

Abstract
In the collective genomes (the metagenome) of the microorganisms inhabiting the Earth’s diverse environments is written the history of life on this planet. New molecular tools developed and used for the past 15 years by microbial ecologists are facilitating the extraction, cloning, screening, and sequencing of these genomes. This approach allows microbial ecologists to access and study the full range of microbial diversity, regardless of our ability to culture organisms, and provides an unprecedented access to the breadth of natural products that these genomes encode. However, there is no way that the mere collection of sequences, no matter how expansive, can provide full coverage of the complex world of microbial metagenomes within the foreseeable future. Furthermore, although it is possible to fish out highly informative and useful genes from the sea of gene diversity in the environment, this can be a highly tedious and inefficient procedure. Microbial ecologists must be clever in their pursuit of ecologically relevant, valuable, and niche-defining genomic information within the vast haystack of microbial diversity. In this report, we seek to describe advances and prospects that will help microbial ecologists glean more knowledge from investigations into metagenomes. These include technological advances in sequencing and cloning methodologies, as well as improvements in annotation and comparative sequence analysis. More significant, however, will be ways to focus in on various subsets of the metagenome that may be of particular relevance, either by limiting the target community under study or improving the focus or speed of screening procedures. Lastly, given the cost and infrastructure necessary for large metagenome projects, and the almost inexhaustible amount of data they can produce, trends toward broader use of metagenome data across the research community coupled with the needed investment in bioinformatics infrastructure devoted to metagenomics will no doubt further increase the value of metagenomic studies in various environments.
Introduction
The vast majority of the biosphere’s genetic and metabolic diversity is currently locked up within the world’s microbial communities, containing a staggering number of yet uncharacterized microbial genomes [48, 73]. It has become well accepted that the diversity of microorganisms represented in culture collections is highly skewed toward those taxa that are amenable to growing under laboratory conditions, making our discovery of microbial genes through cultivation-dependent conventional genome sequencing equally skewed. Even with the recent success of novel and high throughput culturing strategies [30, 31, 59, 65, 67, 86], we are still unable to mimic most microbial environments sufficiently to induce growth of many environmentally relevant microbes. Recent developments in molecular detection and identification techniques have enabled us to get a glimpse of the huge diversity of the microbial world. However, these techniques have only allowed for fragmentary observations of populations and communities, and a full picture of the structure and the (putative) function of microbial communities is still lacking.
In principle, any study that addresses all the individuals of a community as a single genomic pool can be seen as an exercise in metagenomics. In this regard, the pioneering studies that first delved into microbial diversity by direct cloning of microbial DNA followed by meticulous screening for ribosomal RNA genes [47, 49] should be, and in this study are, considered the first metagenomic studies. By the application of PCR in search of 16S rRNA gene diversity [23] and later diversity in other functional genes, a much more directed interrogation of this part of the metagenome became possible. Although understandable on technical grounds, we generally lost sight of the rest of the metagenome for about a decade in our quest to zoom in on phylogenetic markers and specific functional genes of interest. The wonder of PCR indeed made molecular inventories of microbial communities routine, but biases inherent to PCR amplification and the primers used in this procedure are far from trivial [32]. In this light, it is interesting to note that advances in screening methods and sequence throughput have now made it more feasible to survey rRNA gene diversity without the help of PCR amplification, and such approaches are gaining considerable favor [40, 78].
Improvements in cloning technologies [64] and increased sequencing capacity provide new tools to gain greater access to the functional complexity of the metagenome ([4, 26, 57, 78]; Fig. 1), but how can we gain as much understanding as possible from these endeavors? The goals of researchers venturing into the microbial metagenome vary from directed product discovery to total community characterization, and the phylogenetic complexity of the environments studied can range over orders of magnitude. Likewise, methodologies vary widely in metagenomic studies, and community complexity and research goals are the clear determinants of which metagenomic approaches are most appropriate. A number of excellent reviews have highlighted the numerous breakthroughs in metagenomics [25, 33, 41, 66], and it is not our goal in this study to appraise the breadth of work in this emerging area of research. Rather, we seek to highlight recent breakthroughs in the application of metagenomic approaches to important environments, and to discuss the unique advantages and disadvantages of the various metagenomic approaches used to date. In particular, we aim to identify and evaluate research possibilities and novel approaches that hold promise to advance our ability to gain functional knowledge from pursuits in metagenomics.
Figure 1.
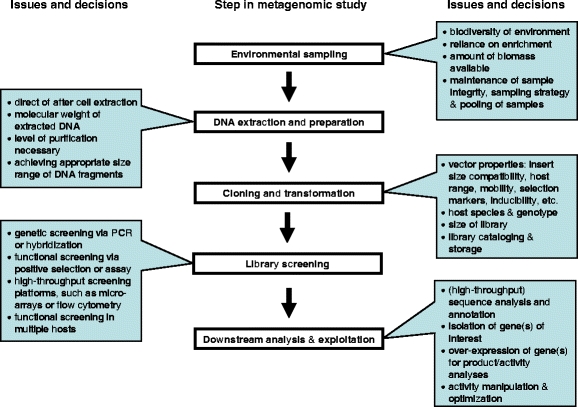
General, common steps in the metagenomic strategy are shown within boxes. Key issues and decisions relevant to each step are shown in the call-out boxes.
Techniques, Approaches, and Examples
A wide range of approaches has been employed to gain access to metagenomes (Fig. 1). The choice of strategy depends on a number of factors, including the complexity of the community, the amount of sample material available, the nature of the substrate, the density of microorganisms in a habitat, and of course the goal, scope, and resources available for the study. For purposes of this discussion, we group metagenomic studies into three classes: (1) shotgun studies that use mass genome sequencing, followed by scaffold reconstruction and gene annotation; (2) product or activity-driven studies that are designed in search of specific microbial activities and the genes encoding them; and (3) studies that attempt to link genome information with phylogenetic markers of microbial groups of interest.
Shotgun analysis of community genomes is a rather simple exercise in terms of wet science. DNA extraction protocols abound that can provide high-quality DNA for the construction of large libraries of clones containing small inserts of environmental DNA, and automated high-throughput methods are implemented to recover and sequence as many clones as necessary or resources will allow. The majority of technical challenges with shotgun metagenomic approaches come in the construction of scaffolds of sequence from vast numbers of unordered short sequences. Advances in assembly methods, stimulated by the human sequencing project, now allow for complex pools of sequences to be assembled if sufficient sequence coverage is available. This last point is most critical to this process and is directly correlated to the complexity of the community under study. Perhaps the most elegant application of community shotgun sequencing (average insert size of 3.2 kb) was presented by Tyson and colleagues [76]. In a relatively modest 100 Mb of sequence, this group was essentially able to reconstruct the genomes of the five dominant organisms composing the biofilms of the acidic mine drainage habitat at Iron Mountain, California, USA, thereby piecing together the metabolic routes of the ecosystem. This sure-to-become classic example shows that simple communities in some ways can be seen as meta-organisms, and as with individual organisms, genome determination opens the door to postgenomic studies to gain further insight into genetic networks and metabolic circuitry in an environment.
