliosidosis. Am J Hum Genet. 1992 May;50(5):1046–1052. [PMC free article] [PubMed] [Google Scholar]