Our ability to master metagenomes decreases dramatically with increased complexity of the community, as demonstrated by the largest metagenomic study published to date [78]. In a monumental project to assess the genomic diversity of the Sargasso Sea, representing over one billion base pairs of sequence, Venter and colleagues found that reasonably large scaffolds could only be assembled for the most dominant community members, including the reconstruction of two nearly complete genomes. Clearly, complete sequencing of such environmental genomes is not an easily attainable goal. Fortunately, it can be argued that this may not be the most relevant goal, as this study exhibited the wealth of genomic information obtained via a variety of analyses into patterns of phylogenetic and functional diversity. Analyses suggested approximately 1,800 different genomic species, with a large number of novel phylotypes. Sequence annotation predicted 1.2 million new genes, including for example 782 rhodopsin-like genes affiliated with a wide range of bacterial taxa. This latter finding suggests that a large fraction of marine bacteria possess chlorophyll-independent light harvesting systems. Numerous other niche-defining genes and pathways were also detected, providing an unprecedented insight into the biogeochemistry of such marine ecosystems.
The problems associated with assembling sequences recovered from shotgun libraries from complex communities become extreme when even more diverse ecosystems are interrogated in this way, as demonstrated by Tringe et al. [74] in their analysis of a soil metagenomic library. Soil-borne microbial communities are thought to be Earth’s greatest source of biodiversity, with estimates ranging from thousands to tens of thousands of species per gram of soil [10, 72]. Indeed, nearly 140 Mb of sequence from a farmland soil revealed less than 1% of sequences showing any overlap, and produced no contigs, indicating that complete sequencing of such habitats is practically unattainable. However, Tringe et al. [74] demonstrated that such an exercise is far from futile. While obviously falling far short of providing an adequate sampling of the genetic diversity of this complex environment, this study did provide a wealth of novel genetic data, revealing hundreds of thousands of new protein-encoding genes, the vast majority of which were only distantly related to known protein sequences. Furthermore, these authors demonstrated that distribution patterns of sequence motifs and clusters of orthologous groups (COGs) of proteins [69, 70] can be used to provide functional fingerprints of environments, which can be compared across disparate habitats.
This brief synopsis of shotgun cloning approaches across a gradient of microbial diversity serves to highlight the power and limitations of such approaches as applied to different environments. As such endeavors expand to include other environments, we can expect that full community genomes will be produced from numerous low-diversity environments such as bioreactors and biofilms [60]. This information will pave the way for postgenomic studies that should help elucidate microbial interactions and pathways, allowing predictive and manipulative management of such economically relevant microbial communities. We predict that numerous genomic scaffolds will be revealed in shotgun clone investigations of important environments of intermediate diversity such as GI tracts [14, 89] and oral cavities [19]. In addition, diversity within gene families of particular relevance to these habitats should be revealed. Within high-diversity habitats such as soil, metagenomic approaches should continue to reveal novel and specialized genes (see also below) and provide comparative insight into the distribution of microbial functions across different habitats.
Product or activity-driven metagenomic studies are often approached from a more applied perspective, with the express goal to discover and exploit useful properties encoded within the metagenome [41]. Given that the majority of natural products are of microbial origin, and that the vast majority of microbial genomes have yet to be explored, it follows that microbial metagenomes contain a great economic potential. Due to their huge diversity and history as sources of commercially valuable molecules with agricultural, chemical, industrial, and pharmaceutical applications [9, 41, 42], soil environments have been the most common subjects of metagenome interrogation in this way [11].
Successful exploitation of microbial activities or metabolic pathways via a metagenomic approach requires a large number of critical steps (Fig. 2). Firstly, the target environment must contain the gene(s) encoding the activity of interest, preferably in a high frequency. Secondly, the DNA extraction and cloning methods must allow for the capture of intact genes or operons. Thirdly, the target genes must be detectable, either genetically or phenotypically. Lastly, once potential target activities have been detected, it must be possible to tailor their expression into viable production schemes. Predicting the success rate can be modeled depending on the nature of the target genes and the proportional abundance of the microorganisms harboring them [22].
Figure 2.
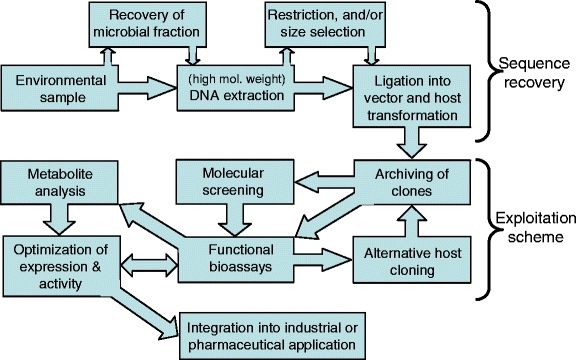
Flowchart showing the experimental steps for the exploitation of genes recovered from environmental metagenomes.
Obviously, one must first start by looking in the right kind of environment, as exemplified by Rhee et al. [55] in their search for thermostable esterases, bearing in mind that not all environments provide easy access to large microbial biomass (see below). Still, except in cases where engineered systems are known to possess high levels of an activity of interest [27], specific target genes will represent only a very small fraction of the total genomic material in environmental samples. One obvious way to stack the deck in favor of detection of a property of interest (e.g., enzyme activity) is to enrich environmental samples for its presence. Metagenomic analysis of enrichment cultures has indeed become a powerful approach to isolation of genes encoding simple functions like biocatalyst or degrading activities [17, 24, 35, 36, 79]. As with other methods that depend on growth of target populations, enrichment procedures before metagenome extraction bias samples toward populations that react particularly well to the specific enrichment conditions. This may severely restrict the diversity and novelty of the target gene pool. Many extremely useful enzyme-encoding genes may occur within populations that respond slowly to enrichment conditions, thereby being masked by potentially less-useful genes that occur within more responsive populations.
Step two in the chain toward metagenome prospecting has for the most part been solved rather well. Numerous DNA extraction and cloning methods are now available, and methods can pretty much be tailored to the sample type and the insert size desired. Insert size and expression background are the key factors when determining cloning strategy, and hinges on the size of the genomic region of interest (i.e., single genes vs full pathways) and the suspected phylogenetic range of target genomes. Choice of cloning strategy is intimately linked with the next link in the discovery chain, namely, identification of clones of interest. In theory, clones of interest can be identified by mass sequencing, where huge amounts of sequence data are examined for “potentially interesting bits” which are then studied in further detail. Alternatively, degenerate nucleotide sequences targeting conserved regions of gene families can be used to screen via various hybridization methods. These examples of screening by “forward genetics” can be effective when target genes belong to a well-defined protein family, but are generally inefficient, and can only detect potentially interesting inserts based upon homology to known motifs.
Functional screening methods potentially provide a means to discover new variants of functions of interest. The efficiency of functional screening of metagenomic libraries relies both on the efficiency and sensitivity of the assay and the compatibility of host’s transcription, translation, and modification machinery to act upon the transgenic DNA in question. Obviously, expansion of host ranges within metagenomic studies [39, 43, 81, 82], even to eukaryote hosts [2], should provide greater access to the expression of a wider range of environmental gene activities, and steps in this direction are already bearing fruit.
In the majority of studies to date, transgenic gene expression has relied on promoter elements intrinsic to the transgenic genomic material. However, the use of vectors that couple inserts to general or specific promoters has also come forward as a useful and highly directed means of probing the metagenome for microbial activities. An example is Substrate-Induced Gene Expression (SIGEX) screening [77, 85]. This novel method clones environmental DNA into GFP-tagged vectors, and libraries are subsequently subjected to the target substrate of interest. Clones expressing GFP in the presence of the target substrate are then sorted and collected by FACS for further cultivation and analysis. This procedure allows one to zoom in on activities that are related to particular substrates or catabolic pathways of interest. Recent advances in vector systems and knowledge of promoter systems are adding to the potential of such directed approaches to functional gene discovery. Several flow cytometric methods have also been devised to examine large metagenomic libraries for activities that can be detected by fluorescent assays (see Diversa patents US 5958672 and 6872526-B2), promising more rapid interrogation of metagenomic libraries for sequences and activities of interest.
A final hurdle in realizing the potential of genes recovered from metagenomic libraries is obtaining high-level expression and incorporation into viable industrial processes. Continued effort to improve well-controlled high-expression systems remains an open research area. Many microbe-derived activities are still less than optimal for implementation in industrial processes. Directed evolution and selection methods [15, 16] are providing fascinating and promising results that may allow researches to mold enzymatic activities to fill their specific needs.
Phylogenetic and large-insert metagenomic approaches provide access to genetic information contained within microbial populations only known to us in the form of specific phylogenetic marker gene sequences [57]. The general strategy is to use 16S rRNA gene markers as phylogenetic handles to identify genomic fragments from not-yet-cultured populations of interest from large-insert libraries [25]. The already classic example of this strategy is the discovery of proteorhodopsin within a genomics fragment belonging to a SAR86 population [4]. The discovery of this niche-defining gene led to further, far-reaching inferences concerning the diversity and extent of phototrophy in the world’s oceans [3], and it serves as the ecological poster child of metagenomics success.
Similar strategies have now been successful in providing insight into other not-yet-cultured organisms including uncultured Acidobacteria [40] and Archaea [51]. Although these successes provide us glimpses into novel genomes, it requires a combination of insight and pure luck to define niches based upon relatively short stretches of genomics information. Indeed, in silico exercises using complete genome sequences can easily demonstrate that it is usually impossible to infer the niche of an organism based upon the 1–2% of the genome adjacent to an rRNA operon. A number of approaches may allow us to glean more functional information from such exercises: (1) Using genes toward the ends of marker-containing inserts as markers for further interrogation of clone libraries would allow one to detect adjacent inserts, thereby expanding the contiguous chromosomal region investigated. Although this sounds highly attractive, the use of such a strategy may only be practical where the target populations represent a considerable proportion of the total community; (2) Using known functional genes of interest instead of phylogenetic markers may provide a more direct route to the discovery of gene clusters of related function. In many cases, prokaryotic phenotypes are the result of the concerted effort of many genes that are often arranged into adjacent operons or super-operonic clusters. Thus, by targeting known genes central to complex phenotypes, the entire metabolic pathway of interest can be captured [25, 56]; (3) Many niche-determining microbial activities reside on relatively mobile genetic elements. Strategies targeting the so-called mobilome [21] provide a means of focusing in an especially interesting subset of microbial activities [44, 45, 68].
As above, a limiting factor in such approaches is our ability to screen libraries for markers or activities of interest, and screening strategies include PCR-based methods, hybridization [38], and several novel approaches such as use of microarrays [61] and flow cytometry [46]. Given that most anchored metagenome approaches rely upon rRNA gene markers, the creation of libraries that are enriched for inserts containing these markers may also prove a useful first step in gaining access to genomic information from defined phylogenetic groups. Homing restriction enzymes may facilitate such approaches. These enzymes target relatively long recognition sites, typically unique within a bacterial genome, and I-CreI for example should theoretically ground metagenomic clones to rRNA gene operons. The prospect of custom-made homing enzymes [58] is especially exciting as these may provide a means of grounding metagenomic libraries to specific genomic sites of choice.
Gleaning Information Out of the Data: Bioformatics and Data Analysis
Metagenomic approaches have the potential to generate tremendous amounts of sequence information. However, the knowledge gleaned from such studies is not proportional to the sequencing effort involved, and it depends on the bioinformatics interpretation of the information obtained. Bioinformatics challenges are encountered at several steps of metagenome analyses, namely: (1) sequence assembly, (2) sequence annotation, and (3) broader use and analysis of metagenomic sequence information.
Algorithms for sequence reconstruction and contig formation have dramatically improved over the last couple of years but still rely to a large extent on principles used for the reconstruction of genomes from single organisms represented with a large coverage. Genome assembly is already complicated when analyzing a single cultured bacterium, and assembly becomes increasingly difficult when the total diversity and structure of a community is not known. Although community genome sequencing projects to date have managed to provide valuable insight into how patterns of sequence coverage and COG recognition can be used to glean important information from incomplete genomic sampling, further progress in this area is essential. For example, building recovered sequence information onto the scaffolds of known genomes is proving to be a highly valuable tool in trying to piece together partial genome sequences recovered from environmental samples. As community genome sequencing efforts continue and novel sequencing methods are introduced, community assembly algorithms will need to place a greater emphasis on unraveling genomic information from partial coverage of genomes and a high abundance of short sequencing reads.
In the ideal scenario, the annotation of gene sequences should depend upon recovery of a full gene sequence, the context of the gene within the genome, sequence homology genes of known function, and experimental evidence of a gene product and function. Even in the analysis of genomes from pure cultures, the last of these criteria is lacking, and assumptions are made based mostly upon sequence homology and recognized sequence motifs, as well as the assumption that past annotations are correct. However, with environmental sequences, the first two criteria are often also lacking, making reliance on pure sequence homology often tenuous at best. Clearly, gene function also relies on context, and conclusions based solely upon sequence similarities should be treated with the appropriate caution. Bearing this in mind, predictions of functional modules and domains based upon dynamic databases of gene families from sequenced genomes, as exemplified for polyketide synthase genes [84], should provide a greater degree of confidence for the annotation of genes recovered directly from the environment.
Due to the costs and infrastructure of large-scale metagenomics efforts, it is clear that such approaches are not yet available to a broad community of scientists. On the other hand, large-scale metagenome projects can produce much more data than any one group can analyze, and initial analyses are typically restricted to general trends of diversity and composition and a selected number of traits of specific interest to the researchers. Of course, recovered sequence information is made available via public databases, but this is often in a less useful form than the original datasets. Opening up metagenomic datasets for interrogation by a broader group of researchers, whose interests span a greater breadth of microbial functions, seems to be a relatively easy step that could greatly increase the understanding gleaned from large-scale metagenomics initiatives.
Practical Aspects and Coordinating Efforts
To date, there has been little broad-scale coordination in efforts to describe environmental metagenomes, and standards of resource management and curation are essentially absent. Who should choose the environments to be studied, and how should they be sampled? Who should decide the best approaches to access these metagenomes? Should cloned material be cataloged and stored, and if so, how and where? What is the most useful form of database management for recovered sequence information, and how should this be implemented? Up to now, the answers to these questions have for the most part been dictated by the specific interests and assets of the researchers spearheading individual metagenome projects. Some recent efforts have been helpful in providing the first coordination in such efforts, as exemplified by the US Department of Energy’s Genomes to Life Program and the Community Sequencing Program sponsored by the Joint Genome Institute. Not only is choice of environment important but also more coordinated funding efforts, better storage and access to cloned material, and standards of annotation and data deposition are necessary. Clearly, greater national and international cooperation in choosing and overseeing such metagenome efforts would help make large-scale metagenomic efforts more valuable, increasing their resource value to the scientific community.
What the Future May Hold
Metagenomics strategies currently followed, and the resources brought to bear in their execution, fit into the category of what might be called “sledgehammer” or “brute force” approaches. Advances in cloning, screening, and sequencing technologies have made such a rough, indirect approach possible, and continued development in these areas will no doubt increase our access to the massive amount of information encoded in uncultivated microorganisms. Still, we may never find genes or assemble genomes originating from relatively low-abundant species or organisms residing in environments with high biodiversity despite their possible keystone roles in their environment or value to man. More focused methods are clearly needed if we wish to increase the efficiency with which we can recover genomic needles of interest from the haystack of environmental microbial diversity.
Why go through the effort of producing and screening large metagenomic libraries for particular genomic fragments of interest if the organisms in question can be cultivated and subjected to genome analysis [75]? A major selling point of metagenomic approaches is that they are not restricted to only culturable microorganisms but also provide access to the “unculturable” majority of microbial communities [83]. Increasingly, the application of the term “unculturable” has proven to be incorrect for many microorganisms, as novel isolation and culturing methods are fueling a new wave of success stories in efforts to culture diverse microbes [30, 31, 54, 59, 65, 67]. Thus, many “unculturable” bacteria are more correctly probably just not-yet-cultured, and investments in culturing efforts may help to reduce the need for indirect and cumbersome metagenomic approaches.
A number of other technologies are emerging that should also help us to focus on particular microbial needles in the haystack. These include: (1) combining metagenome approaches with stable isotope probing methods to focus in on genomes of active community members, (2) increased use of methods that target mRNA to access diversity of expressed genes, (3) zooming in on small sample sizes in particular environments of interest using whole community genome amplification methods to increase DNA quantities, (4) micromanipulation of individual cells for single-cell genome sequencing, and (5) the isolation and sequence determination from single DNA molecules.
Stable isotope probing has become a powerful approach for studying subsets of microbial communities that respond to particular key substrates [52]. Molecular analysis of “heavy-labeled” fractions of microbial communities based upon on phylogenetic and functional gene markers has provided a great impetus in the quest to couple microbial identity and function. However, such methods still focus on individual genes. Application of metagenomic approaches to active fractions of microbial communities offers an obvious route to isolation of important and complex microbial activities. Potential problems in this approach include the recovery of large molecular weight DNA, if large-insert approaches are required, and the limited amount of labeled nucleic acid available for subsequent analysis. Amplification of the labeled fraction may provide a solution to this latter problem as discussed below.
Metagenomic approaches focus on genomic potential as opposed to realized activities, and a greater focus on gene expression in the environment is urgently needed. While gene expression of individual genes are providing insight into particular processes of interest [8], mRNA-based studies targeting numerous microbial activities simultaneously may hold the key to understanding the functioning of microbial consortia [7, 18]. In this respect, DNA-based metagenome studies should be coupled with environmental transcriptomics approaches to gain insight into the genes that are actually active in the environment [50].
Numerous methods (DOP-PCR, IPEP, MDA, OmniPlex) have recently been developed for the amplification of genomic DNA without knowledge of sequence content [12, 62, 71, 88]. Such whole-genome amplification strategies have typically been employed in the analysis of trace amounts of human DNA for analytical purposes [37]. However, the recent use on low-density cultures has opened up the ability to obtain genomic sequences from organisms for which extensive high-density culturing is not yet possible (i.e., genome sequencing has been performed on as little as ∼1,000 cells after MDA; [13]). Similarly, genome amplification methods hold great promise to assist in the analysis of environmental samples that lack sufficient biomass for convenient application of metagenomic methodologies. Whole-genome or metagenome amplification methods will not only allow for the analysis of low-biomass environments but will also allow for the analysis of microbial communities at scales that are more appropriate for elucidating microbial functioning. For instance, many soil processes may best be understood at the level of microbial aggregates and bioreactors at the level of individual flocs.
Taken a step further, such amplification technologies provide access to microbial genomes at the level of a single microbial cell [53, 87, 88]. The ability to gain genome sequence information from a single cell will finally fully bypass our need to culture organisms to gain access to their full genomic potential. Combining single-cell sequencing methods with in situ methods of cell identification and new techniques for the isolation and characterization of single prokaryotic cells [6, 20] presents the possibility of examining microbial community genomes and activities one cell at a time. Why put all the genomes of an ecosystem into a mixer and try to piece the genomes back together again afterward when genomic information can be directly obtained from the individual community members? Such methods will not only open the door to the study of individual cells belonging to phylogenetic groups that are resistant to culturing methods and/or that occur at low frequencies, but will also provide a means of conducting bacterial population biology [63].
As with other methods, such amplification methods also carry a number of potential drawbacks, especially biases introduced by selective amplification [12], production of relatively short DNA fragments, and risks of contamination. Although whole-genome amplification methods provide access to the vast majority of genomic DNA present, and methods are being improved [28], amplification bias will remain an issue for the foreseeable future in the application of such procedures to environmental DNA. The production of relatively short fragments hampers the prospect of recovery of intact genes or operons although methods for larger fragment recovery upon amplification are becoming available [34]. Whole-genome amplification methods, especially the Multiple Displacement Amplification (MDA) method [1, 12, 28], are superior to PCR-based method in recovering large DNA fragments from very limited amount of materials. However, when applied to single cells, the issue of background amplification with MDA is not trivial, as exemplified by Raghunathan et al. [53], who found up to 70% of amplicons to be contaminants. In addition, the amplification by strand displacement creates a complex, repeated forked structure of DNA that may hamper downstream manipulations [80]. Most recently, two methods have been developed to reduce background amplification: one based on nanoliter-scale reaction volumes [29], and the other involving careful experimental procedures coupled by real-time monitoring of amplification kinetics [87]. With these improvements in background amplification, as well as a new sequencing library construction protocol to deal with the unusual hyperbranched DNA structures generated by MDA, Zhang et al. [87] demonstrated amplification of single Prochlorococcus cells and recovered approximately two-thirds of the genome at the sequencing depth of 3.5 ∼ 4.7×. Due to amplification bias on single-cell amplifications, it was estimated that a sequencing depth of ∼15× would be required to recover 90% of the genome, with the filling of remaining gaps best dealt with via PCR-based methods. Nevertheless, this study represents a significant technological advance in obtaining genome information from single cells in environmental samples without lab culturing. Further developments, such as reducing amplification bias and improving sequencing coverage, as well as implementation of high-throughput screening platforms, are required to tackle the highly complex microbial communities in the environment. Before the single-cell genome sequencing method can be robustly and cost-effectively implemented in regular research labs, metagenomic sequencing will remain as an attractive complementary method in the coming years.
Recent technological advances indicate that the analysis of small nucleic acid samples can be taken to the extreme, namely, single DNA or perhaps even RNA molecules [5]. Single molecule sequencing technologies are not yet applicable to the study of environmental samples but, if rendered feasible, hold the potential to open the door to microbial community genomics at the subcellular level.
Conclusions
Metagenomic approaches offer the unique ability to examine directly the genomic content of microbial communities, and recent advances in cloning, sequencing, and screening technologies are rapidly increasing the speed and efficiency with which community genomes can be analyzed. However, the immense microbial diversity of this planet precludes a simple strategy of sequencing everything, and clever choices and coordination in environment selection, screening methods, and data analysis will be key to deriving maximal knowledge and utility from available resources. The greatest advances in accessing community genome pools will probably come not from course improvements in metagenome library construction, but rather in methods to interrogate metagenomes for important microbial functions. Despite the hype of metagenomic approaches, emerging technologies and a revival in culturing efforts may make metagenomic approaches unnecessary in many cases. Thus, while metagenomic approaches can provide unique and unprecedented glimpses into microbial community function, they should not be seen as a means in and of themselves, but rather one impressive tool within the integrated approaches becoming available to tackle the diversity of Earth’s microbial functions.
Acknowledgments
The synthesis of this manuscript was initiated during the MicroEnGen workshops on microbial environmental genomics http://www.nioo.knaw.nl/PROJECTS/microengen/) which is part of the scientific program of the Scientific Committee on Problems in the Environment (SCOPE, http://www.icsu-scope.org/). The authors thank the other metagenomics discussion group members, Jan Dirk van Elsas, John Heidelberg, Eddie Rubin, Jose de la Torre, Liz Wellington, and Hongxun Zhang, for their valuable input. Publication 3984, NIOO-KNAW Netherlands Institute of Ecology.
References
- 1.Abulencia, CB, Wyborski, DL, Garcia, JA, Podar, M, Chen, W, Chang, SH, Chang, HW, Watson, D, Brodie, EL, Hazen, TC, Keller, M (2006) Environmental whole-genome amplification to access microbial populations in contaminated sediments. Appl Environ Microbiol 72: 3291–3301 [DOI] [PMC free article] [PubMed]
- 2.Al-Hasani, K, Simpfendorfer, K, Wardan, H, Vadolas, J, Zaibak, F, Villain, R, Ioannou, PA (2003) Development of a novel bacterial artificial chromosome cloning system for functional studies. Plasmid 49: 184–187 [DOI] [PubMed]
- 3.Beja, O, Spudich, EN, Spudich, JL, Leclerc, M, DeLong, EF (2001) Proteorhodopsin phototrophy in the ocean. Nature 411: 786–789 [DOI] [PubMed]
- 4.Beja, O, Suzuki, MT, Koonin, EV, Aravind, L, Hadd, A, Nguyen, LP, Villacorta, R, Amjadi, M, Garrigues, C, Jovanovich, SB, Feldman, RA, DeLong, EF (2000) Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ Microbiol 2: 516–529 [DOI] [PubMed]
- 5.Braslavsky, I, Hebert, B, Kartalov, E, Quake, SR (2003) Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci USA 100: 3960–3964 [DOI] [PMC free article] [PubMed]
- 6.Brehm-Stecher, BF, Johnson, EA (2004) Single-cell microbiology: tools, technologies and applications. Microbiol Mol Biol Rev 68: 538–559 [DOI] [PMC free article] [PubMed]
- 7.Brzostowicz, PC, Walters, DM, Thomas, SM, Nagarajan, V, Rouviere, PE (2003) mRNA differential display in a microbial enrichment culture: simultaneous identification of three cyclohexanone monooxygenases from three species. Appl Environ Microbiol 69: 334–342 [DOI] [PMC free article] [PubMed]
- 8.Burgmann, H, Widmer, F, Sigler, WV, Zeyer, J (2003) mRNA extraction and reverse transcription-PCR protocol for detection of nifH gene expression by Azotobacter vinelandii in soil. Appl Environ Microbiol 69: 1928–1935 [DOI] [PMC free article] [PubMed]
- 9.Courtois, S, Cappellano, CM, Ball, M, Francou, FX, Normand, P, Helynck, G, Martinez, A, Kolvek, SJ, Hopke, J, Osburne, MS, August, P, Nalin, R, Guerineau, M, Jeannin, P, Simonet, P, Perdonet, JL (2003) Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol 69: 49–55 [DOI] [PMC free article] [PubMed]
- 10.Curtis, TP, Sloan, WT, Scannell, JW (2002) Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA 99: 10494–10499 [DOI] [PMC free article] [PubMed]
- 11.Daniel, R (2005) The metagenomics of soil. Nat Rev Microbiol 3: 470–478 [DOI] [PubMed]
- 12.Dean, FB, Hosono, S, Fang, L, Wu, X, Faruqi, AF, Bray-Ward, P, Sun, Z, Zong, Q, Du, Y, Du, J, Driscoll, M, Song, W, Kingsmore, SF, Egholm, M, Lasken, RS (2002) Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA 99: 5261–5266 [DOI] [PMC free article] [PubMed]
- 13.Detter, JC, Jett, JM, Lucas, SM, Dalin, E, Arellano, AR, Wang, M, Nelson, JR, Chapman, J, Lou, YI, Rokhsar, D, Hawkins, TL, Richardson, PM (2002) Isothermal strand-displacement amplification applications for high-throughput genomics. Genomics 80: 691–698 [DOI] [PubMed]
- 14.Eckburg, PB, Bik, EM, Bernstein, CN, Purdom, E, Dethlefsen, L, Sargent, M, Gill, SR, Nelson, KE, Relman, DA (2005) Diversity of the human intestinal microbial flora. Science 308: 1635–1638 [DOI] [PMC free article] [PubMed]
- 15.Eggert, T, Funke, SA, Rao, NM, Acharya, P, Krumm, H, Reetz, MT, Jaeger, KE (2005) Multiplex-PCR-based recombination as a novel high-fidelity method for directed evolution. Chembiochem 6: 1062–1067 [DOI] [PubMed]
- 16.Eggert, T, Leggewie, C, Puls, M, Streit, W, van Pouderoyen, G, Dijkstra, BW, Jaeger, K-E (2004) Novel biocatalysts by identification and design. Biocatal Biotrans 22: 139–144
- 17.Entcheva, P, Liebl, W, Johann, A, Hartsch, T, Streit, WR (2001) Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl Environ Microbiol 67: 89–99 [DOI] [PMC free article] [PubMed]
- 18.Fleming, JT, Yao, WH, Sayler, GS (1998) Optimization of differential display of prokaryotic mRNA: application to pure culture and soil microcosms. Appl Environ Microbiol 64: 3698–3670 [DOI] [PMC free article] [PubMed]
- 19.Foster, JS, Palmer, RJ-Jr, Kolenbrander, PE (2003) Human oral cavity as a model for the study of genome–genome interactions. Biol Bull 204: 200–204 [DOI] [PubMed]
- 20.Fröhlich, J, König, H (2000) New techniques for the isolation of single prokaryotic cells. FEMS Microb Rev 24: 567–572 [DOI] [PubMed]
- 21.Frost, LS, Leplae, R, Summers, AO, Toussaint, A (2005) Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 3: 722–732 [DOI] [PubMed]
- 22.Gabor, EM, Alkema, WBL, Janssen, DB (2004) Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol 6: 879–886 [DOI] [PubMed]
- 23.Giovannoni, SJ, Britschgi, TB, Moyer, CL, Field, KG (1990) Genetic diversity in Sargasso Sea bacterioplankton. Nature 345: 60–63 [DOI] [PubMed]
- 24.Gupta, R, Beg, QK, Lorenz, P (2002) Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Microbiol Biotechnol 59: 15–32 [DOI] [PubMed]
- 25.Handelsman, J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68: 669–684 [DOI] [PMC free article] [PubMed]
- 26.Handelsman, J, Rondon, MR, Brady, SF, Clardy, J, Goodman, RM (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. J Biol Chem 5: R245–R249 [DOI] [PubMed]
- 27.Healy, FG, Ray, RM, Aldrich, HC, Wilkie, AC, Ingram, LO, Shanmugam, KT (1995) Direct isolation of functional genes encoding cellulases from the microbial consortia in a thermophilic, anaerobic digester maintained on lignocellulose. Appl Microbiol Biotechnol 43: 667–674 [DOI] [PubMed]
- 28.Hosono, S, Faruqi, AF, Dean, FB, Du, YF, Sun, ZY, Wu, XH, Du, J, Kingsmore, SF, Egholm, M, Lasken, RS (2003) Unbiased whole-genome amplification directly from clinical samples. Genome Res 13: 954–964 [DOI] [PMC free article] [PubMed]
- 29.Hutchison, CA, III, Smith, HO, Pfannkoch, C, Venter, JC (2005) Cell-free cloning using {phi}29 DNA polymerase. Proc Natl Acad Sci USA 102: 17332–17336 [DOI] [PMC free article] [PubMed]
- 30.Janssen, PH, Yates, PS, Grinton, BE, Taylor, PM, Sait, M (2002) Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl Environ Microbiol 68: 2391–2396 [DOI] [PMC free article] [PubMed]
- 31.Kaeberlein, T, Lewis, K, Epstein, SS (2002) Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296: 1127–1129 [DOI] [PubMed]
- 32.Kanagawa, T (2003) Bias and artifacts in multitemplate polymerase chain reactions (PCR). J Biosci Bioeng 96: 317–323 [DOI] [PubMed]
- 33.Keller, M, Zengler, K (2004) Tapping into microbial diversity. Nat Rev Microbiol 2: 141–150 [DOI] [PubMed]
- 34.Kittler, R, Stoneking, M, Kayser, M (2002) A whole genome amplification method to generate long fragments from low quantities of genomic DNA. Anal Biochem 300: 237–244 [DOI] [PubMed]
- 35.Knietsch, A, Bowien, S, Whited, G, Gottschalk, G, Daniel, R (2003) Identification and characterization of coenzyme B12-dependent glycerol dehydratase-and diol dehydratase-encoding genes from metagenomic DNA libraries derived from enrichment cultures. Appl Environ Microbiol 69: 3048–3060 [DOI] [PMC free article] [PubMed]
- 36.Knietsch, A, Waschkowitz, T, Bowien, S, Henne, A, Daniel, R (2003) Construction and screening of metagenomic libraries derived from enrichment cultures: Generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl Environ Microbiol 69: 1408–1416 [DOI] [PMC free article] [PubMed]
- 37.Lasken, RS, Egholm, M (2003) Whole genome amplification: abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol 21: 531–535 [DOI] [PubMed]
- 38.Leveau, JHJ, Gerards, S, de Boer, W, van Veen, JA (2004) Phylogeny-function analysis of (meta)genomic libraries: screening for expression of ribosomal RNA genes by large-insert library fluorescent in situ hybridization (LIL-FISH). Environ Microbiol 6: 990–998 [DOI] [PubMed]
- 39.Li, Y, Wexler, M, Richardson, DJ, Bond, PL, Johnston, AWB (2005) Screening a wide host-range, waste-water metagenomic library in tryptophan auxotrophs of Rhizobium leguminosarum and of Escherichia coli reveals different classes of cloned trp genes. Environ Microbiol 7: 1927–1936 [DOI] [PubMed]
- 40.Liles, MR, Manske, BF, Bintrim, SB, Handelsman, J, Goodman, RM (2003) A census of rRNA genes and linked genomic sequences within a soil metagenomic library. Appl Environ Microbiol 69: 2684–2691 [DOI] [PMC free article] [PubMed]
- 41.Lorenz, P, Eck, J (2005) Metagenomics and industrial applications. Nat Rev Microbiol 3: 510–516 [DOI] [PubMed]
- 42.MacNeil, IA, Tiong, CL, Minor, C, August, PR, Grossman, TH, Loiacono, KA, Lynch, BA, Phillips, T, Narula, S, Sundaramoorthi, R, Gilman, M, Holt, D, Osburne, MS (2001) Expression and isolation of antimicrobial small molecules from soil DNA libraries. J Mol Microbiol Biotechnol 3: 301–308 [PubMed]
- 43.Martinez, A, Kolvek, SJ, Yip, CLT, Hopke, J, Brown, KA, MacNeil, IA, Osburne, MS (2004) Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 70: 2452–2463 [DOI] [PMC free article] [PubMed]
- 44.Nield, BS, Willows, RD, Gillings, MR, Holmes, AJ, Nevalainen, KMH, Stokes, HW, Mabbutt, BC (2004) New enzymes from environmental cassette arrays: functional attributes of a phosphotransferase and a RNA-methyltransferase. Protein Sci 13: 1651–1659 [DOI] [PMC free article] [PubMed]
- 45.Neild, BS, Holmes, AJ, Gillings, MR, Recchia, GD, Mabbutt, BC, Nevalainen, KMH, Stokes, HW (2001) Recovery of new integron classes from environmental DNA. FEMS Microbiol Lett 195: 59–65 [DOI] [PubMed]
- 46.Nielsen, JL, Schramm, A, Bernhard, AE, van den Engh, GJ, Stahl, DA (2004) Flow cytometry-assisted cloning of specific sequence motifs from complex 16S rRNA gene libraries. Appl Environ Microbiol 70: 7550–7554 [DOI] [PMC free article] [PubMed]
- 47.Olsen, GJ, Lane, DJ, Giovannoni, SJ, Pace, NR, Stahl, DA (1986) Microbial ecology and evolution: a ribosomal RNA approach. Annu Rev Microbiol 40: 337–365 [DOI] [PubMed]
- 48.Pace, NR (1997) A molecular view of microbial diversity and the biosphere. Science 276: 734–740 [DOI] [PubMed]
- 49.Pace, NR, Stahl, DA, Lane, DJ, Olsen, GJ (1986) The analysis of natural microbial populations by ribosomal RNA sequences. Adv Microb Ecol 9: 1–55
- 50.Poretsky, RS, Bano, N, Buchan, A, LeCleir, G, Kleikemper, J, Pickering, M, Pate, WM, Moran, MA, Hollibaugh, JT (2005) Analysis of microbial gene transcripts in environmental samples. Appl Environ Microbiol 71: 4121–4126 [DOI] [PMC free article] [PubMed]
- 51.Quaiser, A, Ochsenreiter, T, Klenk, HP, Kletzin, A, Treusch, AH, Meurer, G, Eck, J, Sensen, CW, Schleper, C (2002) First insight into the genome of an uncultivated crenarchaeote from soil. Environ Microbiol 4: 603–611 [DOI] [PubMed]
- 52.Radajewski, S, McDonald, IR, Murrell, JC (2003) Stable-isotope probing of nucleic acids: a window to the function of uncultured microorganisms. Curr Opin Biotechnol 14: 296–302 [DOI] [PubMed]
- 53.Raghunathan, A, Ferguson, HR, Bornarth, CJ, Song, WM, Driscoll, M, Lasken, RS (2005) Genomic DNA amplification from a single bacterium. Appl Environ Microbiol 71: 3342–3347 [DOI] [PMC free article] [PubMed]
- 54.Rappe, MS, Connan, SA, Vergin, KL, Giovannoni, SJ (2002) Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418: 630–631 [DOI] [PubMed]
- 55.Rhee, JK, Ahn, DG, Kim, YG, Oh, JW (2005) New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl Environ Microbiol 71: 817–825 [DOI] [PMC free article] [PubMed]
- 56.Riesenfeld, CS, Goodman, RM, Handelsman, J (2004) Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol 6: 981–989 [DOI] [PubMed]
- 57.Rondon, MR, August, PR, Bettermann, AD, Brady, SF, Grossman, TH, Liles, MR, Loiacono, KA, Lynch, BA, MacNeil, IA, Minor, C, Tiong, CL, Gilman, M, Osburne, MS, Clardy, J, Handelsman, J, Goodman, RM (2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66: 2541–2547 [DOI] [PMC free article] [PubMed]
- 58.Rosen, LE, Morrison, HA, Masri, S, Brown, MJ, Springstubb, B, Sussman, D, Stoddard, BL, Seligman, LM (2006) Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res. DOI dx.doi.org//10.1093/nar/gkl645 [DOI] [PMC free article] [PubMed]
- 59.Sait, M, Hugenholtz, P, Janssen, PH (2002) Cultivation of globally-distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys. Environ Microbiol 4: 654–666 [DOI] [PubMed]
- 60.Schmeisser, C, Stöckigt, C, Raasch, C, Wingender, J, Timmis, KN, Wenderoth, F, Flemming, H-C, Liesegang, H, Schmitz, RA, Jaeger, K-E, Streit, WR (2003) Metagenome survey of biofilms in drinking water networks. Appl Environ Microbiol 69: 7298–7308 [DOI] [PMC free article] [PubMed]
- 61.Sebat, JL, Colwell, FS, Crawford, RL (2003) Metagenomic profiling: microarray analysis of an environmental genomic library. Appl Environ Microbiol 69: 4927–4934 [DOI] [PMC free article] [PubMed]
- 62.Shendure, J, Mitra, RD, Varma, C, Church, GM (2004) Advanced sequencing technologies: methods and goals. Nat Rev Genet 5: 335–344 [DOI] [PubMed]
- 63.Shendure, J, Porreca, GJ, Reppas, NB, Lin, X, McCutcheon, JP, Rosenbaum, AM, Wang, MD, Zhang, K, Mitra, RD, Church, GM (2005) Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309: 1728–1732 [DOI] [PubMed]
- 64.Shizuya, H, Birren, B, Kim, U, Mancino, V, Slepak, T, Tachiiri, Y, Simon, M (1992) Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci USA 89: 8794–8797 [DOI] [PMC free article] [PubMed]
- 65.Simon, HM, Jahn, CE, Bergerud, LT, Sliwinski, MK, Weimer, PJ, Willis, DK, Goodman, RM (2005) Cultivation of mesophilic soil crenarchaeotes in enrichments from plant roots. Appl Environ Microbiol 71: 4751–4760 [DOI] [PMC free article] [PubMed]
- 66.Streit, WR, Schmitz, RA (2004) Metagenomics—the key to the uncultured microbes. Curr Opin Microbiol 7: 492–498 [DOI] [PubMed]
- 67.Stevenson, BS, Eichorst, SA, Wertz, JT, Schmidt, TM, Breznak, JA (2004) New strategies for cultivation and detection of previously uncultured microbes. Appl Environ Microbiol 70: 4748–4755 [DOI] [PMC free article] [PubMed]
- 68.Stokes, HW, Holmes, AJ, Neild, BS, Holley, MP, Nevalainen, KMH, Mabbutt, BC, Gillings, MR (2001) Gene cassette PCR: sequence-independent recovery of entire genes from environmental DNA. Appl Environ Microbiol 67: 5240–5246 [DOI] [PMC free article] [PubMed]
- 69.Tatusov, RL, Koonin, EV, Lipman, DJ (1997) A genomic perspective on protein families. Science 278: 631–637 [DOI] [PubMed]
- 70.Tatusov, RL, Fedorova, ND, Jackson, JD, Jacobs, AR, Kiryutin, B, Koonin, EV, Krylov, DM, Mazumder, R, Mekhedov, SL, Nikolskaya, AN, Rao, BS, Smirnov, S, Sverdlov, AV, Vasudevan, S, Wolf, YI, Yin, JJ, Natale, DA (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4: 41 [DOI] [PMC free article] [PubMed]
- 71.Telenius, H, Carter, NP, Bebb, CE, Nordenskjold, M, Ponder, BA, Tunnacliffe, A (1992) Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13: 718–725 [DOI] [PubMed]
- 72.Torsvik, V, Goksøyr, J, Daae, FL (1990) High diversity in DNA of soil bacteria. Appl Environ Microbiol 56: 782–787 [DOI] [PMC free article] [PubMed]
- 73.Torsvik, V, Øvreås, L, Thingstad, TF (2002) Prokaryotic diversity: magnitude, dynamics, and controlling factors. Science 296: 1064–1066 [DOI] [PubMed]
- 74.Tringe, SG, von Mering, C, Kobayashi, A, Salamov, AA, Chen, K, Chang, HW, Podar, M, Short, JM, Mathur, EJ, Detter, JC, Bork, P, Hugenholtz, P, Rubin, EM (2005) Comparative metagenomics of microbial communities. Science 308: 554–557 [DOI] [PubMed]
- 75.Tyson, GW, Banfield, JF (2005) Cultivating the uncultivated: a community genomics perspective. Trends Microbiol 13: 411–415 [DOI] [PubMed]
- 76.Tyson, GW, Chapman, J, Hugenholtz, P, Allen, EE, Ram, RJ, Richardson, PM, Solovyev, VV, Rubin, EM, Rokhsar, DS, Banfield, JF (2004) Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43 [DOI] [PubMed]
- 77.Uchiyama, T, Abe, T, Ikemura, T, Watanabe, K (2005) Substrate-induced gene-expression screening of environmental metagenomic libraries for isolation of catabolic genes. Nat Biotechnol 23: 88–93 [DOI] [PubMed]
- 78.Venter, JC, Remington, K, Heidelberg, JF, Halpern, AL, Rusch, D, Eisen, JA, Wu, DY, Paulsen, I, Nelson, KE, Nelson, W, Fouts, DE, Levy, S, Knap, AH, Lomas, MW, Nealson, K, White, O, Peterson, J, Hoffman, J, Parsons, R, Baden-Tillson, H, Pfannkoch, C, Rogers, YH, Smith, HO (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304: 66–74 [DOI] [PubMed]
- 79.Voget, S, Leggewie, C, Uesbeck, A, Raasch, C, Jaeger, KE, Streit, WR (2003) Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol 69: 6235–6242 [DOI] [PMC free article] [PubMed]
- 80.Vora, GJ, Meador, CE, Stenger, DA, Andreadis, JD (2004) Nucleic acid amplification strategies for DNA microarray-based pathogen detection. Appl Environ Microbiol 70: 3047–3054 [DOI] [PMC free article] [PubMed]
- 81.Wang, GY, Graziani, E, Waters, B, Pan, W, Li, X, McDermott, J, Meurer, G, Saxena, G, Andersen, RJ, Davies, J (2000) Novel natural products from soil DNA libraries in a streptomycete host. Org Lett 2: 2401–2404 [DOI] [PubMed]
- 82.Wexler, M, Bond, PL, Richardson, DJ, Johnston, AWB (2005) A wide host-range metagenomic library from a waste water treatment plant yields a novel alcohol/aldehyde dehydrogenase. Environ Microbiol 7: 1917–1926 [DOI] [PubMed]
- 83.Whitman, WB, Coleman, DC, Wiebe, WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95: 6578–6583 [DOI] [PMC free article] [PubMed]
- 84.Yadav, G, Gokhale, RS, Mohanty, D (2003) SEARCHPKS: a program for detection and analysis of polyketide synthase domains. Nucleic Acids Res 31: 3654–3658 [DOI] [PMC free article] [PubMed]
- 85.Yun, J, Ryu, S (2005) Screening for novel enzymes from metagenome and SIGEX as a way to improve it. Microb Cell Fact 4: 8 [DOI] [PMC free article] [PubMed]
- 86.Zengler, K, Toledo, G, Rappé, M, Elkins, J, Mathur, EJ, Keller, M (2002) Cultivating the uncultured. Proc Natl Acad Sci USA 99: 15681–15686 [DOI] [PMC free article] [PubMed]
- 87.Zhang, K, Martiny, AC, Reppas, NB, Barry, KW, Malek, J, Chisholm, SW, Church, GM (2006) Sequencing genomes from single cells via polymerase clones. Nat Biotechnol 24: 680–686 [DOI] [PubMed]
- 88.Zhang, L, Cui, X, Schmitt, K, Hubert, R, Navidi, W, Arnheim, N (1992) Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci USA 89: 5847–5851 [DOI] [PMC free article] [PubMed]
- 89.Zoetendal, EG, Collier, CT, Koike, S, Mackie, RI, Gaskins, HR (2004) Molecular ecological analysis of the gastrointestinal microbiota: a review. J Nutr 134: 465–472 [DOI] [PubMed]